




Abstract
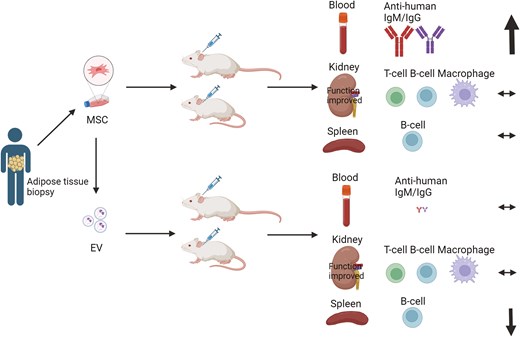
Renal artery stenosis (RAS) is the leading cause of secondary hypertension worldwide. However, current medical and surgical treatment modalities provide minimal benefits for kidney injury. Recent preclinical RAS models have demonstrated promising potential of human mesenchymal stem cells (MSC) and their daughter extracellular vesicles (EV) in improving murine renal function and attenuating inflammation. However, the extent and mechanisms underlying immune rejection of xenogeneic MSCs or EVs are yet undetermined. Therefore, adipose tissue was harvested from adult healthy patients. Adipose-derived MSCs were extracted and cultured, and EVs were isolated from their supernatants via ultra-centrifugation. Then, mice randomly assigned to RAS or sham surgery were divided into 6 groups: sham surgery, RAS, sham + MSC, RAS + MSC, sham + EV, and RAS + EV. Two weeks after intra-aortic injection of MSCs (5 × 105) or EVs (20 µg protein), we compared the intrarenal T-cell and macrophage accumulation, splenic B-cell numbers, circulating cytokines and anti-human antibodies levels among the groups. MSCs and EVs did not influence intrarenal immune cell infiltrations. However, MSCs significantly increased circulating anti-human antibodies. In the spleen, RAS + EV mice showed higher memory IgM+ B-cells but reduced CD19+ B-cells compared to RAS + MSC. In vitro T-cell recall assay showed that both MSCs and EVs exhibited reduced IFN-γ release upon re-stimulation, indicating an immunosuppressive effect. Therefore, xenogeneic MSCs induced a greater humoral response in mice, while EVs triggered a splenic cellular response, but neither elicits discernible kidney rejection. Our results provide key insights into the immunomodulatory mechanisms of MSCs and EVs and immune mechanisms underlying xenograft rejection.
Non-autologous MSCs and EVs instigate disparate immune rejection mechanisms, potentially because of different immunomodulatory components. We found that MSCs more potently than EVs trigger systemic antibody production, which might hinder their usage due to potential damage transplanted MSCs, especially after repeated doses. Contrarily, EVs altered splenic B-cell levels compared to their MSC counterparts, further solidifying their immune-modulatory effects. Overall, injecting human MSCs or EVs into mice is a valuable experimental approach for studying their therapeutic potential and biological behavior. Besides considering their potential immune rejection in experimental design, preclinical data gathered using these approaches can inform potential therapeutic applications in humans.
Introduction
Renal artery stenosis (RAS) is the leading cause of secondary hypertension worldwide.1,2 Although RAS is associated with significant increase in renal and cardiovascular disease and overall mortality, its treatment has been largely unfruitful in regards to patient outcomes.2 Recent advances in regenerative medicine have opened the possibility of using mesenchymal stem/stromal cells (MSCs) for kidney repair, owing to their anti-inflammatory and proliferative properties.3 Autologous MSCs have been shown to reduce kidney injury and improve glomerular filtration rate in animal models and human subjects with RAS,4-7 but might be confounded by exposure to unhealthy milieu in the cell donor. However, concerns have been raised surrounding the availability and safety of allogenic or xenogeneic MSCs.3
MSCs release a large number of extracellular vesicles (EVs), microvesicles of endosomal or membranous origin containing messenger ribonucleic acid (RNA), microRNA, and proteins of the parent cells, and which partly mediate the protective effects of MSCs observed in animal models.8 Our team has previously demonstrated that adipose tissue-derived MSCs and EVs both decreased renal inflammation and improved medullary oxygenation and fibrosis in RAS,3 although by slightly different mechanisms.9 MSCs are more effective in improving the renal microvasculature and decreasing inflammation, whereas EVs better preserve renal cellular integrity by more significant enhancement in angiogenic signaling and decreasing necroptosis.9 Yet, porcine adipose tissue-derived EVs also decrease renal inflammation in RAS,3 and umbilical cord MSC-derived EVs also blunt the inflammatory reaction and improve kidney function in patients with late-stage chronic kidney disease.10
Indeed, MSCs and EVs both exert immunomodulatory effects and reduce the levels of a wide spectrum of pro-inflammatory cytokines.9,11 Furthermore, they are capable of modulating the proliferation, maturation, and differentiation of immune cells.12,13 Because of these properties, MSCs have been considered to be relatively immuno-evasive and allogeneic regimens have been developed.14 Nevertheless, allosensitization and rejection remain concerns for such approaches.
