



Abstract
Three-dimensional (3D) cell migration is an integral part of many physiologic processes. Although being well studied in the context of adult tissue homeostasis and cancer development, remarkably little is known about the invasive behavior of human stem cells. Using two different kinds of invasion assays, this study aimed at investigating and characterizing the 3D migratory capacity of human amniotic fluid stem cells (hAFSCs), a well-established fetal stem cell type. Eight hAFSC lines were found to harbor pronounced potential to penetrate basement membrane (BM)-like matrices. Morphological examination and inhibitor approaches revealed that 3D migration of hAFSCs involves both the matrix metalloprotease-dependent mesenchymal, elongated mode and the Rho-associated protein kinase-dependent amoeboid, round mode. Moreover, hAFSCs could be shown to harbor transendothelial migration capacity and to exhibit a motility-associated marker expression pattern. Finally, the potential to cross extracellular matrix was found to be induced by mTORC1-activating growth factors and reduced by blocking mTORC1 activity. Taken together, this report provides the first demonstration that human stem cells exhibit mTORC1-dependent invasive capacity and can concurrently make use of mesenchymal and amoeboid 3D cell migration modes, which represents an important step toward the full biological characterization of fetal human stem cells with relevance to both developmental research and stem cell-based therapy.

The array of established human stem cells contains cells of various sources, developmental stages, and differentiation capacities. Still, their effective use in stem cell-based therapies is limited. Although being first described in 2003 and meanwhile well studied, the in vivo biological function and the therapeutic application of human amniotic fluid stem cells (hAFSCs) are still elusive. This study reports on the identification and characterization of the three-dimensional migratory capacity of hAFSCs, a so far undescribed hAFSC trait which ultimately governs cell function. These data represent an important step toward the full biological characterization of fetal human stem cells with implications for research and stem cell-based therapy.
INTRODUCTION
In 2003, the first description of a highly proliferative, Oct-4-positive stem cell type in human amniotic fluid was published.1 Based on this finding, many studies thereafter confirmed the existence of human amniotic fluid stem cells (hAFSCs) and demonstrated their differentiation potential upon, for example, neurogenic, osteogenic, chondrogenic, adipogenic, hematopoietic, renal, and hepatic lineages.2-8 From the mixture of different cells contained in human amniotic fluid, hAFSCs can be isolated via c-Kit-positive immunoselection. Descending from such an isolated single hAFSC, it is possible to induce differentiation into cells of all three embryonic germ layers. But still, hAFSCs do not induce teratoma formation in SCID mice.9 On the other hand, monoclonal human hAFSCs harbor the potential to form three-dimensional (3D) multicellular aggregates, called embryoid bodies. Embryoid body formation is known to be accompanied by a decrease of stem cell marker expression and by the induction of differentiation into different lineages and is normally seen as a representative feature of pluripotent stem cells.10 Since hAFSCs do not raise ethical concerns, can efficiently be grown in vitro in monoclonal cultures, are genomically stable, are perfectly amenable to genetic modifications11 and can be differentiated into a wide variety of cell types, they form the basis for a very promising and fast growing stem cell research field. All these characteristics together with the fact that nontreated AFSCs are nontumorigenic, make them also promising candidates for safe transplantation therapies. Since human amniotic fluid also contains other progenitor cells with different committed properties, it is important to note that only c-Kit- and Oct-4-positive AFSCs established by immunoselection exhibit all the features described above.9,12,13 In the here presented study, we have exclusively included clonal cell lines representing this broadly multipotent stem cell type for which we use the definition “hAFSCs” throughout the entire manuscript.
3D migration, which is defined as the capacity to penetrate extracellular matrix (ECM) barriers, enables cells to spread within tissues, to cross basement membranes (BMs), and to enter and exit the vasculature to infiltrate neighboring tissues and distant organs. Cellular invasion by individual cells or cell clusters (collective migration) is an indispensable element during development, tissue homeostasis and repair. For example, the formation of the three primary tissue layers during gastrulation depends on migration events involving several distinct cell populations. In the course of wound healing, 3D migration of immune cells and fibroblasts is essential and, for example, leukocytes extravasating from the bloodstream into the tissue find their functional localizations via navigation through the 3D environment. And importantly, 3D migration is a hallmark of cancer initiation and progression.14,15 Cells can use one or more different modes of migration. The major types of 3D migration are mesenchymal, amoeboid, and lobopodial, of which the latter can be described as a hybrid between mesenchymal and amoeboid. 3D migration following the mesenchymal mode depends on matrix metalloprotease (MMP)-mediated proteolytic matrix degradation and is characterized by an elongated cell shape with membrane protrusions. In case of the Rho-associated protein kinase (ROCK)-dependent, MMP-independent amoeboid 3D migration, rounded or ellipsoid cells slide through matrix pores with little or no adhesion to the matrix.14-18 Despite its enormous relevance for cell fate and function, the question regarding the invasive properties of human stem cells has remained mainly unaddressed so far.
In mammalian cells, the serine/threonine kinase mammalian target of Rapamycin (mTOR) exhibits its activity as part of two protein kinase complexes, named mTORC1 and mTORC2. mTORC1 consists of the mTOR protein, the kinase domain stabilizing mammalian lethal with SEC13 protein 8 (mLST8) and the scaffold protein regulatory-associated protein of mTOR (RAPTOR), which controls the subcellular localization and substrate recruitment of mTORC1. In this protein complex, DEP-domain-containing mTOR-interacting protein (DEPTOR) and proline-rich AKT substrate 40 kDA (PRAS40) negatively regulate mTORC1 activity. mTORC1 activity is affected by stress, oxygen, energy, nutrients, and growth factors. mTORC1 has many different upstream regulators and downstream targets. Via its effects, for example, on the eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1), the ribosomal protein S6 kinase beta-1 (S6K1, also known as p70S6 kinase), the programmed cell death 4 protein (PDCD4), the CAD protein composed of the distinct regions carbamoyl phosphate synthetase II, aspartate transcarbamoylase and dihydroorotase, or the ribosomal protein S6, it is critically involved in the regulation of protein synthesis (translation and ribosome biogenesis) and metabolism (nucleotide synthesis and lipid synthesis). Besides, mTORC1 regulates, for example, catabolism, autophagy, cell differentiation or immune cell function.19,20 Recently, we found that human stem cells regulate the invasive potential of somatic cells via paracrine activation of mTORC1 to induce hypoxia inducible factor 1-alpha (HIF-1α)-dependent MMP expression.21
In the here presented study, we show that hAFSCs exhibit mTORC1-dependent 3D migration potential and concurrently use mesenchymal and amoeboid migration modes. We discuss the implications of these findings for the full biological characterization of fetal human stem cells, the putative in vivo role of hAFSCs, and the application of hAFSCs to study the molecular bases of 3D migration modes and to develop new stem cell-based therapeutic strategies.
