



Abstract
Graft-vs-host disease (GvHD) limits successful outcomes following allogeneic blood and marrow transplantation (allo-BMT). We examined whether the administration of human, bone marrow-derived, multipotent adult progenitor cells (MAPCs™) could regulate experimental GvHD. The immunoregulatory capacity of MAPC cells was evaluated in vivo using established murine GvHD models. Injection of MAPC cells on day +1 (D1) and +4 (D4) significantly reduced T-cell expansion and the numbers of donor-derived, Tumor Necrosis Factor Alpha (TNFα) and Interferon Gamma (IFNγ)-producing, CD4+ and CD8+ cells by D10 compared with untreated controls. These findings were associated with reductions in serum levels of TNFα and IFNγ, intestinal and hepatic inflammation and systemic GvHD as measured by survival and clinical score. Biodistribution studies showed that MAPC cells tracked from the lung and to the liver, spleen, and mesenteric nodes within 24 hours after injection. MAPC cells inhibited mouse T-cell proliferation in vitro and this effect was associated with reduced T-cell activation and inflammatory cytokine secretion and robust increases in the concentrations of Prostaglandin E2 (PGE2) and Transforming Growth Factor Beta (TGFβ). Indomethacin and E-prostanoid 2 (EP2) receptor antagonism both reversed while EP2 agonism restored MAPC cell-mediated in vitro T-cell suppression, confirming the role for PGE2. Furthermore, cyclo-oxygenase inhibition following allo-BMT abrogated the protective effects of MAPC cells. Importantly, MAPC cells had no effect on the generation cytotoxic T lymphocyte activity in vitro, and the administration of MAPC cells in the setting of leukemic challenge resulted in superior leukemia-free survival. Collectively, these data provide valuable information regarding the biodistribution and regulatory capacity of MAPC cells, which may inform future clinical trial design.

Graft-vs-host disease (GvHD) is a difficult-to-treat side effect of blood and marrow transplant; most therapies involve significant immunosuppression. Multipotent adult progenitor cells may provide an alternative to those therapies as treatment and prophylaxis against GvHD.
INTRODUCTION
Allogeneic blood and marrow transplantation (allo-BMT) is the only curative therapy for many cancers and disorders of the hematopoietic and immune systems. The therapeutic potential of allo-BMT for hematologic malignancies relies on graft-vs-leukemia (GvL) effects, which eradicate residual malignant cells via immunologic mechanisms.1,2 Successful outcomes following allo-BMT are limited by several life-threatening complications; graft-vs-host disease (GvHD) and malignant relapse are the two primary contributors to mortality, which remains unacceptably high.3 Unfortunately, GvL effects are closely associated with acute GvHD in both the acute and the chronic settings.4 Standard immunosuppressive strategies to prevent or treat GvHD are often therapeutically suboptimal and predispose to opportunistic infections and relapse.5 Thus, the development of novel strategies that reduce GvHD, preserve GvL, facilitate immune reconstitution, and enhance survival remains the most significant challenge to improve outcomes after allo-BMT.
The pathophysiology of acute GvHD is complex.6,7 Immune dysregulation is hypothesized to occur in distinct phases involving diffuse damage and activation of host tissues by BMT conditioning regimens, the activation of donor T cells, and the culmination of target organ damage induced by soluble and cellular effectors.8-10 When allogeneic “targets” are residual host cancer cells, the final phase contributes to GvL activity. While components of this paradigm have been challenged and refined,8 the hypothesis identifies areas where novel agents can be explored. To this end, subsets of nonhematopoietic, bone marrow-derived, regenerative stromal cells (RSCs) that can generate connective tissues such as bone, cartilage, and fat have recently been discovered.11,12 These RSCs are not a homogeneous population; differences in phenotype, regenerative capacity, and plasticity make these cell populations distinct. Both human mesenchymal stem cells (MSCs) and multipotent adult progenitor cells (MAPCs) fall within this category.13 In addition to producing soluble proteins that critically support hematopoietic stem cell homeostasis and engraftment,14,15 a body of experimental data has revealed that both subsets possess powerful immunoregulatory properties.16-20 These findings identify opportunities to use novel, cellular, strategies that are noncross reactive with immunosuppressive approaches to prevent or treat GvHD.21,22 Maziarz and collaborators recently demonstrated that MultiStem™, a clinical-grade MAPC cell product, can be delivered in serial dosing regimens following allo-BMT.23 The work presented herein was completed in parallel with this clinical study with the intent on illuminating the biodistribution and possible mechanisms of action of these cells when infused following allo-BMT.
METHODS
Murine experiments and assessment of GvHD severity
Female C57BL/6J (B6; H-2b), B6D2F1 (F1; H-2bxd), B6.C-H2< bm1 >/ByJ (BM1; H-2b), and B6.CH2< bm12 >KhEg (BM12; H-2b), mice aged 8 to 12 weeks were purchased from Jackson Laboratory (Bar Harbor, Maine). Mice received allo-BMT as previously described.20,24,25 In studies using the B6 → BM1 system (isolated major histocompatibility complex [MHC] class I mismatch), recipient mice received 13 Gy Total Body Irradiation (TBI) followed by the infusion of 5 × 106 BM and 1.8 × 106 to 2.0 × 106 autoMACS-purified (>85% purity) CD8+ T cells. In the B6 → BM12 system (isolated MHC class II mismatch), BM12 mice received 11 Gy TBI and 0.25 × 106 autoMACS-purified (>90% purity) CD4+ T cells. These changes in BMT parameters were made to ensure that sufficient animals would be available for analysis at time points when significant GvHD is present and any differences between experimental groups could be assessed as previously described.26-29
Cryopreserved MAPC cells were provided by Athersys, Inc (Cleveland, Ohio). Specified groups of BMT recipients received two infusions of 0.25 × 106 to 1 × 106 MAPC cells on day +1 (D1) and +4 (D4).