While allogeneic MSC therapies are far more clinically relevant, xenogeneic models may provide important insights into how the immune system interacts with MSCs and their EVs that generate a wide array of immune responses. CD3+ T-cells, for example, quickly infiltrate xenotransplanted cell grafts,15 promoting antibody production and enhancing cytotoxic effects of other immune cells.16 With co-activation by T-cells, CD19+ B-cells produce antibodies,17 which activate natural killer cells and macrophages and directly damage donor cells.16 Innate immune responses, largely mediated by macrophages, are also key players in both acute and chronic immune xenograft rejection,18,19 and complement C4d depositions may be observed in the xenograft during periods of xenograft failure.20 Notably, EVs have been proposed to be less immunogenic than MSCs21 due to lower expression of major histocompatibility complex22 and higher expression of phosphatidylserine.23 Important contributors to xenogenic cell rejection include a potential role of the spleen in mediating systemic immune responses, where B-cells produce antibodies that can lead to systemic rejection, and tissue-specific immune responses, such as local T-cell and macrophage infiltration at the graft site. Therefore, xenogeneic models constitute a robust experimental platform to compare the vigor of immune responses triggered by transplanted products.
However, immune activation by xenogenic MSCs and their daughter EVs has not been directly compared, and whether EVs more effectively evade the immune system than MSCs is not yet fully elucidated. Therefore, this study aimed to investigate the activation and mechanisms of immune rejection imposed by human MSCs and their daughter EVs injected in mice. We hypothesized that xenogeneic EVs would be less prone to immune rejection than xenogeneic MSCs because of their low risk of immunogenicity.
Methods
Subject recruitment and MSC extraction
As previously described, informed consent was obtained from all participants, including healthy adult patients undergoing kidney donation surgery at Mayo Clinic Rochester, recruited following approval by the Mayo Clinic Institutional Review Board (14-007506)24 in accordance with the Declaration of Helsinki. Additionally, blood samples were collected from healthy volunteers at the Division of Transfusion Medicine, Mayo Clinic Rochester. Abdominal subcutaneous adipose tissue (SAT) was collected from study participants during surgery due to its accessibility and ethical feasibility, avoiding additional invasive procedures.5 Briefly, harvested SAT was minced and digested by adipose tissue dissociation enzyme kit (130-105-808, Miltenyi, Germany) on a gentalMACS OcTo Dissociator (Miltenyi Biotec, Germany) at 37°C for 45 minutes. After adding serum-containing media, the suspension was filtered through a 100 μm cell strainer, centrifuged, and the cellular pellet was resuspended, after which the MSCs were cultured and expanded three times. To mitigate any potential surgical stress effects on MSCs, cells were expanded to passage 3 before experimentation, as this allows stabilization of their phenotype.25
Expanded third-passage MSCs were identified via flow cytometry by positive expression of CD73 (46-0739, eBioscience), CD90 (ab124527, Abcam), and CD105 (ab135528, abcam), negative expression of CD34 and CD45, and their capability to differentiate into adipocytes, chondrocytes, and osteocytes (sc006, R&D Systems).26 In addition, MSCs were prepared and studied by digital electron microscopy (JEOL 1200 EXII; Mayo Clinic’s Electron Microscopy Core) to confirm the morphology of released EVs. EVs isolated from supernatants of MSCs through ultra-centrifugation were identified by expression of CD9 (ab61873, Abcam), CD81 (46-0819, eBioscience), CD29 (MCA1189, AbD Serotec), and CD44 (MSC marker, MA1-19277, Thermo Scientific) using FlowSight Imaging flow cytometer (Amnis Corporation), and their size distribution was assessed by nanoparticle tracking analysis (NTA) using NanoSight NS300.27
Mouse experiments
The mouse experiments were approved by the Mayo Clinic Institutional Animal Care and Use Committee (A00001609-16). In brief, 11-week-old male 129-S1 mice (Jackson Lab, Bar Harbor, ME) were acclimatized 2-3 days before surgery and fed Picolab Rodent Diet 20-5053. Mice were divided into 6 groups (n = 6-10): sham, sham + MSC, RAS, RAS + MSC, sham + EV, and RAS + EV. RAS mice were surgically fitted with 0.15 mm plastic cuffs around the right renal artery, while sham mice underwent the same surgery but without any cuff placement.26 Successful RAS surgery was confirmed by a subsequent ratio of right-to-left volume or weight under 0.9.26
Two weeks after surgery, EVs (20 µg protein)28 or MSCs (5 × 105)29 per mouse based on previously published studies, both in 200 µL PBS, were injected into the murine aorta cannulated through the carotid artery.26 Two weeks following injection, blood pressure was measured using tail cuff, mice were euthanized by carbon dioxide, and their blood, kidneys, and spleens were harvested to evaluate renal function (serum creatinine, Scr, and urea nitrogen, BUN) and immune cells profile.
