





Abstract
Divergent therapeutic outcomes on different disease domains have been noted with IL-23 and IL-17A-blockade in PsA. Therefore, elucidating the role of RORγt, the master regulator of type 17 immune responses, is of potential therapeutic interest. To this end, RORγt inhibition was assessed in combined skin, joint and gut inflammation in vivo, using a PsA model.
We tested the efficacy of a RORγt antagonist in B10.RIII mice challenged with systemic overexpression of IL-23 by hydrodynamic injection of IL-23 enhanced episomal vector (IL-23 EEV). Clinical outcomes were evaluated by histopathology. Bone density and surface erosions were examined using micro-computed tomography. Cytokine production was measured in serum and by intracellular flow cytometry. Gene expression in PsA-related tissues was analysed by qPCR.
RORγt-blockade significantly ameliorated psoriasis, peripheral arthritis and colitis development in IL-23 EEV mice (improvement of clinical scores and weight loss respectively by 91.8%, 58.2% and 7.0%, P < 0.001), in line with profound suppression of an enhanced type IL-17 immune signature in PsA-affected tissues. Moreover, inflammation-induced bone loss and bone erosions were reduced (P < 0.05 in calcaneus, P < 0.01 in tibia). Sustained IL-23 overexpression resulted in only mild signs of sacroiliitis. Gamma-delta (γδ)-T cells, the dominant source of T cell-derived IL-17A and IL-22, were expanded during IL-23 overexpression, and together with Th17 cells, clearly countered by RORγt inhibition (P < 0.001).
RORγt-blockade shows therapeutic efficacy in a preclinical PsA model with protection towards extra-musculoskeletal manifestations, reflected by a clear attenuation of type 17 cytokine responses by γδ-T cells and Th17 cells.
RORγt inhibition demonstrated effectiveness in all disease domains in experimental PsA.
A broad neutralization of type 17 responses, including IL-17A, IL-17F and IL-22, was achieved.
γδ-T cells, dominant producers of type-17 cytokines, and Th17 cells were strongly suppressed upon RORγt-blockade.
Introduction
Spondyloarthritis (SpA) represents a heterogeneous family of arthritic diseases, with PsA and AS as two prototypic subtypes. Typically, extra-musculoskeletal organs such as the skin and gut are involved and a spectrum of cytokines underlies the pathogenesis. Type 17 responses, characterized by cytokines of the IL-23/IL-17 axis, play a prominent role [1–5], although individual targeting of these cytokines showed variable effectiveness [6–9]. Additionally, there is still an unmet medical need in PsA as evidenced by non-responders to current therapies [2, 5, 10].
Differentiation of type 17 cells is controlled by the master transcription factor retinoid-related orphan receptor gamma-t (RORγt) [11]. Inhibition of RORγt affects several downstream mediators such as IL-17A, IL-17F and IL-22, resulting in a broader type 17 immune cell targeting compared with single IL-23 or IL-17A-blockade. Consequently, RORγt inhibition has been explored as an attractive alternative therapy, with strong efficacy in several murine models of psoriasis [12, 13], RA [13] and IBD [14, 15].
Next to conventional type 17 T-helper (Th17) cells, other more innate-like T cells such as γδ-T cells can produce IL-17 [10, 11, 16], both dependently and independently of IL-23 [4, 17]. These IL-23 independent IL-17-mediated pathways have been hypothesized to underlie the contrasting therapeutic outcome with IL-17 and IL-23 blockers in IBD and AS [4]. Intriguingly, RORγt-expressing γδ-T cells have been identified in skin, joint and gut tissue and could, next to Th17 cells and other innate-like (T) cells such as invariant natural killer T (iNKT) cells, mucosal-associated invariant T (MAIT) cells and innate lymphoid cells (ILC), contribute to SpA, PsA and psoriasis development [3, 4, 10, 11, 17–22]. Moreover, several murine arthritis and dermatitis models strongly depend on γδ-T cells as evidenced by diminished disease in T-cell receptor-δ-deficient (TCRδ-/-) mice [2, 10, 16].