Survival was monitored daily, and the severity of systemic GvHD was assessed weekly using a semi-quantitative scoring system as previously described.30 Animals were euthanized at D42-59 posttransplant or earlier based upon GvHD severity as directed by Institutional Animal Care and Use Committee (IACUC) standard of practice guidelines. Acute GvHD was also assessed by histopathology as previously described,31 serum cytokine analysis on D7 and T-cell expansion and activation on D10. In indicated experiments, indomethacin (20 μg ≈ 1 mg/kg, Sigma-Aldrich, St. Louis, Missouri) was administered daily as an intraperitoneal injection (100 μg/mL) for 7 days starting on D1. Control mice treated with MAPC cells received injections of diluent only. Pilot experiments (conducted without MAPC cell infusions) using this dose and schedule demonstrated that indomethacin had no significant effects on survival or small and large bowel pathology when administered to allogeneic and syngeneic BMT recipients compared with controls in each group (data not shown). All animal studies were approved by the IACUC at Case Western Reserve University (protocol 2010-0076) or Johns Hopkins University School of Medicine (protocol MO16M262).
Flow cytometry
Splenocytes harvested from either naïve donor mice or BMT recipients were analyzed pretransplant or on D10, respectively. unless otherwise specified. Splenic cell suspensions were stained for surface T-cell (CD4, CD8) and activation markers (CD25, CD28, CD62L, CD69) along with intracellular Tumor Necrosis Factor Alpha (TNFα) and Interferon Gamma (IFNγ) and then analyzed by flow cytometry using a four-color C6 FlowCytometer (Accuri, Ann Arbor, Michigan) as previously described.20 To analyze Forkhead Box P3 (FoxP3) expression, splenocytes were stained with anti-CD4 and anti-CD25, fixed and permeabilized, and then stained with intracellular anti-FoxP3 (eBioscience, San Diego, California). All monoclonal antibodies were purchased from BD Biosciences Pharmingen (San Diego, California) or eBioscience. Irrelevant fluorochrome rat isotype immunoglobulins were used as negative controls. Data were analyzed using CFlow software (Accuri) as previously described.20,25
MAPC cell product
Details regarding the preparation of the MAPC cell product have been described previously.32-36 MAPC cells isolated from a single qualified donor were expanded ex vivo in cell factories to create a cryopreserved master cell bank under current good manufacturing practice.
Enzyme-linked immunosorbent assay
Peripheral blood was collected on D7 from BMT recipients in 1.1-mL Z-Gel serum separation microtubes (Sarstedt, Newton, North Carolina). Following serum separation, enzyme-linked immunosorbent assay (ELISA) was performed for IFNγ, TNFα, and Prostaglandin E2 (PGE2). Mixed leukocyte culture (MLC) supernatants were harvested at 48 or 72 hours as indicated and then assayed for mouse cytokines (interleukin [IL]-10, Transforming Growth Factor Beta (TGFβ), IFNγ, TNFα, and PGE2) according to the manufacturer instructions (R&D Systems, Inc. Minneapolis, MN and eBioScience).
Mixed leukocyte culture
Spleens harvested from individual mice were enzymatically digested into single-cell suspensions. Purified splenic T cells (B6 mice) or dendritic cells (B6D2F1 mice) were suspended in supplemented 10% fetal calf serum/Roswell Park Memorial Institue medium (RPMI). B6 T cells (2 × 105) were cultured in 96-well plates in the presence of purified, irradiated (2000 cGy), B6 or B6D2F1, splenic, CD11c+ dendritic cells (2 × 104 dendritic cells [DCs]) at 37°C in a humidified incubator supplemented with 7% CO2. After 72 or 96 hours, supernatants were obtained from individual wells and subsequently analyzed for cytokines by using ELISA. Proliferation was measured by incorporation of tritiated thymidine [3H] (3.7037 × 104 Bq) during the last 24 hours of incubation using a 1205 Betaplate reader (Wallac, Turku, Finland). In indicated experiments, MAPC cells were added to MLC wells at MAPC cells-to-murine T-cell ratios ranging from 1:1 through 1:32 with and without indomethacin (1 μg/mL, Sigma-Aldrich), the E-prostanoid 2 (EP2) agonist, butaprost (BT, 2 μg/mL, Cayman Chemicals, Ann Arbor, MI), and the EP2 antagonist, PF-04418948 (PF, 2 μg/mL, obtained from Pfizer with permission). PF and BT were used in MLCs at a MAPC cell-to-T-cell ratio of 1:8.
Cryo-imaging and biodistribution
All animals used in cryo-imaging experiments received alfalfa-free chow for 2 weeks prior to BMT to reduce background fluorescence in the gastrointestinal tract. Human MAPC cells were fluorescently labeled with red quantum dots (Q-dot; 70% efficiency verified by flow cytometry), and 1 M cells were infused on D1 following allogeneic and syngeneic BMT as described above. Four experimental groups were defined: (a) syngeneic mice, no MAPC cells (“syn”); (b) syngeneic mice given MAPC cells (“syn + MAPC cell”); (c) allogeneic mice, no MAPC cells (“allo”); and (d) allogeneic mice given MAPC cells (“allo + MAPC cell”). Mice were sacrificed at 1, 6, 24, and 48 hours, embedded en bloc in optimal cutting temperature (OCT) compound, snap frozen in liquid nitrogen, and imaged as previously described.20
The cryo-imaging device (CryoViz, BioInVision, Inc.) is a fully automated system that provides three-dimensional (3D), tiled, microscopic anatomical bright field and molecular fluorescence images.37 Mice were sectioned at 40 μm and imaged at 10.5 × 10.5 μm in plane resolution. With sensitivity for detection of single cells, cryo-imaging permits unique quantitative analyses of fluorescently labeled cells.38 We used specialized algorithms to allow detection of both fluorescent green T cells and red MAPC cells.39 Filters were used to extract features, and a machine-learning, “bagged decision tree”, classifier was used to detect cells.20 In some experiments, Tc-99 Single photon emission computer tomography (SPECT) imaging was compared with cryo-imaging in our GvHD model. Briefly, cells were labeled with Tc-99 or quantum dots and injected intravenously. SPECT signals were measured and calibrated and compared against the number of cells counted by cryo-imaging.