Tissue studies
We assessed the tissue rejection of MSCs or EVs using immunofluorescence staining of immune cells accumulated in the kidneys. T-cells were identified by CD3 (MAB4841, R&D, MN) positivity, and those double-positive for CD3 and granzyme-B (44153, Cell Signaling) were considered active T-cells.30 F4/80 (ab6640, abcam) co-staining with inducible nitric oxide synthase (iNOS; SC-7271, Santa-Cruz) or CD206 (HPA004114-100UL, Sigma-Aldrich) identified murine M1 and M2 macrophages, respectively. Complement activation in the kidney was studied using immunohistochemistry staining for C4d (ab183311, Abcam) as percentage of positive area using ImageJ.31 B-cells were studied in splenic samples using IgM (ab190369, Abcam) and CD19 (NBP2-25196, Novus Biologicals) immunofluorescence staining as percent of tissue area.
Systemic xenoreactive antibodies
To quantify xenoreactive antibodies developing in mice in response to human MSCs or EVs, we used an in vitro human MSC reaction assay to detect murine anti-human antibodies bound to MSC. For this purpose, we adopted an assay developed to detect the generation of primate anti-swine antibodies.32 Briefly, 50µl murine serum was collected from all groups, heat inactivated (56°C 30min) and incubated with human MSCs (100,000 cells in 50µl PBS). The number of MSCs bound to anti-human mouse IgG and IgM were then assessed using flow cytometry32 on FlowSight Imaging flow cytometer (Amnis Corporation) and quantified as the percentage of positive MSCs that bound IgG and IgM out of total MSCs in the assay.
In vitro recall assay
The antigen-recall response described the antigen-specific reaction of T-cells upon encountering previously-recognized antigens and is useful to assess the effect of MSCs and EVs on memory and activation capacity of T-cells. On a 12-well plate, human peripheral blood mononuclear cells (PBMCs, 2 × 106) isolated from healthy donors using Ficoll-Paque plus were seeded in wells with MSCs (1 × 105) or EVs (30 µg). Then Dynabeads Human T-Activator CD3/CD28 (25ul beads/1 × 106 T-cells) were added into each well and incubated for 72 hours for a priming phase. PBMCs were collected and washed, resuspended in fresh RPMI-1640 medium and plated in a new 96-well plate (4 × 105 PBMCs/250 µL medium/well) and restimulated with Dynabeads Human T-Activator CD3/CD28. After 72 hours supernatants were collected for ELISA to measure interferon (IFN)-γ to assess the subset of activated T-cells.
In vitro complement lysis assay
This assay can be used to study immune responses and evaluate the potency of therapeutic antibodies. To determine whether anti-human antibodies generated are complement-fixing, we isolated PBMCs from donor blood using Ficoll-Paque and resuspended them in RPMI 1640 at 1 × 10⁶ cells/mL, seeded in a 24-well plate. Mouse serum (1:10) from each experimental group was added into each well incubated for 60 minutes, following another 60 minutes incubation with human complement serum (1:10, Innovative Research). A heat-inactivated complement was used as a control. Complement-mediated cell toxicity was evaluated by measuring PBMC viability, which was assessed using trypan blue on a Countess-II FL (Life Technologies) cell counter.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 10.2.0 (GraphPad Software). Shapiro-Wilk tests were used to determine if the data followed a normal distribution. One-way or two-way ANOVA (Tukey or Dunnett’s T3 post-hoc tests based on equality of variation) were used to test among groups. Statistical significance was set at P ≤ .05.
Results
MSCs and EVs characterizations
MSCs phenotypes were confirmed by their expression of surface markers CD73, CD90, and CD105, and their differentiation into adipocytes, chondrocytes, and osteocytes (Figure 1A). EVs were identified by the expression of EV markers CD9, CD81, and CD29, and their parent MSC marker CD44 which indicated their source. NTA analysis showed that EVs peak size distribution was around 159 nm (Figure 1B). Electron microscopy confirmed the typical cup-shape morphology of EVs9 released by their parent MSCs (Figure 1C).