To further elucidate the therapeutic potential of RORγt-blockade on combined PsA-related skin, joint and gut pathology, we tested the selective RORγt inhibitor PF-06747711 [12] in a mouse model of systemic IL-23 overexpression using IL-23 enhanced episomal vectors (EEV) [23]. Additionally, we evaluated how RORγt inhibition affected γδ-T cells, given their major contribution to IL-17 secretion, next to conventional Th17 cells. Targeting the master type 17-regulator might provide crucial insights in the differential involvement of type 17 cells in the SpA disease spectrum.
Materials and methods
Animals
Male B10.RIII mice [B10.RIII-H2r H2-T18b/(71NS)SnJ, #000457] were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and were 9–10 weeks old during IL-23 EEV injection. Female Lewis rats were purchased from Charles River Laboratories (Wilmington, MA, USA) and were 7–9 weeks old at AIA-induction. TNFΔARE mice [24] and littermate wild type controls (10 weeks old) were used to benchmark histopathological evaluation of sacroiliac joints.
Induction of PsA-like disease by IL-23 EEV
Injection of 75 ng IL-23 EEV (System Biosciences, Palo Alto, CA, USA) or empty CAGS EEV was done by hydrodynamic delivery (HDD) [18]. One day post-HDD, daily treatment with PF-06747711 (30 mg/kg) or vehicle via oral gavage was initiated. After 4 weeks mice were euthanised, followed by collection of blood, spleen, inguinal and popliteal lymph nodes, paws, pelvis, skin and colon.
Rat adjuvant-induced arthritis model (AIA)
Female rats were immunized via three 50 μl intradermal injections at the tail base with complete Freund’s adjuvant [15 mg/ml Mycobacterium tuberculosis (Difco, BD, Franklin Lanes, NJ, USA) in incomplete Freund’s adjuvant (Sigma-Aldrich, Saint Louis, MO, USA)]. Treatment was initiated between day 10–13, when the change in paw volume by plethysmography (Ugo Basile, Gemonio, Italy) reached the threshold of 0.2 ml. Rats were enrolled into groups with equal paw swelling and orally gavaged for seven days after disease onset with 10, 30 or 100 mg/kg of PF-06747711 or vehicle, once daily, after which they were euthanised and blood was collected.
Serum cytokine measurement
IL-23 was measured using ELISA MAX™ Deluxe-Set Mouse IL-23 (Biolegend, San Diego, CA, USA). LEGENDplex™ MU-Th17-Panel (7-plex) (Biolegend) was used for determination of IL-17A, IL-17F, IL-22, IFN-γ, TNF-α and IL-6.
Histopathology
Skin (ear), colon, ankles and pelvis were fixed in 4% formaldehyde. Ankles and pelvis were decalcified in 5% formic acid. Paraffin-embedded sections of 7 μm were stained with haematoxylin and eosin (H&E) or Safranin O.
Flow cytometry
Single-cell suspensions from spleen and lymph nodes were re-stimulated ex vivo with PMA and calcium-ionomycin. Intracellular FACS staining was performed using the antibodies in Supplementary Table S1, available at Rheumatology online. The gating strategy is depicted in Supplementary Fig. S1, available at Rheumatology online.
qRT-PCR
RNA was extracted from colon, ear, ankle and knee synovium using the RNeasy Mini Kit (Qiagen, Hilden, Germany). cDNA was synthesized with the QuantiTect Reverse Transcription kit (Qiagen) and qPCR was performed with LightCycler 480, using the primers listed in Supplementary Table S2, available at Rheumatology online. Data were analysed with qbase+ (Biogazelle).
Statistics
Statistical analysis was done with GraphPad Prism. Depending on normality testing, a two-tailed unpaired t test/Mann–Whitney test was used when comparing two groups, one-way ANOVA/Kruskal–Wallis with correction for multiple testing (Bonferroni/Dunn’s test) was used for multi-group analysis, and Pearson/Spearman coefficients were calculated for correlation analysis. Clinical scores and weight loss were analysed with repeated measurements using the residual maximum likelihood (REML) approach in Genstat v19.
Ethics
All experimental procedures involving animals were approved by the Ethics Committee of Laboratory Animal Welfare of Ghent University (ethical approval code ECD 19–76) or the Pfizer Institutional Animal Care and Use Committee (ethical approval code KSQ-2013–00905). All experiments were conducted in accordance with local, national and ethical regulations.