JAM assay
B6 cytotoxic T lymphocytes (CTLs) were generated as described.20,25 B6 T cells (2 × 106) were stimulated with F1 splenic DCs (2 × 105) in primary bulk MLCs (24-well plates, BD Biosciences Pharmingen), isolated by Ficoll gradient, and subsequently added to 96-well plates (starting with 4 × 104 T cells/well and then serial twofold dilutions) with 3H-labeled target cells (1 × 103 cells with starting 40:1 CTL-to-target ratio) with and without MAPC cells. After 4 hours, cells were harvested, assayed for 3H release, and percent cytotoxicity was calculated as previously described.20
In vivo GvL experiments
P815 (H-2d, CD45.2) is a mastocytoma cell line derived from DBA/2 mice. Injection of P815 cells into animals (H-2d mice) that cannot reject P815 is uniformly lethal and results in massive nodular tumor infiltration of the liver and spleen.24 In GvL experiments, B6Ly5.2 (CD45.1) mice were used as allogeneic BM donors and 500 P815 tumor cells were added to the BM inoculum on D0. Specified groups received two infusions of 1 × 106 human MAPC cells on D1 and D4. Survival was monitored daily, and postmortem examination defined the cause of death after BMT as either GvHD or tumor. Death from P815 was defined by nodular enlargement of the liver and spleen, whereas GvHD death was defined as the absence of tumor and the presence of GvHD as determined by the clinical scoring system. This GvL mouse model has been used extensively in the translational application of several novel agents for the treatment or prevention of GvHD.20,25,40-43 The administration of tumor cells at the time of bone marrow infusion is a widely accepted approach. We specifically chose to infuse cells at the time of transplant to remove any effect TBI would have on tumor growth and thereby rely on the cytolytic effects of activated donor T cells.
PGER2 (EP2) mRNA expression in GvHD organs after BMT
Transcript analysis of PGE2R (EP2) was performed on mRNA extracted from splenic and intestinal (small and large bowel) tissue lysates using Trizol (Life Technologies) and quantitated by spectrophotometry following the manufacturer's protocol. Reversel Transcription Polymerase Chain Reaction (RT-PCR) was performed to make cDNA, which was subsequently subjected to real-time PCR using Taqman assays (Life Technologies, Carlsbad, CA) on an Applied Biosystems 7300 Real-Time PCR System. PGE2R expression was normalized to GAPDH before the fold change for PGE2R was calculated by comparing values obtained from individual Allo or Allo + MAPC cell recipients to the average value measured in syngeneic controls. Target expression in syngeneic mice was given an arbitrary value of 1.
Statistics
All values are expressed as the mean ± SEM. Statistical comparisons between groups were completed using the Mann-Whitney test (nonparametric data) and unpaired t test (parametric data). For survival curves, the Mantel-Cox log-rank test was used. Statistical significance was defined as a P value less than .05.
RESULTS
Human, bone marrow-derived, MAPC cell infusions reduce GvHD severity, attenuate T-cell expansion and pro-inflammatory cytokine release, and improve survival in a dose-dependent manner after experimental allo-BMT
To interrogate the in vivo effects of MAPC cell therapy after allo-BMT, correlative animal studies were performed using an established, haplo-identical, murine, allo-BMT system (B6 → B6D2F1) as described in Section 2. Recipients of syngeneic BMT (B6D2F1 → B6D2F1) served as controls. In preliminary experiments, increasing numbers (0.25-1.0 × 106) of human MAPC cells were administered via tail-vein injection on D1 and D4 after BMT. As shown in Figure 1A, a dose-dependent effect on D35 survival was observed. Follow-up experiments using 1 million MAPC cell per injection confirmed initial results and showed improvements in survival (Figure 1A), target organ GvHD (Figure 1B), and clinical score (data not shown). The reduction in systemic GvHD severity was associated with reductions in serum levels of IFNγ and TNFα on D7 and decreases in total splenic T-cell numbers on D10 (Figure 1C). Further examination of splenic T-cell populations showed diminished expansion of the CD4+ and CD8+ compartments (Figure 1D). Moreover, numbers of donor-derived IFNγ, TNFα, and IL-10 producing CD4+ and CD8+ cells were also significantly decreased at D10 (Figure 1D). These findings directly correlated with the changes in systemic cytokine levels (Figure 1C) and were associated with a reduction in cell surface activation makers on both CD4+ and CD8+ cells (Figure S2A,B). While the percentage of CD4+, CD25+, FoxP3+ cells in the spleen at D10 was increased in mice receiving MAPC cell, total numbers of splenic Tregs were not (data not shown). Collectively, these findings demonstrate that MAPC cell infusions significantly abrogated the development of systemic GvHD and had profound immune modulatory effects on alloreactive, donor-derived, T-cell expansion early after allo-BMT.