MATERIALS AND METHODS
Antibodies, reagents, and cellular treatments
Primary antibodies and reagents used for immunoblotting and immunofluorescence include anti-E-cadherin (BD Transduction Laboratories, US, #610181), anti-N-cadherin (CST, US, #13116), anti-vimentin (CST, #5741), anti-Snail (CST, #3879), anti-Slug (CST, #9585), anti-ZEB1 (CST, #3396), anti-MMP14 (Abcam, UK, #51074), anti-α-tubulin (Calbiochem, Merck Millipore, US, #CP06), Phalloidin (for F-Actin) (CST, #12877), anti-S6 S240/4 (CST, #5364), anti-PDCD4 (CST, #9535), anti-4E-BP1 (CST, #9644), anti-Akt S473 (CST, #4060), anti-p44/42 T202/T204 (CST, #9106), anti-GAPDH (Trevigen, US, #2275), anti-p70S6K T389 (CST, #9234), and anti-CAD S1859 (CST, #12662). Experiments with the broad spectrum MMP-inhibitor Marimastat (Tocris, UK), the ROCK-inhibitor Y27632 (Tocris), and the mTORC1-inhibitor Rapamycin (Calbiochem) were performed in the absence (dimethyl sulfoxide, DMSO, vehicle control) or presence of the drug at concentrations of 10 μM (Marimastat, Y27632) and 100 nM, respectively, unless otherwise indicated. For immunoblotting experiments involving growth factor stimulation, cells were first starved in serum-free medium for 12 to 16 hours and then treated with 10 nM insulin (Merck Millipore), 100 ng/mL insulin-like growth factor (IGF-1, Peprotech, UK), 100 ng/mL phorbol-12-myristate-13-acetate (PMA, Merck Millipore), 10% fetal bovine serum (Hyclone, US), or 100 ng/mL hepatocyte growth factor (HGF, Peprotech) for another 20 minutes before harvest.
Cell culture
hAFSC lines AFS-A1, -H1, -Q1, -CB3, -C3M6, and -C3M7 were provided by Anthony Atala and Colin Bishop (Wake Forest Institute for Regenerative Medicine, Winston-Salem, North Carolina), hAFSC lines AFS-SSC.I and -CD117/2 were established in-house and described previously.9,10,22-24 All cell lines were generated through immunoselection of c-Kit (CD117)-positive cells from human amniotic fluid via magnetic cell sorting. With the exception of AFS-CD117/2, hAFSCs represent monoclonal cell lines established through minimal dilution. AFS-SSC.I is a single-cell clone derived from AFS-CD117/2. The comprehensive characterization of the stem cell marker expression and the cellular characteristics of these hAFSC lines have been reported earlier9-11,21-24 and is summarized in Figure S1. Human AFSCs were maintained on tissue culture-treated plates in minimal essential medium (MEM)-α (Invitrogen, US) supplemented with 15% fetal bovine serum (Hyclone), 18% Chang B, 2% Chang C (both Irvine Scientific, US), and 2 mM l-glutamine (Lonza, Switzerland). Cells were grown at 37°C and 5% CO2 and were routinely passaged with trypsin-ethylenediaminetetraacetic acid (EDTA) every 2 to 3 days at a ratio of 1:3 to 1:8. To avoid spontaneous differentiation, cells were never kept too sparse nor allowed to overgrow (≥100% confluency). Continuous culture was limited to 7 days (~2-3 passages). Primary and immortalized/transformed cells analyzed in parallel include AF.I and AF.III, human amniotic fluid cells (sample nos I and III) obtained through amniocentesis performed for routine prenatal diagnosis. Amniotic fluid was provided by Gabor Joo (Semmelweis University Medical School, Budapest, Hungary); IMR-90, primary human lung fibroblasts (ATCC, US, #CCL-186); CCD-1079Sk, primary human foreskin fibroblasts (ATCC, #CRL-2097); HFF-1, primary human foreskin fibroblasts (ATCC, #SCRC-1041); HCF, primary human cardiac fibroblasts isolated from the ventricle of an adult heart (Promocell, Germany, #C-12375); HCH, primary human chondrocytes isolated from normal human articular cartilage from the femoral head (Promocell, #C-12710); NIH/3T3, immortalized mouse embryonic fibroblasts (ATCC, #CRL-1658); HT1080, human fibrosarcoma cells (ATCC, #CCL-121); MCF7, human mammary carcinoma cells (ATCC, #HTB-22); MDA-MB-231, human mammary carcinoma cells (ATCC, #HTB-26); HCC-1.2 and HCC-1.1, human hepatocellular carcinoma cells25; HUVEC, primary human umbilical vein endothelial cells (Promocell, #C-12200); Hep G2, human hepatocellular carcinom cells (ATCC, #HB-8065); and ARPE-19, human retinal pigmented epithelium cells (ATCC, #CRL-2302). Human amniotic fluid cell samples AF.I and III were essentially propagated as described for hAFSCs, all other cells were maintained following the protocol provided by the manufacturer or founder.25 All cells, including hAFSCs, were periodically screened for the absence of mycoplasma (EZ-PCR Mycoplasma Detection Kit, Biological Industries, Israel).
Immunoblotting
For preparation of total cell lysates for subsequent sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting, cells were washed with ice-cold 1X phosphate-buffered saline (PBS, Lonza), harvested by gentle scraping and pelleted by brief centrifugation. Cell pellets were then washed twice with ice-cold 1X PBS and lysed in chilled Triton X-100 buffer containing 40 mM Hepes pH 7.5, 120 mM NaCl, 1 mM EDTA, 10 mM β-glycerophosphate, 50 mM NaF, 1% (vol/vol) Triton X-100, supplemented with 0.5 mM phenylmethanesulfonyl fluoride, 2 μg/mL aprotinin, 2 μg/mL leupeptin, 0.3 μg/mL benzamidine hydrochloride, and 10 μg/mL trypsin inhibitor (all Merck) for 20 minutes on ice. Supernatants containing soluble proteins were collected by centrifugation at 20000g for 20 minutes at 4°C and stored at −80°C. Equal amounts of denatured samples were then resolved by SDS-PAGE and transferred to nitrocellulose following a standard protocol. After blocking in 5% nonfat, dry milk for 1 hour at room temperature, the membrane was incubated with primary antibodies diluted in 5% bovine serum albumine (BSA)/1X Tris-buffered saline-Triton X-100 (TBS-T) for 12 to 16 hours (overnight) at 4°C and horseradish peroxidase (HRP)-linked secondary antibodies (CST) diluted in 5% nonfat dry milk/1X TBS-T for 1 hour at room temperature. Signals were detected with the enhanced chemiluminescence method (Pierce, Thermo Fisher Scientific, US).21 Uncropped images of immunoblots are presented in Figures S2 and S4.