More details can be found in the supplementary information available at Rheumatology online.
Results
Systemic inflammatory cytokine release upon IL-23 overexpression is strongly impacted by RORγt inhibition
To assess the effect of RORγt inhibition on IL-23/IL-17 driven combined skin, joint and gut inflammation, we used the IL-23 EEV model showing features of PsA (Fig. 1A). Hydrodynamic injection of IL-23 EEV (Fig. 1B) in B10.RIII mice induced a stable overexpression of serum IL-23 (Fig. 1C), resulting in weight loss and development of psoriasis-like skin disease and arthritis. The magnitude of these pathological features correlated well with serum IL-23 levels (Fig. 1D). Dosing with RORγt antagonist PF-06747711 led to unbound plasma compound concentrations of 150 nM, ∼7.5-fold higher than the compound’s IC50 for inhibition of IL-17 production by murine T cells in vitro (20 nM) [12] (Supplementary Fig. S2, available at Rheumatology online). IL-23 EEV injected mice had prominent elevation of serum cytokines IL-17A, IL-22, IL-17F, TNF-α and IFN-γ, which was strongly suppressed upon PF-06747711 treatment (Fig. 1E).
![Systemic inflammatory cytokine release upon IL-23 overexpression is strongly impacted by RORγt inhibition. (A) Male B10.RIII mice received IL-23 EEV or empty CAGS EEV (non-diseased control). After one day, treatment was initiated with RORγt inhibitor PF-06747711 or methylcellulose vehicle as placebo (IL-23 EEV n = 10–11/group, CAGS EEV n = 3/group). (B) Structure of the IL-23 EEV. (C) Quantification of serum IL-23 levels on day 0, 8 and 28 (in ng/ml). (D) Correlation between serum IL-23 and clinical scores for dermatitis and peripheral joint inflammation and weight loss, at endpoint for vehicle-treated mice (IL-23 EEV n = 10, CAGS EEV n = 3) (Pearson correlation coefficient). (E) Effect of RORγt inhibition on serum cytokines [mean (s.e.m.)]. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/rheumatology/62/9/10.1093_rheumatology_kead022/2/m_kead022f1.jpeg?Expires=1747881667&Signature=W5VQHZdEp9L68NSlHUdh8gGV05cCYWa3yb-xtBHIDgCQ1MIV4Oc6Gj3cC45p-MDo0FTUQzflZEJEsuh5ID~dtpwqz~O--ZUyMnscVb0FCn7rSa2nl1A9xvL2bqdrvhAbBNg3xarZSx~7kaT1D4D2k~~VYRXSDNXbsFod8IPKnwNlXOqYlt~JQFvQQAzscpgkXwvw5ea2IgO7N3WoYagjh6Rt9fbwmZP4QAY0yaqQo0oJNquYaI2T91tg72tKNajb-98E1RAOwIrQG1uFJxc6iEFz6s3~B27mul-0oW71aO4OsXkBXgSKS7QDy7uqwjhXr4UyUhpsNO31ihDj38td6w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Systemic inflammatory cytokine release upon IL-23 overexpression is strongly impacted by RORγt inhibition. (A) Male B10.RIII mice received IL-23 EEV or empty CAGS EEV (non-diseased control). After one day, treatment was initiated with RORγt inhibitor PF-06747711 or methylcellulose vehicle as placebo (IL-23 EEV n = 10–11/group, CAGS EEV n = 3/group). (B) Structure of the IL-23 EEV. (C) Quantification of serum IL-23 levels on day 0, 8 and 28 (in ng/ml). (D) Correlation between serum IL-23 and clinical scores for dermatitis and peripheral joint inflammation and weight loss, at endpoint for vehicle-treated mice (IL-23 EEV n = 10, CAGS EEV n = 3) (Pearson correlation coefficient). (E) Effect of RORγt inhibition on serum cytokines [mean (s.e.m.)]. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
RORγt-blockade efficiently suppresses inflammatory and erosive peripheral arthritis, enthesitis and dermatitis but does not improve mild sacroiliitis
After confirming marked suppression of type 17 related cytokines in serum of IL-23 overexpressing mice by PF-0674771, we subsequently evaluated its impact on disease manifestations. Peripheral arthritis was seen in ankles and wrists, next to dactylitis and mild enthesitis of the Achilles tendon. Additionally, the snout, ears and paws showed prominent psoriasis-like dermatitis. Mice treated with PF-06747711 clearly showed reduced clinical scoring, for both prevalence (Supplementary Fig. S3, available at Rheumatology online) and severity (Fig. 2A) of these symptoms. The latter was validated by histopathology of ankle and ear cutis sections (Fig. 2B).