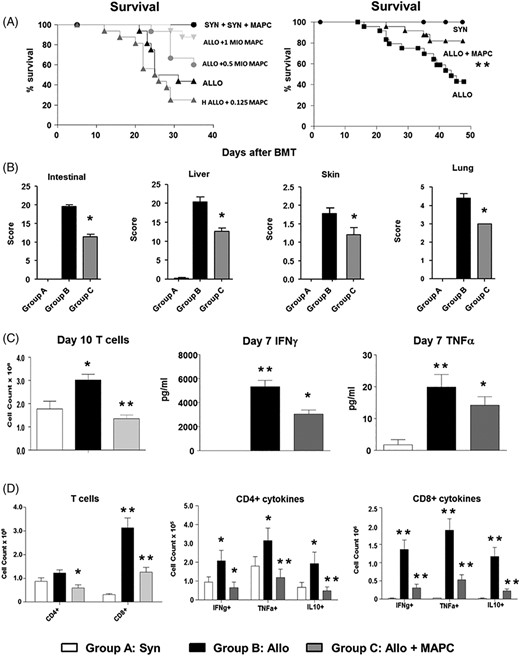
Multipotent adult progenitor cell (MAPC) infusions improve survival, reduce graft-vs-host disease (GvHD) severity, and attenuate T-cell expansion and pro-inflammatory cytokine induction after experimental allogeneic blood and marrow transplantation (BMT). Irradiated B6D2F1 mice received syngeneic or allogeneic BMT and subsets of allogeneic mice received MAPC infusions on D1 and D4 as described in Methods. A, MAPC infusions improve survival following allogeneic BMT in a dose-dependent manner. Data represent three combined transplant experiments (n = 15-30 mice per experimental group; **P < .01 allo-control vs allo + MAPC by Mantel-Cox log-rank test). B, Next, organs harvested from transplant recipients on D10 were analyzed for GvHD severity. Improved survival associates with decreases in target organ histopathology (n = 4-6 mice per experimental group; *P < .05 by Mann-Whitney test). C, Spleens (D10) and whole blood (D7) were collected from transplant recipients. MAPC infusions result in decreased donor T-cell expansion and serum levels of IFNγ and TNFα (n = 4-6 mice per group; *P < .05; **P < .01 by Mann-Whitney test). D, Further evaluation of splenic T-cell subsets showed that MAPCs regulate the expansion and cytokine expression of both CD4+ and CD8+ populations. Data are expressed as mean ± SEM and are from one of three representative independent experiments (n = 4-6 mice per group; *P < .05; **P < .01 by Mann-Whitney test)
Human MAPCs modulate in vitro murine T-cell activation and cytokine secretion
To define the capacity of and mechanisms responsible for the immunomodulatory properties of MAPC cells in this system, we co-cultured human MAPC cells with B6 T cells and either B6 or B6D2F1 DCs in MLCs as described in Section 2. MAPC cells significantly inhibited murine T-cell proliferation in a concentration-dependent manner, a finding that was associated with significant reductions in inducible levels of TNFα and IFNγ in MLC supernatants (Figure 2A). Suppression of proliferation was not seen with the addition of human fibroblast confirming that immunomodulation was unique to MAPC cells (data not shown) and as previously described.20 Evaluation of immunomodulatory cytokines revealed that while levels of supernatant IL-10 levels were reduced in the presence of MAPC cell, levels of PGE2 and TGFβ1 were significantly elevated by nearly 4- and 10- to 25-fold, respectively (Figure 2B).
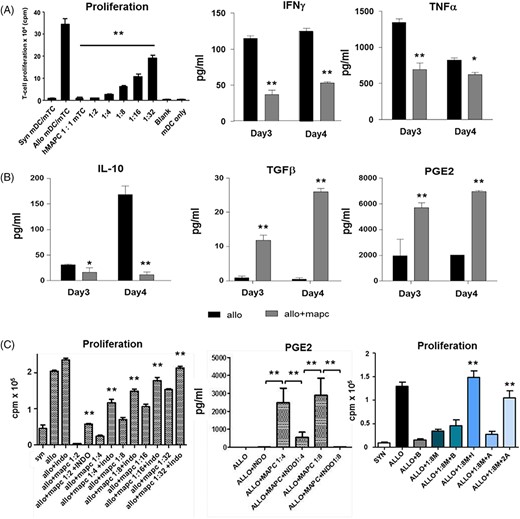
PGE2 and immune modulatory effects of multipotent adult progenitor cells (MAPCs) on T-cell alloreactivity in vitro. MLCs were prepared as described using increasing mixed T-cell (mTC) to MAPC ratios. A, MAPCs inhibit murine T-cell alloreactivity in a dose-dependent fashion and associate with reductions in the inducible cytokines TNFα and IFNγ. B, MAPC-mediated suppression of in vitro mTC proliferation is associated with reductions of interleukin (IL)-10 levels and significant increases in PGE2 and TGFα production. C, Effects of MAPC-mediated suppression of TC alloreactivity are both COX- and EP2-dependent. B6 splenic mTCs were co-cultured with syngeneic (B6) and allogeneic (B6D2F1) splenic-derived dendritic cells (DCs) in the presence or absence of MAPCs. Indomethacin (IM, 1 μg/mL) the EP2 antagonist, PF-04418948 (PF, 2 μg/mL), the EP2 agonist, butaprost (BT, 2 μg/mL), or diluent were added to wells at defined MAPC-to-mTC ratios and proliferation was measured at 72 hours. Proliferation and cytokine secretion were measured in triplicate. Data are expressed as means ± SEM from one of three independent experiments (*P < .05 and **P < .01 for indicated comparisons by Mann-Whitney test). MAPC, multipotent adult progenitor cell; ML, mixed leukocyte cultures
The role of PGE2 in human MAPC cell-mediated suppression of mouse T-cell alloreactivity and survival advantage following allo-BMT
We examined the role PGE2 played in MAPC cell-mediated immune regulation observed in our in vitro and in vivo experimental systems. First, the addition of indomethacin, a cyclooxygenase (COX) inhibitor known to block PGE2 production, to MLCs significantly reversed the MAPC cell-induced suppression of T-cell proliferation (at all MAPC cell:T cell ratios tested) and resulted in expected reductions in PGE2 levels in cell culture supernatants (Figure 2C). PGE2 signals through four different receptors (EP1, 2, 3, and 4), which have variable affinity for PGE2. In this regard, the EP2 antagonist, PF-04418948 (PF), and agonist, BT, were used to further delineate the effects of PGE2. As shown in Figure 2C, the addition of indomethacin again restored T-cell proliferation in the presence of MAPC cells. PF-induced EP2 blockade also significantly reversed suppression of T-cell proliferation by MAPC cells, albeit not to the same degree as direct COX inhibition. Next, we found that the addition of BT to MLCs potently suppressed T-cell proliferation and fully reversed the effects of indomethacin (Figure 2C). These data suggest that direct ligation of the EP2 receptor was sufficient to mediate MAPC cell effects on T-cell proliferation. Given these observations, we determined whether similar effects would be observed in vivo. PGE2 serum levels measured on D7 were significantly higher in allo-BMT recipients receiving MAPC cells compared with allo-BMT controls (Figure 3A) and associated with increased mRNA expression of PGE2 in the spleen and the bowel (both small and large) critical organs for early T-cell activation and injury, respectively, (Figure 3B-D). Administration of Indomethacin for 7 consecutive days (D1-D7) to MAPC cell treated allo-BMT recipients significantly reduced D7 PGE2 serum levels (Figure 3E), and this finding was associated with abrogation of the survival advantage consistently observed following MAPC cell injections (Figure 3F). These results demonstrate that the observed in vitro and in vivo effects of MAPC cells on alloreactive T-cell proliferation in our murine system are mediated in large part through PGE2.