Gelatin zymography
Gelatin zymography for the analysis of secreted MMP2 and MMP9 was essentially performed following standard protocols.26 Briefly, conditioned medium of cells grown in serum-free medium for 24 hours was resolved by 10% SDS-PAGE containing 0.2% gelatin (Merck Millipore) in nonreducing sample buffer. Following electrophoresis, SDS was washed out by incubating the gels in renaturation buffer (2.5% Triton X-100) with gentle agitation for 30 minutes at room temperature. Gels were rinsed three times in distilled water followed by incubation in developing buffer (50 mM Tris·HCl, 200 mM NaCl, 5 mM CaCl2, 0.02% Brij 35) for 30 minutes at room temperature and another 16 hours at 37°C. In a final step, gels were stained with 0.5% Coomassie Blue-R-250 for 30 minutes followed by destaining in 10% methanol/5% acetic acid and image acquisition using a standard gel documentation system. Samples were normalized to cell numbers, and recombinant human MMP2 and MMP9 (GE Healthcare, US) were co-analyzed as controls. Uncropped images of gels are presented in Figure S3.
Transwell invasion assay
Migration through a thin layer of reconstituted BM (rBM) was assessed via Fluoroblok 24- or 96-multiwell inserts at 8-μm pore size precoated with standard Matrigel (Corning, US) for the subsequent detection of invasive cells with calcein AM. Alternatively, transparent inserts (Corning) for the detection of crystal violet-stained cells were used. Transwell top chambers were seeded with 2.5 × 104 (24-well) or 7.5 × 103 (96-well) cells resuspended in serum-free basal medium, bottom chambers were filled with basal medium supplemented with serum (complete growth medium) or defined growth factors. After 22 hours of incubation at 37°C and 5% CO2, invasive cells on the transwell bottom membrane of the Fluoroblok inserts were stained with 4 μg/mL calcein AM (BD Biosciences) in Hanks' balanced salt solution (HBSS) (Lonza) for 1 hour at 37°C and 5% CO2 and were quantified with a plate reader and/or imaged and counted using an inverted microscope and Adobe Photoshop. For transparent inserts, noninvasive cells in the top chamber were removed with cotton swabs, and invasive cells on the transwell bottom membrane were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, US) and stained with 0.05% crystal violet (Merck Millipore). Stained cells were scored under the microscope. Independent invasion experiments were performed in duplicate or triplicate wells.
In vitro plug invasion assay
The plug invasion assay is based on the vertical gel invasion assay27 and was performed as previously described28,29 with minor modifications. Briefly, standard Matrigel HC (Corning) was thawed overnight at 4°C and diluted with cold basal medium containing 2X serum supplement (w/gradient condition) or basal medium (w/o gradient condition) at a ratio of 1:1. 40 to 70 μL of this mixture was added to one well of a 96-well plate or alternatively to a cloning cylinder (Corning) placed into a 96-well plate. For even Matrigel distribution, plates were centrifuged at 100g for 5 minutes at room temperature followed by a 1-hour incubation at 37°C and 5% CO2. Cells were resuspendend in serum-reduced medium (10% of the standard concentration used for regular maintenance and expansion), seeded on top of the Matrigel plugs at a density of ~0.5 to 1 × 105 cells/well and allowed to invade for 72 hours at 37°C and 5% CO2. For analysis, Matrigel plugs were fixed in 2% paraformaldehyde (Electron Microscopy Sciences)/1% glutaraldehyde (Merck Millipore) for 30 to 60 minutes at room temperature and stained with 0.1% toluidine blue (Merck Millipore) or left unstained. Using insulin syringes (BD Biosciences), fixed plugs were removed from the plates or cloning cylinders, transferred onto a microscope slide preplated with 10 to 20 μL of water and cut into 1 to 2 even slices (~1 to 2 mm thick). Vertical cross sections were imaged with an inverted microscope, and the average length of the invasive sprouts per sample was quantified using Adobe Photoshop and is indicated as the average invasion distance in μm. Invasion was assessed in duplicate or triplicate wells.
Transendothelial migration assay
The transendothelial migration assay for the assessment of extravasation in vitro was performed following a published protocol30 and is based on a transwell invasion assay set-up. Briefly, 5.0 × 104 HUVEC resuspended in complete growth medium were plated into a transparent 24-multiwell insert at 8-μm pore size precoated with standard Matrigel (Corning) and maintained for 12 to 24 hours at 37°C and 5% CO2 to form a confluent cell monolayer. The integrity of the monolayer was confirmed by microscopic examination of the transwell apical side and phalloidin staining of HUVEC plated into a matched control well. Transwell top chambers with the HUVEC monolayer inside were then seeded with 2.5 × 104 hAFSCs resuspended in serum-free basal medium and bottom chambers were filled with basal medium supplemented with serum (complete growth medium). After 22 hours of incubation, migrated cells on the transwell bottom membrane were processed for crystal violet staining and quantification as described for the transwell invasion assay. For interpretation of results, a standard transwell invasion assay (without the HUVEC monolayer) was run in parallel.
Immunofluorescence
Cells grown and treated in 96-well plates were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in 1X PBS (Lonza) for 15 minutes, followed by permeabilization in blocking buffer (1% BSA in 1X PBS) containing 0.3% Triton X-100 for 1 hour and incubation with primary and secondary antibodies (CST) diluted in 1% BSA/0.3% Triton X-100 in 1X PBS for 16 to 24 hours at 4°C and 60 minutes at room temperature, respectively. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (2 μg/mL) (Merck Millipore) for 3 minutes at room temperature before image acquisition on an Olympus IX51 inverted microscope.
Densitometry
Densitometry of immunoblots for the quantification of full-length vimentin and its inactive ~48-kD cleavage fragments31,32 was performed using ImageJ following standard protocols. Total vimentin includes the 57-kD full-length protein and the ~48-kD cleavage fragments. The percentage of cleavage fragments is presented as % cleaved vimentin.
Statistical analysis
Student's t-test (unpaired, two-tailed) was used to compare two groups for independent samples. Values are shown as mean ± SD of biological replicates from one representative experiment or as mean ± SEM of biological replicates from independent experiments. Results documented by immunoblots, zymography gels or micrographs are representative of experiments that were repeated independently.
RESULTS
hAFSCs harbor a mesenchymal phenotype and express motility-associated markers
All eight hAFSC lines used in this study were originally established from native human amniotic fluid cell samples by immunoselection for CD117 (c-Kit, the receptor for stem cell factor) through magnetic cell sorting. With the exception of the polyclonal line CD117/2, all hAFSC lines were generated as monoclonal cell lines by limited dilution-based single-cell cloning and the expansion of so derived clones. The comprehensive characterization of these AFSC lines with regard to embryonic and mesenchymal and/or neural marker expression and cellular characteristics has been published earlier (Figure S1).9-11,21-24 These cell lines exhibit logarithmical proliferation without the need of feeder layers, with stable normal karyotypes and without evidence for spontaneous differentiation or apoptosis9-11,21-24 (see also Figure 1A). The here studied hAFSC lines were found to display comparable mesenchymal-like cell morphology and growth properties, such as scattered, cell–cell adhesion-independent cell growth (Figure 1A).