(A) MSCs were identified via flow cytometry by positive expression of CD73, CD90, and CD105 and their capability to differentiate into adipocytes, chondrocytes, and osteocytes. (B) EVs were identified by expression of CD9, CD81, and CD29 upon flow cytometry, and their peak size distribution was 159 nm. (C) Electron microscopy confirmed the typical cup-shape morphology of EVs released by their parent MSCs
MSCs and EVs do not induce intrarenal immune cell infiltration
Neither MSCs nor EVs affected the number of intrarenal CD3+ T-cells in RAS mice. Sham + EV exhibited significantly fewer T-cells compared to sham and sham + MSC (Figure 2), but there was no difference between MSCs and EVs in the RAS groups (P = .138). Furthermore, the numbers of granzyme+CD3+ T-cells were not different among the groups (Figure 2). Similarly, intrarenal F4/80+ macrophage infiltration was not different among the groups (Figure 3), and C4d staining showed no complement activation in kidneys from either the MSC- or EV-treated groups.
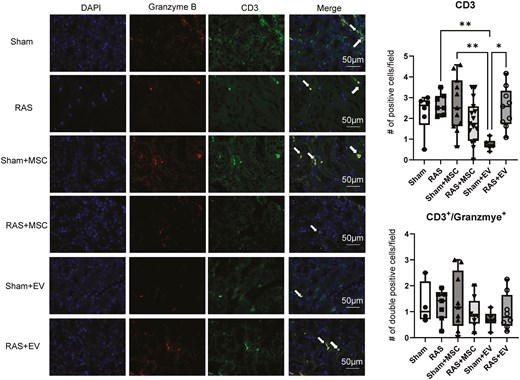
Identification of CD3+ and CD3+/granzyme-B+ mice T-cells in mouse kidneys. Sham + EVs mice had less CD3+ T-cells than sham + MSC mice. There was no difference between the intervention groups regarding CD3+ and CD3+/granzyme+ T-cells compared to sham. *P < .05 vs. RAS + EV, **P < 0.01 vs. sham + MSC and RAS. One-way ANOVA with Tukey’s multiple comparison test, n = 6-15/group.
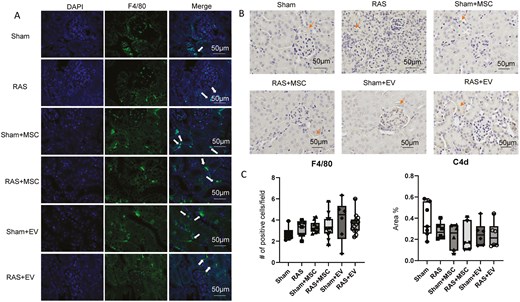
(A) Identification of F4/80+ murine intrarenal macrophages (arrows) and C4d (B. arrows) complement in sham and RAS kidneys. (C) MSCs and EVs did not impact macrophage accumulation and complement activation compared to sham, as assessed using the one-way ANOVA test, n = 6-15/group.
EVs alter splenic IgM+ or CD19+ B-cells
Neither MSC nor EV increased the number of CD19+ or IgM+ B-cells in the spleen of sham mice. Interestingly, RAS + EV had a larger number of IgM+ cells than RAS + MSC, but a smaller number of CD19+ cells than either RAS + MSC or Sham (Figure 4).
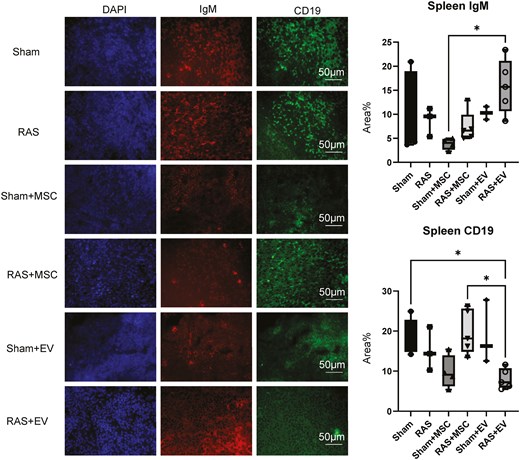
Classification of IgM+ and CD19+B-cells in the spleens of experimental mice. MSCs had no effect on IgM+ or CD19+cells compared to sham, while RAS + EV had more IgM+ cells than Sham + MSC, and fewer CD19+ cells than RAS + MSC and Sham mice. *P < .05. One-way ANOVA with Tukey’s multiple comparison test, n = 3-5/group.
MSCs but not EVs increase serum anti-human antibodies
Compared to untreated sham or RAS, both MSC-treated mice groups had significantly increased serum anti-human IgM antibody levels (P < .001) and anti-human IgG levels, as indicated by their bonding to human MSC. Contrarily, EVs had no significant effect on either serum anti-human IgM or IgG antibodies compared to sham. Sham + MSC also increased both anti-human IgM and IgG significantly compared to sham + EV (P < .0001), and RAS + MSC compared to RAS + EV (P = .0021) (Figure 5).