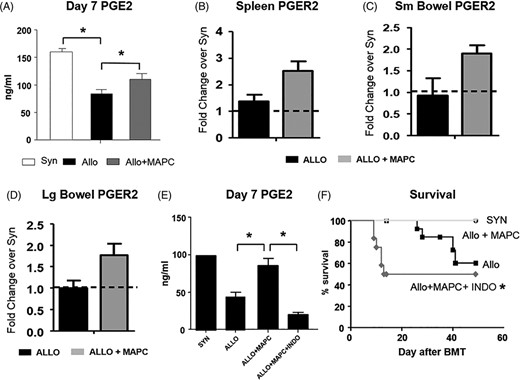
Multipotent adult progenitor cell (MAPCs) increase PGE2 concentration in the serum and PGER2 expression in target organs of graft-vs-host disease (GvHD) after experimental allogeneic blood and marrow transplantation (BMT). Contribution of PGE2 to MAPC immune regulation in vivo. A, PGE2 levels are significantly elevated at D7 in allogeneic BMT recipients receiving MAPCs, which correlates with reduced systemic and target organ GvHD (Figure 1). B-D, MAPC administration is also associated with a trend in increased mRNA expression of PGER2 in the spleen and the small and large bowel. E, Administration of IM (20 μg via intraperitoneal injection) on D1 to D7 after BMT to allogeneic BMT recipients receiving 1 × 106 MAPC on D1 and D4 significantly reduces PGE2 levels on D7, and, F, abrogates the survival advantage associated with MAPC infusions. Data shown are combined from one of two separate (PGE2) or two combined (survival) independent BMT experiments (Data are expressed as means ± SEM for PGE2 levels. n = 4-6 per group for PGE2 and 8-16 for survival experiments, *P < .05 and **P < .01.)
In vitro tumor cell lysis and GvL activity in allo-HCT mice are maintained in the presence of human MAPCs
We determined the impact MAPC cell infusions would have on GvL activity after allo-BMT. First, the presence of MAPC cells during CTL generation had no effect on the ability of B6 CTLs to lyse P815 tumor targets on a per cell basis (Figure 4A). Similarly, allo-BMT recipients receiving MAPC cell infusions maintained a survival advantage over untreated allo-controls even after in vivo challenge with P815. Consistent with our previous work and that of multiple other groups, syngeneic recipients receiving P815 succumbed to fulminant tumor progression by D21.20,25 By contrast, allo-BMT recipients effectively rejected P815 cells but ultimately died from GvHD at a rate greater than animals receiving MAPC cells (Figure 4B,C).
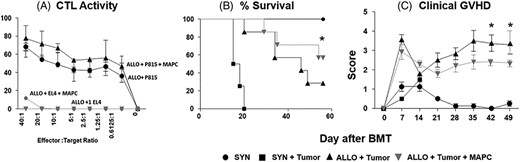
Multipotent adult progenitor cell (MAPCs) attenuate graft-vs-host disease (GvHD) without decreasing graft-vs-leukemia (GvL) activity in vitro and in vivo. B6 cytotoxic T lymphocytes (CTLs) were mixed with 3H-labeled target cells with and without MAPCs. CTL activity was calculated from spontaneous 3H release at various ratios of CTL to 3H-labeled target cell. A, Co-culture with MAPCs has no effect on cytolytic activity of activated B6 CTL. Irradiated B6D2F1 mice received syngeneic or allogeneic blood and marrow transplantation (BMT) and subsets of allogeneic mice received P815 mastocytoma cells (H-2d) in the BMT inoculum on D0 followed by MAPC infusions (or saline) on D1 and D4 as described in Methods. B, MAPC infusions improve leukemia-free survival following allogeneic BMT. C, MAPC infusions reduce systemic GvHD following allogeneic BMT as measured by GvHD clinical scores. Data are expressed as mean ± SD (CTL assay) or SEM (clinical score) and represent one of three similar experiments (CTL assay) or two combined transplant experiments (n = 10-20 mice per experimental group; *P < .05 allo-control vs allo + MAPC by Mantel-Cox log-rank test)
Human MAPCs redistribute from the lungs to the liver and secondary lymphoid organs within 24 hours after infusion
Little is known about the biodistribution of RSCs following intra-venous administration early after BMT. Secondary lymphoid organs (SLO) like the spleen and lymph nodes are critical sites for donor T cells to encounter and respond to host antigen following allo-BMT.44,45 Furthermore, MAPC cell treatment significantly reduced splenic T-cell expansion and activation in the early posttransplant period (Figure 1C,D). We used SPECT and cryo-imaging technology to track the biodistribution of MAPC cells after intravenous infusion and to determine if MAPC cells might ultimately co-localize with alloreactive T cells in SLO during GvHD induction.37 Mice received TBI followed by allo- or syn-BMT as described in Section 2. In initial experiments, MAPC cells (~0.57 × 106 cells) were labeled either with fluorescent (Qtracker 625 red quantum dots) or radio-labeled (~0.57 × 106 cells) with Technetium (Tc99) and infused at D1 in select animals. Control mice in each group received unlabeled MAPC cells. Animals were then imaged by SPECT analysis for Tc99 distribution at approximately 1 and 24 hours after infusion. Animals were snap frozen and subjected to cryo-imaging in order to analyze the distribution of fluorescent cells at each time point. As shown in Figure 5A, the 3D SPECT image reveals that the majority of signal was found in lung (yellow arrow). A fluorescent, 3D, cryo-image (Figure 5B) shows a distribution nearly identical to the SPECT image; with single-cell resolution, the majority of labeled MAPC cells were found in the lung with relatively few dispersed in other locations. The absence of detectable cells in the bladder on cryo-imaging strongly suggests that the signal seen in the bladder on SPECT imaging likely represents “free” tracer that is being excreted in the urine. Computer analysis demonstrated that nearly 90% of cells infused could be accounted for by cryo-imaging at this early time point. At 24 hours postinjection, radio-labeled MAPC cells could no longer be detected by SPECT imaging, consistent with the rapid and expected decay of Tc99 (data not shown). By contrast, redistribution of cells from the lung to the liver, spleen, and mesenteric lymph nodes was apparent in cryo-images at 24 hours as demonstrated in computer-generated 3D imagery (Figure 5C) and summarized in Figure S3A. By comparing the signal intensity in control mice vs that observed in MAPC cell-treated mice, the false-positive rate was negligible (Figure S3A). To study the biodistribution of injected MAPC cells, we completed additional experiments with fluorescently labeled MAPC cells and a MAPC cell density (cells/volume) analysis in several key organs was completed at 1, 6, 24, and 48 hours following injection. After a peak cell density in the lung at 1-hour postinfusion, cell density in the lung steadily decreased over time (data not shown), and this correlated with redistribution of cells and an increase in MAPC cell density in other major organs (Figure S3A,B). Computer-generated 3D renderings capture this redistribution of cells from the lungs to the liver and spleen (Figure 5D-F). This cellular relocation was readily apparent in cryo-images as well, where upon closer examination, MAPC cells were specifically observed in the marginal zones of the spleen and in the paracortex of mesenteric lymph nodes (Figure 5G-J).