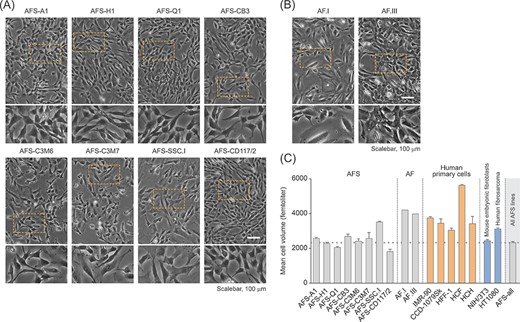
Human amniotic fluid stem cell (hAFSC) characterization for cell morphology and cell size. A,B, Phase contrast imaging for the evaluation of cell morphology in c-Kit-selected hAFSC lines (A) and low passage human amniotic fluid cells obtained via amniocentesis performed for routine prenatal diagnosis (B). The bottom panels represent a twofold magnification of the corresponding image section marked with a rectangle. C, Volume-based cell size distribution of logarithmically growing hAFSCs (AFS). For comparison, human amniotic fluid cells (AF) and primary as well as immortalized/transformed cells of mesenchymal origin were co-analysed. The latter include fetal/neonatal primary human lung and foreskin fibroblasts (IMR-90 and CCD-1079Sk, HFF-1, respectively), adult primary cardiac fibroblasts and chondrocytes (HCF and HCH, respectively), mouse embryonic fibroblasts (NIH/3T3) and human fibrosarcoma cells (HT1080). “AFS-all” represents the average of all values obtained for the hAFSC lines shown
Both cell shape and cell size are of significant relevance in the context of cell migration.33 Accordingly, we next analyzed the cell volume of the hAFSC lines included in this study. Probably with the exception of the SSC.I line, the cell size of the different hAFSCs was found to be very comparable. Compared to other human cells, such as primary amniotic fluid cells obtained through amniocentesis performed for routine prenatal diagnosis (AF.I, AF.III), primary lung fibroblasts (IMR-90), primary foreskin fibroblasts (CCD1079Sk, HFF-1), primary human cardiac fibroblasts (HCF), primary human chondrocytes (HCH), and human fibrosarcoma cells (HT1080), hAFSCs exhibit a smaller average size (Figure 1A-C).
In the course of epithelial-mesenchymal transition (EMT) E-cadherin is downregulated and N-cadherin expression, which promotes invasiveness, is induced.34 The matched pair of epithelial and mesenchymal hepatocellular carcinoma cells (HCC-1.2 and HCC-1.1) is a useful tool to investigate these molecular events associated with EMT.25 In contrast to epithelial cells, mesenchymal cells exhibit expression of the invasion-mediating mesenchymal marker vimentin, the E-cadherin-repressing transcription factors Snail, Slug and ZEB1 and the ECM-degrading protease MMP14.32,35-38 Having confirmed the reliability of our immunoblot setting to analyze these markers in epithelial and mesenchymal cells (Figure 2A), we applied this approach to hAFSCs.
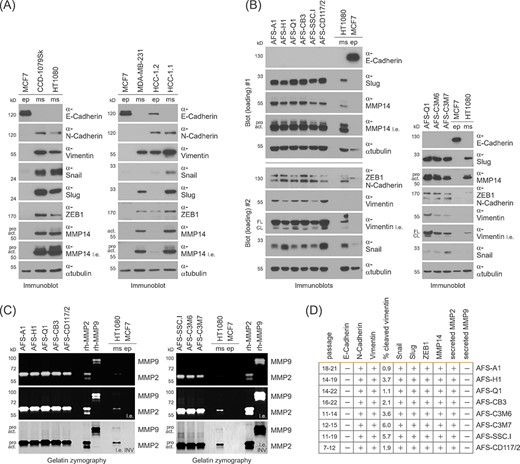
The epithelial-mesenchymal transition (EMT)-related marker profile of human amniotic fluid stem cells (hAFSCs). A, Immunoblotting for the detection of EMT-related marker proteins in epithelial (ep) and mesenchymal (ms) cells of fibroblast (CCD-1079Sk, HT1080), mammary (MCF7, MDA-MB-231), and hepatic (HCC-1.2, HCC-1.1) origins. l.e., long exposure; pro, pro-form; act, active form. B, Immunoblotting for the analysis of EMT-related marker proteins in hAFSCs. Epithelial and mesenchymal cells were co-analysed as a reference. For data presented in the left panel, the same lysates were separated on two different gels (Blot#1 and Blot#2). l.e., long exposure; pro, pro-form; act, active form; FL, full-length; CL, cleaved. C, Gelatin zymography for the detection of secreted MMPs in hAFSCs. Recombinant human MMP2 and MMP9, and cells of epithelial and mesenchymal origin were co-analyzed as controls. For better visualization of results, a color-inverted version of the long exposure was included (l.e. INV). D, Summary of results obtained in B and C including the hAFSC passage numbers and the quantification results for inactive, cleaved vimentin
In the course of these experiments, we found all hAFSCs to be negative for E-cadherin and to exhibit abundant expression of N-cadherin, vimentin (we co-evaluated its inactive cleavage fragments of ∼48-kD), Snail, Slug, ZEB1, and MMP14 (Figure 2B). Since mesenchymal 3D migration depends on MMP-mediated proteolytic matrix degradation, we next sought to answer the question whether hAFSCs secrete active MMPs. Gelatin zymography of conditioned medium revealed that hAFSCs secrete active MMP2, but not MMP9 (Figure 2C,D). Taken together, our findings define a mesenchymal cell morphology and shape, a small cell size and the expression of motility-associated proteins as a uniform characterization of hAFSCs.
hAFSCs exhibit mesenchymal and amoeboid 3D migration potential
To characterize hAFSCs for their invasive capacity, we carefully selected assay conditions that preserve the stem cell state and do not accidentally offer any subtle differentiation cues that inwardly distort the results. We used the transwell invasion assay and the plug (or vertical gel) invasion assay to study individual, and collective and individual invasion, respectively.14-18,21 In these assays the quantification of the 3D migration potential is presented as the percentage of cells on the transwell bottom membrane, which crossed the Matrigel-coated porous membrane (we evaluated the amount of migrated cells in relation to the amount of seeded cells), or as the distance of invasion into the Matrigel plug (Figure 3A). All eight analyzed hAFSC lines showed comparable pronounced invasive potential in the transwell invasion assay (NIH/3T3 and HT1080 cells were co-analyzed as poorly and highly invasive controls, respectively) (Figure 3B,C). Detailed microscopic analyses revealed that hAFSCs exclusively exhibited elongated, mesenchymal cell morphology under standard cultivation conditions and on the transwell top as long as they did not invade into the Matrigel (Figure 3D, panel 1 and panel 2). Upon 3D migration, hAFSCs showed both, elongated, and rounded cell morphology (Figure 3D, panels 3 and 4; Figure 3E). The average doubling time of hAFSCs has been described to be between 24 and 36 hours.9-11 The approach to evaluate transwell invasion assay results after 6, 9 and 12 hours showed that 3D migration of hAFSCs is independent of cell proliferation (Figure 3F). Due to the separated chambers, transwell invasion assays create optimal conditions to investigate the role of growth factor-mediated chemotactic effects on the studied 3D migration processes. Growth factor variation experiments involving media supplemented with varying concentrations of fetal calf serum demonstrated hAFSC invasion to follow an inherent potential independent of chemotactic signaling (Figure 3G).