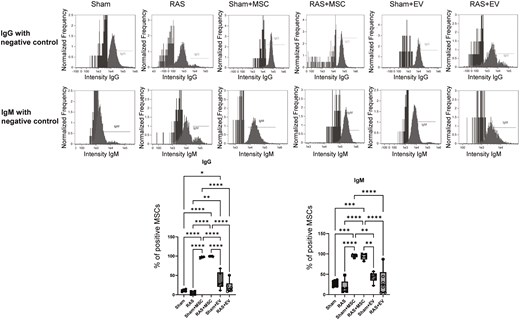
Quantification of anti-human IgG and IgM antibodies in the systemic circulation of mice. MSC-treated groups demonstrated significantly increased circulating anti-human IgG and IgM antibodies. *P < .05, **P < .01, ***P < .001, ****P < .001. One-way ANOVA with Tukey’s multiple comparison test, n = 4-6/group.
MSC and EVs improved renal function in RAS
All mice in our study had similar body weights. Compared with Sham, RAS significantly increased blood pressure which remained significantly elevated in RAS + MSC, but not in RAS + EV, indicating that EVs were slightly more anti-hypertensive than MSCs, although blood pressure in RAS + EV did not fall compared to RAS. RAS also significantly increased Scr and BUN in the untreated group which were improved after both MSC and EV treatments. Neither MSCs nor EVs influenced mean arterial pressure and renal function in sham animals, suggesting that they have no effects on these systemic parameters in normal animals (Table 1, Figure 6).
Systemic characteristics of sham/RAS mice injected with MSC or EV.
| Sham | RAS | Sham + MSC | RAS + MSC | Sham + EV | RAS + EV | |
|---|---|---|---|---|---|---|
| N | 7 | 8 | 6 | 7 | 6 | 6 |
| Body weight (gr) | 24.6 ± 2.0 | 26.1 ± 1.1 | 24.2 ± 2.2 | 24.5 ± 2.8 | 25.1 + 0.6 | 24.1 ± 1.4 |
| MAP (mmHg) | 128.8 ± 21.6 | 151.3 ± 19.6* | 129.5 ± 26.9 | 152.7 ± 36.2* | 126.1 ± 19.7 | 143.7 ± 48.5 |
| Scr (g/dL) | 0.17 ± 0.01 | 0.57 ± 0.19* | 0.23 ± 0.13 | 0.35 ± 0.22 | 0.11 ± 0.04 | 0.31 ± 0.03 |
| Sham | RAS | Sham + MSC | RAS + MSC | Sham + EV | RAS + EV | |
|---|---|---|---|---|---|---|
| N | 7 | 8 | 6 | 7 | 6 | 6 |
| Body weight (gr) | 24.6 ± 2.0 | 26.1 ± 1.1 | 24.2 ± 2.2 | 24.5 ± 2.8 | 25.1 + 0.6 | 24.1 ± 1.4 |
| MAP (mmHg) | 128.8 ± 21.6 | 151.3 ± 19.6* | 129.5 ± 26.9 | 152.7 ± 36.2* | 126.1 ± 19.7 | 143.7 ± 48.5 |
| Scr (g/dL) | 0.17 ± 0.01 | 0.57 ± 0.19* | 0.23 ± 0.13 | 0.35 ± 0.22 | 0.11 ± 0.04 | 0.31 ± 0.03 |
Abbreviations: MAP: mean arterial pressure, Scr: serum creatinine.
*P < 0.05 vs. Sham.
Systemic characteristics of sham/RAS mice injected with MSC or EV.
| Sham | RAS | Sham + MSC | RAS + MSC | Sham + EV | RAS + EV | |
|---|---|---|---|---|---|---|
| N | 7 | 8 | 6 | 7 | 6 | 6 |
| Body weight (gr) | 24.6 ± 2.0 | 26.1 ± 1.1 | 24.2 ± 2.2 | 24.5 ± 2.8 | 25.1 + 0.6 | 24.1 ± 1.4 |
| MAP (mmHg) | 128.8 ± 21.6 | 151.3 ± 19.6* | 129.5 ± 26.9 | 152.7 ± 36.2* | 126.1 ± 19.7 | 143.7 ± 48.5 |
| Scr (g/dL) | 0.17 ± 0.01 | 0.57 ± 0.19* | 0.23 ± 0.13 | 0.35 ± 0.22 | 0.11 ± 0.04 | 0.31 ± 0.03 |
| Sham | RAS | Sham + MSC | RAS + MSC | Sham + EV | RAS + EV | |
|---|---|---|---|---|---|---|
| N | 7 | 8 | 6 | 7 | 6 | 6 |
| Body weight (gr) | 24.6 ± 2.0 | 26.1 ± 1.1 | 24.2 ± 2.2 | 24.5 ± 2.8 | 25.1 + 0.6 | 24.1 ± 1.4 |
| MAP (mmHg) | 128.8 ± 21.6 | 151.3 ± 19.6* | 129.5 ± 26.9 | 152.7 ± 36.2* | 126.1 ± 19.7 | 143.7 ± 48.5 |
| Scr (g/dL) | 0.17 ± 0.01 | 0.57 ± 0.19* | 0.23 ± 0.13 | 0.35 ± 0.22 | 0.11 ± 0.04 | 0.31 ± 0.03 |
Abbreviations: MAP: mean arterial pressure, Scr: serum creatinine.