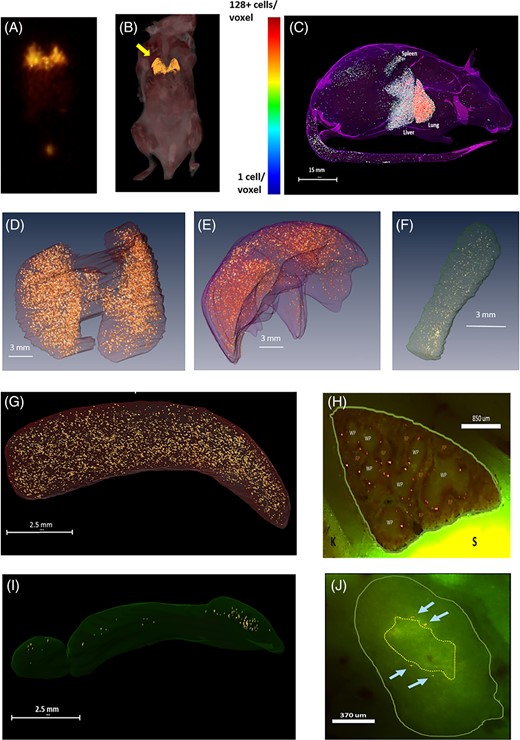
Biodistribution of multipotent adult progenitor cells (MAPCs) and murine T-cells (mTCs) within allogeneic and syngeneic transplant-recipient spleens. Irradiated B6D2F1 mice received syngeneic or allogeneic blood and marrow transplantation (BMT) as described in Figure 1, and subsets of allogeneic mice received MAPC infusions on D1. Using cryo-imaging, red Qdot-labeled MAPCs (rendered in gold) and SPECT scanning, the biodistribution of MAPCs was analyzed at 1, 6, 24, and 48 hours following tail vein injection as described in Section 2. A,B, As anticipated, the majority of labeled cells are found in lung (yellow arrow) 1 hour after intravenous infusion. C-F, By 24 hours postinjection, cells can be seen redistributing from the lung and to the liver, and spleen of allogeneic BMT recipient mice as captured by three-dimensional, computer-generated imagery. G,I, Examination of cryo-microscopic imaging under low power confirms accumulation in the spleen and lymph nodes and high-power view, H,J, demonstrates MAPCs specifically localizing in the splenic marginal zone and the paracortex of the mesenteric lymph nodes. K, kidney; S, stomach; RP, red pulp; WP, white pulp
DISCUSSION
MAPC cells represent a subset of RSCs that have the potential to differentiate into cells from each of the three germ layers and can be expanded extensively ex vivo while retaining their pluripotency. MAPC cells also possess tissue regenerative and immunomodulatory properties similar to MSCs.12,46,47 In contrast to MSCs, MAPC cells have broader ex vivo pluripotential48 and retain pluripotency longer.49 MultiStem are a clinical-grade human MAPC cell product manufactured under specifications and release criteria by Athersys, Inc. MultiStem cells retain broad differentiation capacity and immunomodulatory activity.32-36 These cells can be cryopreserved prior to use and ultimately administered as a “universal” product without patient matching requirements. MutliStem has been used in preclinical and clinical studies for the treatment of acute respiratory distress syndrome,50 spinal cord injury,51 wound healing,52 GvHD,22 traumatic brain injury,53 myocardial infarction,54 and ischemic stroke.55
The co-administration RSCs in the context of allo-BMT has shown promise in optimizing outcomes (reviewed in Reference 5). Recently, a phase I clinical trial utilizing MultiStem as GvHD prophylaxis following allo-BMT was completed.23 Thirty-six patients received increasing doses of MultiStem cells (1, 5, or 10 × 106 cells/kg) as a single intravenous dose on D2 post-BMT (N = 18) or once weekly for up to five doses of 1 or 5 × 106 cells/kg (N = 18). MultiStem infusions were well tolerated and had no adverse effects on donor neutrophil and platelet count recovery or donor chimerism. The cumulative incidences of grades II-IV and III-IV acute GvHD by day 100 were 37% and 14%, respectively (N = 36). Of the 9 patients receiving the highest single MultiStem dose (10 million cells/ kg) on D2, only 1 (11%) developed grades II-IV GvHD and no patient developed grades III-IV GvHD.23 Importantly, early relapse-free survival and the incidence of infection-related complications by D100 were favorable compared with historical controls.23 While these preliminary data suggest that the ability for MultiStem to control donor T-cell alloreactivity and expansion early after BMT might be critical to their protective effects, challenges related to optimizing the effectiveness of these cells remain.35 Specifically, the biodistribution of MultiStem cells and the specific mechanisms by which they may exert beneficial effects are not fully understood.