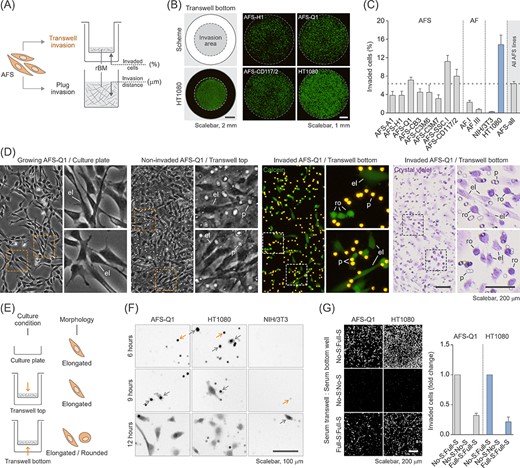
Human amniotic fluid stem cells (hAFSCs) display high invasive capacity and heterogenous cell morphology in the transwell invasion assay. A, Illustration of the two different types of invasion assays used in this study including the measured parameters. Data in B-G show results obtained with the transwell invasion assay. rBM, reconstituted basement membrane. B, Representative calcein stainings of invaded hAFSCs on the transwell bottom membrane after 22 hours of incubation. The scheme indicates the invasion area in relation to the total area of the transwell membrane which was quantified as a whole to obtain the percentage of invaded cells. HT1080 cells were co-analyzed as a positive control. C, Transwell invasion assay of all eight hAFSC lines (AFS) and two human amniotic fluid cell samples (AF). Poorly invasive NIH/3T3 cells and highly invasive HT1080 cells were co-analyzed as controls. “AFS-all” represents the average of all values obtained for the hAFSC lines shown. D, Morphological comparison of AFS-Q1 grown on a standard cell culture plate (panel 1) and as noninvaded and invaded cells in the transwell invasion assay after 22 hours of incubation (panels 2-4). Invaded cells on the transwell bottom membrane were detected upon calcein staining of living cells (panel 3) or upon crystal violet staining of fixed cells (panel 4). The side panels represent a magnification of the corresponding image sections marked with a rectangle. el, elongated; p, pore; ro, rounded. E, Illustration summarizing the observed morphological phenotypes in D. F, Transwell invasion assay of AFS-Q1 after 6, 9 and 12 hours of incubation. Representative pictures of calcein-labeled cells are shown. HT1080 and NIH/3T3 cells were co-analyzed for comparison. Grey arrows, pores filled with migrating cells; yellow arrows, cells migrating out of the pore onto the transwell bottom membrane. G, Transwell invasion assay of AFS-Q1 upon modification of the serum gradient at 22 hours of incubation. No serum (No-S) in the transwell and full serum (Full-S) (complete growth medium) in the bottom well represents the standard condition used in this study to which results were related. HT1080 cells were co-analyzed for comparison
Performing plug invasion assays we confirmed the invasive potential of all hAFSC lines included in this study (the epithelial human breast cancer cell line MCF7 was co-analyzed as negative control) (Figure 4A-C). Furthermore, microscopic analyses of cell morphology demonstrated again that hAFSCs, exhibiting mesenchymal morphology under normal culture condition, switch to the concurrent appearance of both, elongated and rounded phenotypes, upon 3D migration (Figure 4D-F). Moreover, the comparison of the results obtained with and without the application of growth factor gradients, confirmed the 3D migration capacity of hAFSCs to be independent of chemotactic signaling (Figure 4G). And finally, the approach to evaluate plug assay results after 24, 48, and 96 hours allowed 3D migration of hAFSCs to be confirmatory defined as a linearly growing process, which is independent of cell proliferation (Figure 4G). Overall, these results demonstrate hAFSCs to harbour an inherent invasion potential and provide first evidence that they potentially make use of mesenchymal and amoeboid 3D migration modes.
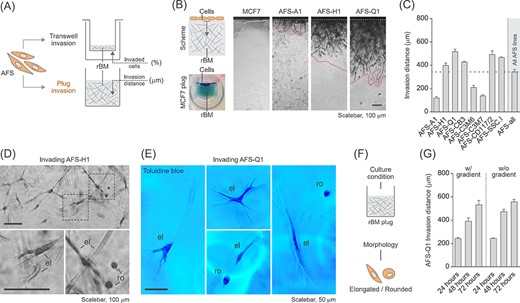
Human amniotic fluid stem cells (hAFSCs) migrating in 3D show mixed morphology of elongated and rounded cells. (A) Illustration of the two different types of invasion assays used in this study including the measured parameters. Data in B-G show results obtained with the plug (vertical gel) invasion assay. rBM, reconstituted basement membrane. B, Representative pictures of hAFSCs invading the rBM plug at 72 hours of incubation. The scheme illustrates the basic assay principle, the toluidine blue staining of non-invasive MCF7 cells co-analyzed as negative control shows the reconstituted basement membrane (rBM) plug as a whole at harvest. The area between the two dotted lines in the plug sections marks the invasion area used for quantification of the invasion distance. C, Plug invasion assay of all eight hAFSC lines. “AFS-all” represents the average of all values obtained for the hAFSC lines shown. D,E, Close-up view of AFS-H1 (D) and AFS-Q1 (E) invading the rBM plug at 72 hours of incubation. The bottom panels in D represent a magnification of the corresponding image sections marked with a rectangle. el, elongated; ro, rounded. F, Illustration summarizing the observed morphological phenotypes in D and E. G, Plug invasion assay of AFS-Q1 upon modification of the growth factor gradient at 24, 48, and 72 hours of incubation. The w/gradient condition represents the standard condition used in this study
The mesenchymal, elongated 3D migration mode is known to depend on MMP activity and to be induced upon ROCK inhibition, whereas the amoeboid, round invasion mode is independent of MMPs and blocked by ROCK inhibition.16,17,39,40 Transwell and plug invasion assays revealed that in the three analyzed hAFSC lines, AFS-Q1, AFS-H1, and AFS-A1, neither blocking MMP activity via the broad spectrum inhibitor Marimastat nor blocking ROCK activity via Y27632 inhibitor treatment fully diminished the invasion potential (Figure 5A-C). Blocking MMP activity reduced 3D migration of hAFSCs by half, whereas ROCK inhibition significantly induced it (Figure 5A,B). Microscopic analyses revealed that compared to the DMSO control migrating cells in the Marimastat condition predominantly acquired a rounded shape, while cells upon ROCK inhibition shifted toward a fully mesenchymal, elongated morphology (Figure 5C-E). And finally, the investigation of the effects of these inhibitors in 24-hour-plug assays with and without growth factor gradients demonstrated that mesenchymal and amoeboid 3D migration of hAFSCs does neither depend on the completion of an entire cell cycle nor on chemotactic signaling (Figure 5F). In summary, these data prove hAFSCs to harbor an inherent potential to make use of both the mesenchymal and the amoeboid migration modes.