*P < 0.05 vs. Sham.
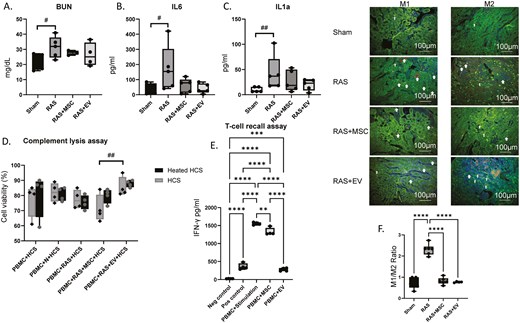
(A) BUN plasma levels were elevated in RAS but improved after MSC or EV treatment. #P = .05 One-way ANOVA with Dunnett’s multiple comparison, n = 4-5. (B) Plasma IL-6 levels were elevated in RAS and reduced post-treatment with both MSCs and EVs. #P = .05 One-way ANOVA with Dunnett’s multiple comparison, n = 5/group. (C) Plasma IL-1α levels in RAS animals tended to increase compared with sham. ##P = .07, One-way ANOVA with Dunnett’s multiple comparison, n = 5/group. (D) An in vitro complement lysis assay showed that antibodies generated against MSCs or MSC-EVs did not activate the complement system, although serum from RAS + MSC tends to induce higher cellular toxicity than from RAS + EV mice (##P = .07). Two-way ANOVA with Tukey’s post-hoc test, n = 4/group. HCS, human complement serum. (E) PBMCs primed with human MSCs exhibited reduced IFN-γ release upon re-stimulation, and EVs further decreased IFN-γ levels compared to MSCs alone, although neither normalized it. Neg, negative; Pos, Positive. (F) Kidney sections co-stained with markers for macrophages (F4/80, red) M1 (iNOS, green) or M2 (MRC1, green). RAS kidneys showed a predominance of M1-macrophages (yellow/orange, F4/80+/iNOS+), and treatment with either MSCs or EVs similarly shifted their polarization in favor of the M2 phenotype (yellow/orange, F4/80+/MCR1+), decreasing the M1/M2 ratio. **P < .01, ***P < .001, ****P < .001. One-way ANOVA with Tukey’s multiple comparison test, n = 4-6/group.
Immunomodulatory effects of MSC and EV
Plasma IL-6 levels were elevated in RAS (P = 0.05 vs. Sham) and IL-1α levels strongly tended to increase (P = 0.066 vs. Sham), but both were reduced post-treatment with both MSCs and MSC-EVs (Figure 6). In vitro, PBMCs primed with human MSCs exhibited reduced IFN-γ release upon re-stimulation, indicating an immunosuppressive effect rather than pro-inflammatory priming. Notably, EVs further decreased IFN-γ levels compared to MSCs alone (Figure 6), suggesting an enhanced immunomodulatory potential of EVs in this context. These observations underscore the immunomodulatory potential of both MSCs and their daughter EVs.
MSCs or MSC-EVs did not activate complement in vitro
The in vitro complement lysis assay showed that PBMCs incubated with serum from either MSC- or MSC-EV-injected mice, in the presence of human complement, did not exhibit reduced viability compared to those incubated with serum from control mice. However, the RAS + MSC group displayed a trend (Figure 6, P = 0.07) toward reduced viability compared to the EV-treated group, although this has not reached statistical significance. These findings suggest that antibodies generated against MSCs or MSC-EVs did not activate complement, yet EVs may induce a weaker complement response than MSCs, supporting their potential as a less immunogenic alternative to cell-based therapies.
Discussion
In this study, we aimed to compare the extent of immune rejection in MSCs and their daughter EVs in both healthy mice and mouse models of RAS. We found that neither EVs nor MSCs increased the numbers of immune cells infiltrating sham or RAS murine kidneys, but EVs reduced the splenic CD19+ B-cell count of treated RAS mice. Furthermore, EVs generated less stimulation of the systemic antibody immune response and thus appear to be less immunogenic than MSCs. In vitro, EVs had a greater immunomodulatory effect than MSC. Yet, both MSCs and EVs successfully improved renal function in RAS mice.