To this end, we used an established and clinically relevant murine model to elucidate the in vivo immunomodulatory effects of MAPC cells on donor T-cell responses after allo-BMT. Murine allo-BMT models have helped define mechanisms contributing to stem cell engraftment, immune reconstitution, GvHD, and GvL activity (reviewed in Reference 56). Murine xenogeneic transplant models should be useful in defining in vivo immunoregulatory capacity of human RSCs since these cells have low immunogenicity, lack MHC class II and co-stimulatory molecule expression, and fail to activate T-cells in vitro,20,57 all key features that allow their use in MHC-mismatched transplantation.58 We recently studied the biological effects of human MSCs in our haplo-identical preclinical GvHD model. We found that the infusion of MSCs early after BMT could similarly mitigate GvHD severity and that the homing of MSCs to the marginal zone was critical to their ability to suppress donor T-cell expansion in the splenic white pulp.20
The data in this report extend previous observations to a commercial stem cell product ripe for future application. We demonstrate that human MAPC cells decrease mouse T-cell proliferation in a reproducible, dose-dependent, fashion in vitro; this effect was associated with a reduction in T-cell activation and inflammatory cytokine secretion as well as robust increases in the concentrations of PGE2 and TGFβ in cell culture supernatants. Intravenous administration of MAPC cells on D1 and D4 after allo-BMT significantly reduces splenic T-cell expansion and numbers of donor-derived, TNFα and IFNγ-producing CD4+ and CD8+ cells at D10 compared with allo-controls. These findings were associated with reductions in serum levels of TNFα and IFNγ on D7 and ultimately in systemic GvHD as measured by survival, clinical score, and target organ inflammation. While we did not specifically enumerate donor T cells in GvHD target organs, we were able to breakout parameters in our histopathology scores that focus on cellular infiltration into the intestine (small and large) and liver. Semi-quantitative, subset analyses show that mononuclear cell infiltration into both target organs was greater in allo-BMT controls compared to allo-BMT animals receiving MAPC cells (intestine: 6.6 ± 0.4 vs 2.4 ± 0.6; liver: 5.0 ± 0.3 vs 2.0 ± 0.3, P < .05). It remains unresolved whether a reduction of lymphocytes into the liver and intestinal track is secondary to diminished donor T-cell expansion in the spleen or a direct effect of MAPC cells at the level of inflamed tissue. The results from our biodistribution studies (Figure 5G-J) support the former. MAPC cells had no effect on the generation of CTL activity against P815 tumor cells in vitro. Further evaluation using an established GvL model demonstrated that the administration of MAPC cell resulted in superior leukemia-free survival; allogeneic mice treated with MAPC cell had less GvHD but effectively eradicated tumor challenge. Our findings are in contrast to a previously published report showing that systemic infusion of murine MAPC cells on D-1 did not significantly affect survival following allo-BMT.59 This underscores how dose and schedule of MAPC cell infusions may optimize their ability to mitigate GvHD. Direct injection of MAPC cells into the spleen, an organ we (Figure 5G-H) and others have identified as a critical site following redistribution of cells from the lung20 did, however, result in protective, immunoregulatory effects during GvHD induction in that murine system.59
MAPC cells modulate immune responses by several mechanisms. In our experiments, inhibition of PGE2 production by indomethacin abrogated the immunomodulatory effects of human MAPC cells resulting in the restoration of T-cell proliferation in vitro and mitigated the protective effects of MAPC cells on GvHD mortality in vivo. This inhibitory effect could be reversed in vitro by the administration of an EP-2 agonist, BT, supporting the role of not only PGE2 in the immunomodulatory action of MAPC cell, but also a specific PG receptor. Immune suppressive effects of MAPC cell on T-cell proliferation and activation have been shown by others13 and PGE2 has been identified as a critical contributor to these effects in many36,59,60 but not all circumstances.61 While PGE2 is a known mediator of inflammation, it has been widely demonstrated to promote potent suppressive effects on both innate and antigen-specific immunity.62 PGE2 has been shown to negatively regulate a variety of T-cell responses including altering the polarization of T-helper cells and enhancing the induction and differentiation of FOXP3 + CD4 + CD25+ regulatory T cells.13,36,59,60 The ability of PGE2 to specifically suppress T-cell expansion/proliferation and activation has been demonstrated by others62-64 and is dependent in large part on the EP2 receptor63 as supported by our data (Figure 2). Specifically, PGE2 has been shown to suppress the activation and expansion of antigen specific T cells by inhibiting both IL-2 responsiveness and the production of IL-2,62 a cytokine known to play a role in the expansion of alloreactive donor T cells after BMT. While we did not pursue strategies to block TGFβ after BMT, nor did we test for NO or IDO levels in vitro or in vivo, we do recognize that other soluble factors could be contributing to the immune modulatory effects of MAPC cells in this and other models.13,35
PGE2 signals through four different receptors (EP1, EP2, EP3, and EP4). EP2 and EP4 are G-protein-coupled receptors that are present on lymphocytes. When bound to PGE2, both receptors lead to increases in cyclic adenosine monophosphate (cAMP) and activation of protein kinase A (PKA).62 EP4 is rapidly downregulated in the presence of PGE2, while EP2 is not.65 This relative resistance to ligand-induced desensitization suggests that EP2 signaling may remain operative over prolonged periods of inflammation.65 Activation of the EP2 receptor is also associated with decreased production of IFNγ and decreased T-cell proliferation.63 It is also worth noting that EP2 receptors are essential for preventing radiation-induced apoptosis of crypt epithelial cells at the jejunum in mice.66 In our studies, expression of the PGE2 receptor was significantly increased in both the small and the large bowel where apoptosis (another parameter in our semi-quantitative score) was diminished (3.6 ± 0.4 vs 2.4 ± 0.2, P < .05). However, the relevance of these findings in humans remains to be determined. In aggregate, these results support a role for PGE2 as a mechanism of suppression of T-cell activation by MAPC cells as have been observed by other investigators.60
We found that MAPC cells regulate the expansion, cytokine secretion, and activation of donor-derived, CD4+ and CD8+ populations (Figures 1 and S2A,B). In a final set of experiments, we explored the effects of MAPC cells on T-cell proliferation induced by isolated MHC class I (CD8+ T cells) or class II (CD4+ T cells) differences. First, MAPC cells were added to either B6 CD8+ T cells and BM1 DCs or B6 CD4+ T cells and BM12 DCs in MLCs as described in Section 2. MAPC cells significantly inhibited murine T-cell proliferation in a concentration-dependent manner, but suppression was more pronounced in the isolated class I disparate, CD8+ T-cell-driven MLC (Figure S4A,B). With these data in hand, we determined the in vivo effects of MAPC cells on GvHD induced specifically by CD8+ or CD4+ T cells using the strain combinations noted above in BMT experiments. The administration of human MAPC cells on D + 1 and D + 4 resulted in a significant albeit modest, reduction in systemic GvHD severity (survival and clinical score) in the B6 → BM1 (isolated class I) strain combination (Figure S4C,D). By contrast, this protective effect was not observed in the B6 → BM12 (isolated class II) model (Figure S4E,F).