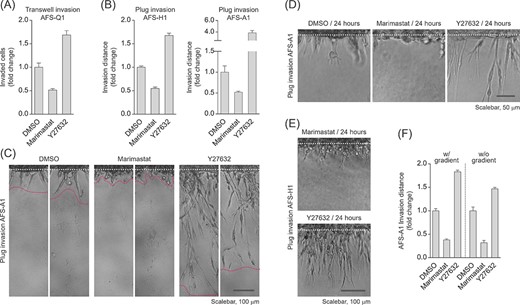
Inhibition of Rho-associated kinase triggers an increase in human amniotic fluid stem cell (hAFSC) invasion and a shift toward a fully mesenchymal phenotype. A, Transwell invasion assay of AFS-Q1 treated with the broad spectrum MMP-inhibitor Marimastat (10 μM) or the ROCK inhibitor Y27632 (10 μM) at 22 hours of incubation. B, Plug invasion assays of AFS-H1 and AFS-A1 at 72 hours of incubation upon inhibition of matrix metalloproteases (MMPs) or Rho-associated protein kinase (ROCK) as described in A. C, Pictures of AFS-A1 invading the reconstituted basement membrane (rBM) plug representative of data in B. Two sections per condition of the same experiment are shown. The area between the two dotted lines marks the invasion area used for quantification of the invasion distance. D,E, Close-up view of Marimastat- or Y27632-treated AFS-A1 (D) and AFS-H1 (E) invading the rBM plug at 24 hours of incubation for the visualization of early effects of inhibitor treatments. (F) Plug invasion assay of 24 hours Marimastat- or Y27632-treated AFS-Q1 upon modification of the growth factor gradient. The w/gradient condition represents the standard condition used in this study
hAFSCs exhibit transendothelial migration potential
Specific cells, such as, for example, leukocytes or tumor cells, harbor the potential to emigrate from the vasculature via crossing the endothelium to subsequently invade the surrounding tissue. This largely mechanical process, known as extravasation, includes the formation of dynamic interactions with the endothelium that trigger significant cytoskeletal changes, transendothelial cell migration and the potential to invade tissues. In the course of this process the migrating cells usually squeeze in amoeboid fashion across the endothelial cells before they then pass between tightly opposing endothelial cells.41-43 Our finding, that hAFSCs can switch to a rounded, amoeboid phenotype and exhibit 3D migration capacity, prompted us to investigate whether they also harbor transendothelial migration potential. To investigate this question, we made use of a well-established transendothelial migration assay based on a transwell set-up, in which the endothelium is mimicked by a confluent monolayer of HUVEC grown on ECM on a porous membrane30,44 (Figure 6A,B). These experiments revealed that hAFSCs harbor transendothelial migration capacity (Figure 6C,D). Fitting the expectations, the HUVEC monolayer slightly reduced hAFSC invasion in the transwell invasion assay (Figure 6E) and transendothelial migration of hAFSCs was accompanied by a significant shift of their morphology from the elongated to the rounded cell phenotype (Figure 6F). Taken together, these data demonstrate hAFSCs to exhibit extravasation capacity.
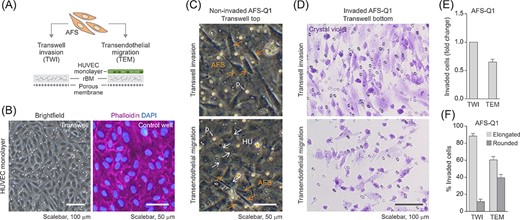
Human amniotic fluid stem cell (hAFSCs) harbor transendothelial migration potential. A, Illustration of the transendothelial migration (TEM) assay. The TEM assay is based on a transwell invasion (TWI) assay set-up (Figure 3) in which the rBM layer is seeded with primary human umbilical vein endothelial cells (HUVEC) to mimic the inner surface of a vessel wall composed of the endothelium sitting on top of a basement membrane. B, Representative brightfield and immunofluorescence images of HUVEC seeded into a transwell (left panel) or stained for F-actin (right panel) to monitor the formation of a compact cell monolayer. For better visualization of staining results, phalloidin labeling for the detection of F-actin was performed in a matched control well. C,D, TWI and TEM assays of AFS-Q1 at 22 hours of incubation. Pictures show non-invaded cells on top of the rBM (TWI) or on top of the HUVEC monolayer (TEM) (C) and invaded cells on the transwell bottom membrane after crystal violet-staining (D). Yellow arrows, hAFSCs (AFS); white arrows, HUVEC (HU); p, pore. E, Quantification of TEM results (number of invaded cells) in relation to TWI results. F, Relative contribution of elongated and rounded cells among invaded cells upon TWI and TEM
mTORC1 regulates 3D migration of hAFSCs
Whereas the crucial role of mTOR signaling for cancer cell invasion has been demonstrated in a variety of different tumor cell models,45-50 the involvement of mTOR in the control of 3D migration of nontransformed cells is not well studied. In an earlier study we found that the invasion potential of primary human fibroblasts depends on mTORC1 and can be induced by hESCs, human induced-pluripotent stem cells (hiPSCs), and hAFSCs cells via paracrine induction of mTORC1 activity.21 And very recently, it was reported that migration of outer radial glial cells, a population of neural stem cells prevalent in the developing human cortex, is regulated by mTORC1.51 Triggered by all these earlier findings, we decided to investigate the role of mTORC1 for the here reported invasive potential of hAFSCs.