The immunogenicity of MSCs and MSC-derived EVs in an allogeneic context has significant clinical implications, particularly for repeated dosing strategies. Previous studies suggest that multiple doses of MSCs may improve therapeutic efficacy compared to a single dose, likely due to sustained paracrine effects rather than engraftment.33 Despite cumulative benefits demonstrated,34,35 the impact of repeated MSC or EV dosing on immune sensitization remains an open question.21 EVs exert therapeutic effects even upon multiple doses without triggering significant immune clearance,36,37 but recent evidence suggests that repeated administration may still trigger some immune activation through the development of anti-EV antibodies.38 Our findings indicate that MSCs trigger a stronger humoral immune response, as evidenced by elevated anti-human IgG and IgM levels, whereas EVs primarily modulate splenic B-cell populations. Given that multiple MSC or EV doses magnify antifibrotic and anti-inflammatory effects in myocardial infarction models,34,35,39 future studies should investigate whether similar immunomodulatory benefits or risks arise in renal injury and transplantation settings.
We compared the immune effects of MSCs and EVs in a murine RAS model. The mechanisms underlying kidney damage in RAS include increased oxidative stress and pro-inflammatory mediators, rendering regenerative stem cell therapies using immunomodulatory MSCs an attractive approach in preclinical animal RAS models and in human subjects.6,7,40 However, due to concerns regarding potential tumor transformation, MSC-derived EVs have gained attention as an effective alternative,3 albeit slightly less anti-inflammatory9 approach.
Autologous MSCs and EVs do not trigger immune rejection but are less cost-effective and more vulnerable to donor disease than allogeneic or xenogeneic products where the donor can be carefully selected, and products are scalable. Nevertheless, the beneficial effects of allogeneic or xenogeneic MSCs and EVs could be hindered by potential immune rejection. To the best of our knowledge, no studies so far directly compared side-by-side the extent of xenogeneic human MSC and EV rejection in mice.
Xenotransplantation has emerged as a robust experimental platform to test the immune responses triggered by transplanted products that activate a strong immune rejection16 by innate and subsequently by adaptive immunity, which may impede cell engraftment, function, and survival. Intra-graft markers of early responding cells like CD3+ T-cells and macrophages, as well as C4d complement, are often applied to detect immune rejection and cellular infiltration after xenotransplantation.16 Interestingly, we did not detect increased immune cell infiltration in the kidney or spleen after MSC or EV injection, suggesting limited tissue rejection of these cell products, likely because MSCs and EVs exert a wide range of immunomodulatory effects. We have previously shown that mice injected with human MSC-derived EVs exhibit reduced tumor necrosis factor-ɑ, IL-1β, and IL-6 compared with untreated RAS.41 Additionally, EVs interact with macrophages during initial immune responses and foster their polarization to an M2 phenotype, promoting an anti-inflammatory state.42 Similarly, MSCs from healthy humans reduce proinflammatory M1 macrophages, increase M2, and reduce other inflammatory markers in mouse kidneys,26 and both MSCs and EVs may directly inhibit immune cell proliferation.43-45 Therefore, whether non-autologous MSCs and EVs elicit a rejection response in animal models of disease may be difficult to determine using tissue infiltration of immune cell as markers.
Contrarily, we found that xenogeneic MSCs substantially increased the levels of anti-human antibodies in the systemic murine circulation. Antibody production by the host is among the greatest obstacles facing modern-day xenotransplantation.16,46 Humoral graft rejection is triggered by antibodies directed against the donor cells that may be pre-formed or produced de novo by B-cells after recognition of xenoepitopes. In mice, the role of anti-graft antibodies remains unclear. Dijovny et al. compared the mechanisms of graft rejection in allotransplantation and rat-to-mice xenotransplantation and associated cytokine profiles,47 and demonstrated that anti-graft IgM antibodies were crucial for xenotransplant rejection. Injection of human tumor antigens in mice also increased anti-tumor antibodies.48 Indeed, a homologous human antigen could be the target of mice antibodies. In the current study we delivered in mice human EVs and the parent MSCs from which they were derived, both at doses typically used experimentally.28,29 It is worth noting that many previous studies focused on larger solid tissue xenografts, rather than the small number of cells or particles used in our study. Hence, our study suggests that the amount of systemic xenoantigen needed to induce an immune response is rather small.
We also found in RAS + EV mice lower splenic CD19+ B-cell numbers compared to RAS + MSC, likely because EVs inhibit B-cell proliferation. While both MSCs and EVs can inhibit immune cell proliferation, the effects of MSCs might be partly mediated through their EVs.44,45 Furthermore, EVs hold the potential for cell-free therapies, reducing risks associated with cell-based therapies (eg, tumor formation). Interestingly, EV-treated mice showed increased splenic IgM+ memory B-cells, which increase in response to blood-borne T-independent antigens.49 This subset of memory B-cells is more likely to proliferate and reenter the germinal center during a secondary immune response.50,51 However, we delivered a single dose of EVs in our mice, and future studies will be needed to determine if repeated xeno-EV injection might ultimately face greater rejection. In our study, given the small size and lower likelihood of EVs getting eliminated by immune cells, their circulation time might have been longer than that of MSCs. Presumably, reducing serum IgM/IgG levels, possibly through splenic B-cell depletion or plasmapheresis, may be useful at least during the early post-xenotransplant period.