![RORγt-blockade efficiently suppresses peripheral arthritis, enthesitis and dermatitis but does not significantly improve mild sacroiliitis. (A) Clinical scoring for IL-23 EEV-injected mice treated with RORγt inhibitor (n =11) or vehicle (n =3). Above: peripheral joint inflammation (peripheral arthritis, dactylitis, enthesitis). Below: dermatitis (ear, snout and blepharitis). (B) Histopathology (IL-23 EEV n =10–11/group, CAGS EEV n =3/group). Above: ankle pathology score (peripheral arthritis and enthesitis); representative H&E sections of enthesis Achilles tendon. Below: skin pathology score (composite score stratum corneum, epidermis and dermis); representative H&E sections of ear. (C) Histopathological scoring of sacroiliitis; representative H&E images. Inflammatory infiltrate is indicated between white brackets ([]). A comparison is made with the TNFΔARE model with established axial inflammation (TNFΔARE/+ n=10, TNF+/+ n=8). All graphs show mean (s.e.m.), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/rheumatology/62/9/10.1093_rheumatology_kead022/2/m_kead022f2.jpeg?Expires=1747881667&Signature=wlKRE211yW6epzMoTN0pOWSp~3PNZfB5b2rG2p7rkM9067GPfWFVbsTdnGzpCUrow4IXQYqtDElNnWXNEKfZkDFX2mecwgwzv4sjApCxzjS3SaQxGBl-zf-bOtq-p2ApWiHn~5E2-S4kc6knbioLL72KOgYewFnmfFGNwDKTznYNPeapOqiQ0ATKj5C-AVsvB4WBFUkyREr107iIsPM6jslb93F6iQ7EWgPTYPIuuBPXC2xC~PdXxywIs~o8SGxMlpZA0MilfqWocMjrCfcI4OD7Cx1spy2E7RPG0nn7ov30DwobeT19IPtOvqvS1CjmOpmD28BnsoOyMGv0ToJfNg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
RORγt-blockade efficiently suppresses peripheral arthritis, enthesitis and dermatitis but does not significantly improve mild sacroiliitis. (A) Clinical scoring for IL-23 EEV-injected mice treated with RORγt inhibitor (n =11) or vehicle (n =3). Above: peripheral joint inflammation (peripheral arthritis, dactylitis, enthesitis). Below: dermatitis (ear, snout and blepharitis). (B) Histopathology (IL-23 EEV n =10–11/group, CAGS EEV n =3/group). Above: ankle pathology score (peripheral arthritis and enthesitis); representative H&E sections of enthesis Achilles tendon. Below: skin pathology score (composite score stratum corneum, epidermis and dermis); representative H&E sections of ear. (C) Histopathological scoring of sacroiliitis; representative H&E images. Inflammatory infiltrate is indicated between white brackets ([]). A comparison is made with the TNFΔARE model with established axial inflammation (TNFΔARE/+ n=10, TNF+/+ n=8). All graphs show mean (s.e.m.), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Additionally, we assessed the effect of RORγt-blockade in the rat adjuvant-induced arthritis (AIA) model, representing an IL-17 dependent preclinical arthritis model [25], without development of dermatitis and colitis. PF-06747711 was administered once daily at either 10, 30 or 100 mg/kg dosage for seven consecutive days after disease onset (Supplementary Fig. S4A, available at Rheumatology online). Assessment of RORγt inhibitor plasma concentrations showed that compound exposure was proportional to dose (Supplementary Fig. S4B, available at Rheumatology online) [12]. A significant reduction of hind paw arthritis, measured by paw swelling, and IL-17A levels were observed at all compound doses (Supplementary Fig. S4C and D, available at Rheumatology online).