We first aimed to clarify whether mTORC1 is active in hAFSCs. mTORC1 is known to promote protein synthesis by phosphorylating the p70S6 kinase at T389,52,53 which subsequently phosphorylates the ribosomal protein S6 at S240/454 and the PDCD4-protein, which is then ubiquitinated and degraded in proteasomes.55 Furthermore, the effects of mTORC1 on protein synthesis are also mediated by phosphorylation of 4E-BP1, which regulates the initiation of translation.56-58 In addition, mTORC1-activated p70S6 kinase is involved in the control of nucleotide synthesis via phosphorylation of CAD, the rate limiting enzyme in pyrimidine biosynthesis, at S1859.59,60 Immunoblot analyses of the functional readouts S6 phosphorylation, PDCD4 protein expression and 4E-BP1 phosphorylation (via the phosphorylation-mediated mobility shift of the total protein) demonstrated mTORC1 to be highly active in the three hAFSC lines AFS-Q1, AFS-A1, and AFS-H1. Whereas the Slug-negative epithelial cell lines MCF7 (human breast cancer), Hep G2 (human liver) and ARPE-19 (human retinal pigment epithelia), obviously do not exhibit mTORC1 activity, the mesenchymal human breast adenocarcinoma cell line MDA-MB-231 was found to exhibit this readout pattern at levels comparable to hAFSCs (Figure 7A).
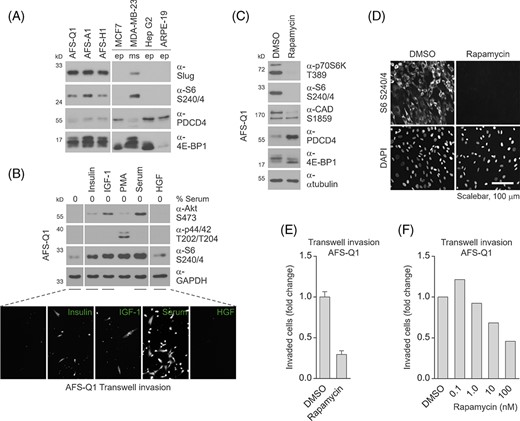
Human amniotic fluid stem cell (hAFSC) invasion is mediated by mTORC1. A, Immunoblotting for the detection of mTORC1 downstream targets S6, PDCD4, and 4E-BP1 in hAFSCs. For comparison, epithelial (ep) and mesenchymal (ms) cells of different origin and the epithelial-mesenchymal transition (EMT)-related transcription factor Slug were co-analyzed. B, Immunoblotting (upper panel) and transwell invasion assay (lower panel) of AFS-Q1 upon stimulation with insulin, insulin-like growth factor (IGF-1), and serum for the evaluation of mTORC1 pathway activation via Akt and S6 and cellular invasion, respectively. PMA and hepatocyte growth factor (HGF) treatments and the detection of p44/42 phosphorylation were co-evaluated as positive and negative controls. C,D, Immunoblotting (C) and immunofluorescence (D) of mTORC1 downstream targets in Rapamycin-treated AFS-Q1 as indicated for verification of mTORC1 pathway inhibition at the population and single-cell level, respectively. E,F, Transwell invasion assays of AFS-Q1 upon Rapamycin treatment as indicated at 22 hours of incubation. For data presented in C-E, Rapamycin was used at a standard concentration of 100 nM
Insulin and IGF-1 are known to activate the enzyme cascade of phosphatidylinositol-3-kinase and phosphoinositide-dependent kinase-1 to phosphorylate and thereby activate Akt at T308. Akt is further phosphorylated at S473 by mTORC2. Activated Akt phosphorylates the tumor suppressor protein tuberin (TSC2) and thereby downregulates its GTPase-activating potential toward Rheb, which is a potent regulator of mTORC1.19,20 To investigate the role of mTORC1-activating growth factors for 3D migration of hAFSCs, their intracellular mTORC1 activity was diminished via serum-deprivation. Treatment of serum-deprived hAFSCs with insulin, IGF-1 and serum, but not with HGF (used as a control), caused a significant induction of mTORC1 activity detected by phosphorylation of S6 at S240/4, but was without effects on the phosphorylation of the p44/42 mitogen-activate protein kinases at T202/T204 (co-analyzed as a control). Transwell invasion assays showed that insulin, IGF-1 and serum, but not HGF, can reactivate the 3D migration potential of hAFSCs (Figure 7B). These data suggest that activation of mTORC1 could drive the invasive potential of hAFSCs.
Rapamycin, binding the prolyl-isomerase FKBP12, is a widely used and potent inhibitor of mTORC1. Importantly, in contrast to mTORC1, Rapamycin treatment does not affect the ability of mTORC2 to phosphorylate its substrates.61-65 Immunoblot analyses of the phosphorylation status of p70S6 kinase, S6, CAD, and 4E-BP1 and of PDCD4 protein expression demonstrated that Rapamycin harbors the potential to efficiently downregulate mTORC1 activity in hAFSCs (Figure 7C). Immunostaining approaches to analyses phosphorylated S6 further showed that the Rapamycin-mediated downregulation of mTORC1 activity was equally distributed over all hAFSCs (Figure 7D). And finally, transwell invasion assays revealed that Rapamycin treatment strongly diminished the 3D migration potential of hAFSCs in a dose-dependent manner (Figure 7E,F). In summary, these data demonstrate mTORC1 to be a potent regulator of the invasive potential of hAFSCs.
DISCUSSION
Currently, fetal stem cells are considered to be of tremendously increasing relevance for both, basic research and the development of new therapeutic strategies. Although they are well characterized in terms of cultivation conditions, self-renewal and differentiation potential, a considerable lack of knowledge on their basic cell biological properties still exists. Filling this gap must be seen to be pivotal for a more comprehensive understanding of the cellular and molecular characteristics of fetal stem cells with regard to their usage for investigations on fundamental biological questions as well as to the development of safe and efficient fetal stem cell-based human therapies.13,66,67 3D migration, defined as the ability to cross BMs and to spread within tissues, is of highest importance for development, tissue homeostasis and repair, and is a hallmark of cancer initiation and progression.14,15,33 Taking all that into account, it seems surprising that in spite of its enormous relevance, the invasive capacity of fetal stem cells remained completely unaddressed so far. In this study, we present the first demonstration that hAFSCs exhibit a 3D migratory potential and consistently express motility markers and secrete active MMPs. In addition, we show that hAFSCs harbor extravasation capacity. This is an important step towards the full biological characterization of hAFSCs with relevance to basic science and implications for the development of optimized stem cell-based human therapy.