The different immunomodulatory properties of MSCs and EVs can potentially be explained by the selective transfer of specific molecules to EVs from their parent MSCs.52 EVs carry various components of their mother MSCs, including miRNA, antigens, chemokines, and major histocompatibility complexes, and show a different expression profile of surface antigens.53 The variance in immunomodulatory components and immunogenic antigens between human MSCs and EVs may underlie the contrasting immune rejection mechanisms seen in our study. However, further studies are needed to illuminate the specific components of humans EVs and MSCs and their roles in altering immune rejection.
Nevertheless, we found that both MSCs and EVs improved renal function without prominently lowering blood pressure in our study. In line with our findings, a meta-analysis of preclinical studies by Wang et al. found no significant changes in blood pressure with the administration of MSCs.54 Similarly, EVs have had little effect on blood pressure RAS swine models in previous studies55 although they were slightly more effective than MSCs in the current study. The renal protective effects of MSCs or EVs may be mediated mainly through their anti-inflammatory properties, as reported in a pooled analysis that found a reduction of Scr in MSC-treated animal models.54
Pertinently, the recently introduced induced pluripotent stem cell-derived MSCs (iPSC-MSCs) might afford a promising alternative to conventional MSC sources, as they exhibit strong immunomodulatory properties and offer a scalable and consistent source of MSCs with reduced batch-to-batch variation.56,57 Indeed, GMP-grade iPSC-MSCs have been successfully utilized in clinical trials for refractory graft-versus-host disease (GVHD),58 and GMP-compliant MSC-derived EV production is currently under investigation.59 These developments highlight the importance of advancing quality control measures for MSC and MSC-EV production to enhance therapeutic consistency and clinical outcomes.
Our study faced a few limitations. First, the assay that we used to measure IgG and IgM antibody levels provides an indirect assessment of the murine immune reaction to human MSCs and their daughter EVs. Because EVs were harvested from the same MSCs, they might have been expected to generate a similar systemic antibody profile as their parent cells, if any. Yet it remains unclear whether anti-human antibodies hinder their immunomodulatory and renoprotective effects. Additionally, the extreme scenario of rejection created following xenotransplantation facilitates discerning the immunomodulatory of MSCs and EVs, yet xenogeneic cell delivery is less likely than allogeneic approaches to be clinically applied, making its therapeutic relevance uncertain. Lastly, the direct immunomodulatory effects of xenogeneic MSCs and EVs offset and make it difficult to elucidate the mechanisms of immune rejection. Nevertheless, the present study remains the first of its kind to compare immune mechanisms involving xenotransplantation of MSCs and their EV progenies. Given the clinical potential of repeated MSC-EV therapy, further investigation into their comparative immunogenicity in allogeneic settings is warranted.
Conclusion and Summary
Xenotransplanted MSCs and EVs instigate disparate immune rejection mechanisms in RAS, potentially because of different immunomodulatory components. Antibody production might present a major obstacle to MSC usage due to their potential to damage transplanted MSCs, especially after repeated doses. In contrast, EVs altered splenic B-cell levels more than their MSC counterpart, further solidifying their immunomodulatory effects. Targeting these mechanisms could provide a potential means to circumvent immune rejection. Further studies are needed to determine if similar differences are observed with the use of allogeneic products. Notably, we found that xenogeneic MSCs elicit a robust systemic, but not tissue response, which may account for their salutary effects observed experimentally.
Overall, injecting human MSCs or EVs into mice is a valuable experimental approach to studying their therapeutic potential and biological behavior. Besides considering their potential immune rejection in experimental design, preclinical data gathered using these approaches can inform potential therapeutic applications in humans.
Acknowledgments
This study was partly supported by NIH grant numbers: DK120292, DK122734, HL158691, and AG062104.
Author contributions
Tarek Ziad Arabi: Data curation, Investigation, Methodology, Validation, Visualization, Writing-original draft. Yazan Almasry: Data curation, Investigation, Methodology, Writing-original draft. Ailing Xue: Data curation, Investigation, Methodology. Alfonso Eirin: Writing-review & editing. Amir Lerman: Writing-review & editing. Xiang-Yang Zhu: Conceptualization, Data curation, Investigation, Methodology, Project administration, Supervision, Validation, Visualization, Writing-original draft. Writing-review & editing. Lilach O. Lerman: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing-original draft. Writing-review & editing
Conflicts of interest
Lerman is an advisor to CureSpec, Cellergy, and Ribocure Pharmaceuticals. The authors declare no conflict.
Data availability
Data available on request. The data underlying this article will be shared on reasonable request to the corresponding author.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}