Recently, there is interest in the role of IL-23 in PsA patients with axial involvement, as controversy exists regarding potential differences with ankylosing spondylitis [26, 27]. Therefore, we assessed sacroiliac joints of IL-23 overexpressing mice via H&E and safranin stainings (Fig. 2C). We observed a mild inflammatory infiltrate, but no signs of cartilage destruction. The development of sacroiliitis was rather mild in IL-23 EEV mice as compared with profound axial features seen in a TNF driven model such as TNFΔARE mice (Fig. 2C; Supplementary Fig. S5, available at Rheumatology online). RORγt inhibition did not significantly improve sacroiliitis.
Because inflammation-induced bone loss is another prominent feature upon IL-23 overexpression [18, 20], we assessed the effect of RORγt-blockade on bone density, cortical and trabecular thickness and the presence of surface erosions by µCT analysis (Fig. 3A–B; Supplementary Fig. S6, available at Rheumatology online). All of these were restored upon PF-06747711 treatment.
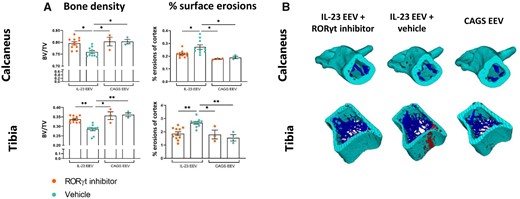
Treatment with RORγt inhibitor restores bone density and reduces bone surface erosions. µCT analysis of tibia and calcaneus bone at day 28 (IL-23 EEV n=10–11/group, CAGS EEV n=3/group). (A) Left: bone density (bone volume/total volume). Right: percentage surface erosions of cortex. All graphs show mean (s.e.m.), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (B) Representative 3D reconstructions of calcaneus and tibial bone with cortex in green, trabeculae in blue and bone surface erosions in red
RORγt inhibition reduces colitis-induced weight loss and improves stool consistency
In addition to arthritis and dermatitis, the IL-23 EEV model is characterized by intestinal inflammation [20, 23]. Indeed, one week after disease induction, mice gradually lost weight (Fig. 4A). Given the conflicting outcomes of IL-23 and IL-17-blockade on gut inflammation, we were particularly interested in the effect of RORγt inhibition. PF-06747711-treated mice were completely protected from weight loss throughout the experiment. Mice with IL-23 overexpression did not develop overt diarrhea but stools were softer and less well-formed. Stool consistency scores improved significantly upon treatment (Fig. 4B).
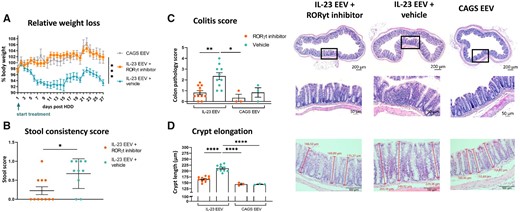
RORγt inhibition reduces colitis-induced weight loss and improves stool consistency. (A) Relative weight loss (% of initial body weight) for IL-23 EEV-injected mice treated with RORγt inhibitor or vehicle (IL-23 EEV n=10–11/group, CAGS EEV n=3/group). (B) Stool consistency score on day 25. (C) Histopathological assessment of colon: colitis score. Representative H&E stained sections. (D) Measurement of crypt length (in µm) in colon (mean of nine measurements per mouse). Representative H&E stained sections. All graphs show mean (s.e.m.), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Weight loss was likely driven by colitis, as histopathological examination demonstrated moderate focal inflammation in the mid and distal part of the colon of IL-23 EEV injected mice (Fig. 4C), which was significantly correlated with the observed weight loss (Supplementary Fig. S7, available at Rheumatology online). Infiltrating immune cells were found in the lamina propria of the mucosa but not in the submucosa. Moreover, clear crypt elongation was measured (Fig. 4D). RORγt-antagonism significantly improved histological colitis scores and reduced crypt length, thereby offering protection towards gut inflammation.