The major and best characterized types of 3D locomotion are the mesenchymal and amoeboid modes. A typical feature of the mesenchymal motility mode is the elongated cell shape with cell polarization to form a leading edge that extends protrusions, associated with adhesive interactions with the substratum. This mesenchymal 3D migration mode, which is used, for example, by fibroblasts and specific malignant cells, is further characterized by high MMP-dependent proteolytic activity triggering ECM remodeling.14-18,68,69 Amoeboid 3D migration, which is used, for example, by leukocytes, dendritic cells and neutrophils, is characterized by a rounded or ellipsoid cell shape. In the course of this MMP-independent, but ROCK-dependent motility mode cells slide through matrix pores with little or no adhesion to the matrix.14-18,70-72Although it is well established that cells can switch between the mesenchymal and the amoeboid migration modes in a context-dependent manner,14,73 only few cell types, including, for example, macrophages and tumor cells, have been proven to exhibit this plasticity so far.14,39,40,73,74 In the here presented study, morphological examinations and inhibitor treatments demonstrate that hAFSCs can concurrently make use of the mesenchymal and amoeboid migration mode. To our best knowledge, this is the first reported evidence for 3D motility mode plasticity in human stem cells. The question whether this is a distinctive feature for specific stem cells definitely deserves a broader characterization, especially since this potential could obviously be essential for the variety of functions stem cells fulfill in different organs and tissues. And it is also of highest relevance with regard to the usage of stem cells in human therapies. Although in the last years the field of regenerative medicine, including cell and gene therapy and tissue engineering, has witnessed a permanent increase in clinical trials, the number of scientifically evaluated and proven therapeutic applications is still low. In most of the cases the major roadblock to the forced translation into clinics is the still insufficient characterization of the used stem cells.13,75 Since in the course of regenerative approaches stem cells eventually must navigate to specific organs or tissues, a detailed characterization of the motility potential of stem cells that are already close to clinical implementation is mandatory.
Although they were discovered almost two decades ago1 and were already used in hundreds of published studies, the in vivo biological relevance of hAFSCs is still unknown. In earlier publications we have highlighted the fact that the currently known spectrum of properties of hAFSCs perfectly matches features which must be designated to a stem cell type to be considered as a candidate for the origin of pregnancy-associated progenitor cells (PAPCs).76,77 Together with cell-free fetal DNA (cffDNA) in the mother's circulation, PAPCs in the tissue of pregnant women, constitute the well-known phenomenon of fetal cell microchimerism (FCM). Because of their plasticity of morphology and diverse phenotypes, PAPCs are consensually considered to be stem cells, with characteristics and potentials between those of pluripotent ESCs and adult stem cells. The stemness of PAPCs is further supported by the fact that they integrate into various maternal tissues, such as, for example, blood, lung, skin, heart, spleen, liver, and even brain.76,78 It has been demonstrated that upon maternal injury fetal microchimeric cell populations can migrate to affected maternal sites and can support tissue repair. Such a beneficial hypothesis draws a picture of healing and regenerative PAPCs, which might even be supportive for the mother's defense against, for example, infections or cancer development. However, it is also discussed that PAPCs could be of disadvantageous consequences for the mother being involved in pregnancy diseases, such as for example, preeclampsia. And finally, it is speculated that fetal microchimerism could simply be a biologically irrelevant, incidental by-product of pregnancy.79-82 However, whereas this pregnancy-induced trafficking of small amounts of fetal cells to the mother has already intensively been studied, the cellular origin of PAPCs is still under discussion. Already some years ago, we argued that hAFSCs exhibit features, such as nonadhesive proliferation and survival, nontumorigenic behavior and the wide differentiation potential, which support their putative role in FCM. But we also argued, that other features, which are indispensable to fulfill the required spectrum of properties of PAPCs, including mainly 3D migration and extravasation potentials, have not been proven for hAFSCs so far.77 PAPCs are known to enter the maternal peripheral blood system, distribute, exit the vasculature and infiltrate maternal tissues and organs.78-82 In this respect, the here proven invasion and transendothelial migration capacities of hAFSCs represent the missing pieces to complete this puzzle in support of their suggested role in FCM.
It is also important to note that in the course of this study we identified hAFSCs as an optimal experimental tool to study the pathways and molecular mechanisms controlling 3D migration. For example, so far the molecular basis of the switch between the mesenchymal and amoeboid motility modes could only be studied in macrophages and tumor cells.14,33,34,68,69 However, such studies are impeded by the fact that macrophages are not easy to isolate and cultivate and tumor cells typically harbor a variety of genomic mutations with supposable effects on the balancing of 3D migration modes. hAFSCs constitute a primary, non-transformed, genomically stable, highly proliferative cell model that can comfortably be cultivated in the absence of spontaneous differentiation or apoptosis.9-11 We here made use of this model to investigate the role of mTORC1 for 3D cell migration. Although the relevance of this kinase complex for the regulation of catabolism, autophagy, cell differentiation or immune cell function has intensively been studied,19,20 the knowledge about mTORC1's involvement in the control of invasion is mainly limited to studies on cancer entities, such as for example, hepatocellular carcinoma cells, metastatic brain tumors cells or melanoma cells.40-45 Using the approach of Rapamycin-mediated enzyme modulation it was shown that 3D migration of human fibroblasts and of outer radial glial cells in the human cortex depends on mTORC1.21,46 Furthermore, the invasive potential of human fibroblasts was demonstrated to be induced by hESCs, hiPSCs, and hAFSCs cells via paracrine induction of mTORC1 activity.21 We here report that the application of serum, insulin or IGF-1, which trigger intracellular mTORC1 activity induce 3D migration of hAFSCs, whereas the growth factor HGF, which is without any effect on mTORC1, cannot promote an induction of invasion. And finally, we made use of invasion assays to demonstrate that specifically blocking mTORC1 activity via Rapamycin prevents hAFSCs from invading and crossing ECM barriers. On the one hand, this is the first description of a role of mTORC1 in fetal human stem cell invasion. And on the other hand, our study testifies hAFSCs to be an optimal human cellular tool for the molecular investigation of 3D migratory processes.
CONCLUSIONS AND SUMMARY
Currently, the array of established human stem cells contains cells of various sources, developmental stages and differentiation capacities. This diversity potentially creates new opportunities for stem cell research and therapy by providing the rationale for the tailored use of stem cells to meet very specific demands while minimizing risks, unwanted effects or other limitations associated with current approaches. For this concept to be valid, however, the selection of one stem cell type over another needs to be based on the profound knowledge of its properties. Genuine hAFSCs, representing an intermediate between embryonic and adult stem cell states, share the pluripotent stem cell property of trilineage differentiation but do not form tumors when injected into mice. Although being studied since almost two decades now, the in vivo biological role and the therapeutic application of hAFSCs are still elusive. This report provides the first demonstration that human stem cells exhibit mTORC1-dependent invasive capacity and can concurrently make use of mesenchymal and amoeboid 3D cell migration modes. In summary, identification of this new hAFSC trait represent an important step towards the full biological characterization of fetal human stem cells which is ultimately linked to their effective use in research and therapy.
ACKNOWLEDGMENTS
We thank Anthony Atala and Colin Bishop (Wake Forest Institute for Regenerative Medicine, Winston-Salem, North Carolina) for providing the human amniotic fluid stem cell lines AFS-A1, -H1, -Q1, -CB3, -C3M6 and -C3M7.
AUTHOR CONTRIBUTION
M.H.: conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript. M.R.: collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
Author notes
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}