Taken together, our data demonstrate that human MAPC cells have significant effects on T-cell alloreactivity. While the effects on CD4+ cells was more profound in vivo—numbers of activated, cytokine producing CD4+ cells were reduced to levels seen in syngeneic controls, the regulatory effect was more evident, but not solely limited to, CD8+ cells in vitro. The biologic underpinnings of these observations have yet to be fully elucidated. As noted above, regulation of T-cell proliferation and activation by MAPC cells has been shown by others.13 Specifically, Plesser et al also found that MAPC cells exerted suppressive effects on CD8+ CD69+ T-cells proliferation in a dose-dependent manner,67 whereas Carty and colleagues showed effects on both CD4+ and CD8+ populations using murine homeostatic and IL-7 proliferation models.60 With regard to the role of PGE2 in our experiments, the EP2 an EP4 receptors have been identified as the primary receptor subtypes on human and murine lymphocytes,63,64,68 but we did not interrogate CD4+ and CD8+ cells for differential expression of these receptors. Moreover, the regulatory effects of MAPC cells on the CD8+ compartment could have important implications with respect to GvL activity, which remained intact in our model. In this context, we found that MAPC cells had no appreciable effect on the expression of perforin and granzyme or markers of immunologic exhaustion (PD1, Lag3, or TIM3) on activated CD8 following allo-BMT (data not shown). Hence, although MAPC cells migrated the lymph nodes and spleen (Figure 5) and reduced T-cell expansion after BMT, they did not influence cytolytic machinery (preforin/granzyme) or activity (Figure 4A) of remaining T cells thereby allowing for residual, and substantial GvL activity. Such a hypothesis is consistent with how traditional (antimetabolite, eg, methotrexate, and calcineurin inhibitors, eg, tacrolimus and cyclosporine) and more contemporary (alkalators, eg, cyclophosphamide) agents are believed to control GvHD while permitting antitumor effects. Furthermore, this remains practical given that the majority of patients proceed to BMT in remission (minimal residual disease negative or otherwise) and rarely with active disease.
Finally, cyro-imaging experiments provide important insights regarding the bio-distribution of MAPC cells in the early post-BMT period. Cells are initially found in the lung after intravenous injection. However, our results refute claims of a previous study suggesting that stromal stem cells get trapped within the pulmonary vasculature and never leave the lung.69 Rather, findings herein are consistent with our previous work20 and the observations of others70 showing that viable stromal cells can redistribute from pulmonary vasculature and to the bone marrow,71 a site we also identified labeled MAPC cells 24 hours after infusion (Figure S3A), and other organs where they associate with decreased target organ GvHD (liver—Figure S3B) and a reduction of donor T-cell activation and proliferation (spleen and lymph nodes—Figure 5G,J) 24 to 48 hours after infusion. The later findings are significant as the spleen has also been identified as a key site of immune modulatory effects of MAPC cells in rodent models of spinal cord injury, stroke72,73 and GvHD20 particularly following intravenous injection.
In conclusion, potent immunomodulatory activity was observed following administration of MAPC cells within the first week of experimental allo-BMT. These effects were characterized by significant reductions in donor-derived, T-cell activation and expansion, systemic inflammation, and GvHD severity while preserving potent cytotoxic antileukemic activity both in vitro and in vivo. Importantly, our data shed light on the biodistribution of human MAPC cells after injection, the optimal dose and timing of administration, and possible mechanisms of action underlying the regulatory effects of MAPC cell therapy. To this end, our exploration of human MAPC cell treatment after allo-BMT is timely and may offer critical insights to optimizing their therapeutic potential in future clinical trials. Given the immune modulatory effects of MAPC cells on T-cell activation observed herein and reported by others, careful monitoring for leukemic relapse and opportunistic infections will be critical in any forthcoming studies.
ACKNOWLEDGMENTS
The authors wish to acknowledge Debashish Roy and Madhusudhana Gargesha at BioInVision, Mayfield Village, Ohio, for their assistance in cryo-imaging. The EP2 antagonist, PF-04418948, was kindly supplied by Pfizer. Research was supported in part by funding from the Center for Stem Cell and Regenerative Medicine at Case Western Reserve University (K.R.C.), the Ohio Board of Regents (K.R.C.), Meredith Cowden Foundation (K.R.C.), and the Stadler Family Charitable Foundation (K.R.C.).
AUTHOR CONTRIBUTIONS
L.M.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; D.W., J.J.A., W.V.H.: conception and design, data analysis and interpretation, final approval of manuscript; Z.H.L.: conception and design, collection and/or assembly of data, final approval of manuscript; R.D.: conception and design, final approval of manuscript; H.M.L.: conception and design, manuscript writing, final approval of manuscript; K.R.C.: conception and design, data analysis and interpretation, manuscript writing, financial support, final approval of manuscript; S.K.E., A.V.W., C.P.: collection and/or assembly of data, final approval of manuscript; P.W., Y.K.: collection and/or assembly of data, data analysis and interpretation, final approval of manuscript; B.D.: data analysis and interpretation, final approval of manuscript.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
How to cite this article:
Author notes
CONFLICT OF INTEREST
J.J.A. declared advisory role with MOREHealth. D.L.W. declared patent holder with CWRU which is licensed to BioInVision and research funding from NIH STTR P1 and an NIH SBIR P2 grant between my lab and BioInVision. R.D. declared leadership and stock interest in Synthego Inc. The other authors declared no potential conflicts of interest.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}