Blocking RORγt prevents γδ-T cell expansion and strongly inhibits T cell-derived IL-17A and IL-22 production
γδ-T cells were previously shown to be drivers of arthritic disease in murine IL-23 overexpression [28, 29]. Furthermore, CD4+ T cells were found to be partially responsible for IL-23-induced bone loss [18]. Therefore, immune profiling of γδ-T cells next to conventional T cells was performed by flow cytometry at day 28. IL-23 overexpression led to an increased prevalence of splenic γδ-T cells, both in terms of frequency (Fig. 5A) and absolute cell counts (Supplementary Fig. S8A, available at Rheumatology online). As a result, conventional T cells were proportionally reduced (Fig. 5A) although absolute cell counts were not affected (Supplementary Fig. S8A, available at Rheumatology online). Remarkably, dosing with PF-06747711 completely inhibited the observed γδ-T cell expansion (Fig. 5A;Supplementary Fig. S8A, available at Rheumatology online).
![Blocking RORγt prevents γδ-T-cell expansion and strongly inhibits T-cell-derived IL-17A and IL-22 production. Immunophenotyping by flow cytometry on day 28 (IL-23 EEV n = 10–11/group, CAGS EEV n = 3/group). (A) Prevalence of splenic γδ-T cells and conventional T cells. Representative pseudocolor plots of γδ-T cell staining. (B) Relative contribution [mean (s.e.m.)] of γδ-T cells vs conventional T cells in cytokine production in spleen, popliteal lymph nodes (PLN) and inguinal lymph nodes (ILN) of IL-23 EEV injected mice treated with vehicle. (C) IL-17A production comparing splenic γδ-T cells vs conventional T cells. Representative pseudocolor plots. (D) Pie diagrams of IL-17A coproduction with IL-22, IFN-γ and TNF-α in spleen (mean). Representative pseudocolor plots. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/rheumatology/62/9/10.1093_rheumatology_kead022/2/m_kead022f5.jpeg?Expires=1747881667&Signature=rCCHoRzDo8mSDVJXF7WpQhsAS3UUaggXPiaodrfK4nIzJq2B~4Pwc63onMx0SO-YhJfZfRZwhbGc9ltvKqr4C9ycvmg3sKApig0ywHIJKMXqRn64BhN8BqzYiB332FWVCacYlkdDtu5TbGeT6MVt9IZiDvMrCbPfw9xKXZcZMv2cp7wwrOOmAednUChf6levw~Yn5EHHDrp4h4VTVYXNGCpUOqxg056R3t0JdZEt4iHK0nMVQU9kr94Fg4SVsdpGx2bLUXRyAhQk5nJegNNaO4sGg6HyK~qZB4vx6w7-UY7KcxDCTza9HLkio0U6blUXNoX2JEi5Bc~sIrwEzWHciA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Blocking RORγt prevents γδ-T-cell expansion and strongly inhibits T-cell-derived IL-17A and IL-22 production. Immunophenotyping by flow cytometry on day 28 (IL-23 EEV n = 10–11/group, CAGS EEV n = 3/group). (A) Prevalence of splenic γδ-T cells and conventional T cells. Representative pseudocolor plots of γδ-T cell staining. (B) Relative contribution [mean (s.e.m.)] of γδ-T cells vs conventional T cells in cytokine production in spleen, popliteal lymph nodes (PLN) and inguinal lymph nodes (ILN) of IL-23 EEV injected mice treated with vehicle. (C) IL-17A production comparing splenic γδ-T cells vs conventional T cells. Representative pseudocolor plots. (D) Pie diagrams of IL-17A coproduction with IL-22, IFN-γ and TNF-α in spleen (mean). Representative pseudocolor plots. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Although γδ-T cells represented a minor population compared with conventional T cells (Supplementary Fig. S8A, available at Rheumatology online), they were responsible for more than half of the T cell-derived IL-17A and IL-22 production in spleen (Fig. 5B). A total of 25.4% of splenic γδ-T cells were producing IL-17A vs only 3.4% of conventional T cells (Fig. 5C). Moreover, in popliteal lymph nodes (PLN) and inguinal lymph nodes (ILN), γδ-T cells represented the dominant IL-17 producing cell type (Fig. 5B). Conversely, conventional T cells were the main source of T cell-derived IFN-γ and TNF-α. RORγt-antagonism inhibited IL-17A production in both cell types, respectively, by 73.5% and 66.1% (Fig. 5C), and also absolute counts of IL-17A+ cells in spleen and joint-draining lymph nodes were significantly diminished (Supplementary Fig. S8B and C, available at Rheumatology online). Of note, IL-17A-producing invariant natural killer T (iNKT) cells, low abundantly present in spleen and lymph nodes, were equally suppressed (data not shown), supporting the broad type 17-neutralisation effect upon in vivo RORγt inhibition.
Next, we examined dual production of cytokines as multi-cytokine producers may have pleiotropic effects [30]. In the IL-23 EEV model, 62.1% of IL-17A + γδ-T cells coproduced IL-22, whereas 35.1% coproduced TNF-α. Only a minor population was coproducing IL-17A and IFN-γ (mean 5.5%). Blockade of RORγt significantly reduced the amount of both IL-17A single positive γδ-T cells and conventional T cells as well as IL-17A+ cells coproducing IL-22, IFN-γ or TNF-α. This was the case both proportionally (Fig. 5D) as for absolute cell counts (Supplementary Fig. S8D, available at Rheumatology online), except for the scarcely present IL-17A+IFN-γ+ conventional T cells. Single IL-22 producers were also inhibited, although this was only statistically significant for γδ-T cells. IFN-γ and TNF-α single producers on the other hand were not affected by RORγt inhibition.
RORγt inhibitor good responders have equal IL-17A suppression but have fewer IL-17A- IFNγ+ conventional T cells compared with partial responders
Remarkably, we observed a duality in clinical response to RORγt inhibition in the IL-23 EEV model, particularly towards joint inflammation. Treated mice could be subdivided into a group of good responders that did not develop notable symptoms and a group of partial responders. The arthritis-dermatitis composite scores of the latter demonstrated a response that was intermediate compared with RORγt inhibitor-good responders and vehicle-treated mice. However, they showed no significant improvement specifically for joint inflammation, while they responded similarly for dermatitis as good responders (Supplementary Fig. S9A, available at Rheumatology online). The differential clinical effect could not be explained by lower serum IL-23 levels in good responders (Supplementary Fig. S9B, available at Rheumatology online).
Intriguingly, mice showing only a partial clinical response had a higher prevalence of splenic IL-17A-IFN-γ+ conventional T cells (P < 0.05) and γδ-T cells (P =0.10) compared with good responders (Supplementary Fig. S9C, available at Rheumatology online). Importantly, no difference was found in frequency of IL-17A+ cells, showing that IL-17A suppression was similarly achieved in both good and partial responders (Supplementary Fig. S9D, available at Rheumatology online).
RORγt inhibition profoundly modulates IL-23-induced type 17 and alarmin gene signatures in PsA-related tissues
After determining the effect of PF-06747711 on T cell-derived cytokine production, we assessed its impact on overall gene expression using qRT-PCR analysis in disease-relevant tissues, being skin, ankle, knee synovium and colon (Fig. 6A;Supplementary Fig. S10, available at Rheumatology online). This confirmed elevation of Il17a and Il22, next to the other pathogenic IL-17 isoform Il17f, in the IL-23 EEV model with subsequent profound suppression by RORγt inhibition (Il17f: P < 0.05 in skin and ankle). Clinical scores for dermatitis, peripheral joint inflammation and weight loss were significantly correlated with Il17a and Il22 gene expression in skin, ankle and colon, respectively (Fig. 6B). In contrast, Il17e was diminished in skin and colon of IL-23 EEV-injected mice. IL-17E is a distinct member of the IL-17 cytokine family that promotes type 2 responses and can have a pro-inflammatory role, but can also be anti-inflammatory by suppressing Th17 function [31]. Indeed, upon IL-23 overexpression a downregulation of Gata3 was noted in skin. Consequently, treatment with PF-06747711 led to significantly increased cutaneous Il17e and Gata3 expression compared with placebo. Overall type 1 responses were upregulated in the IL-23 EEV model as evidenced by a moderate increase in Tbx21 (Tbet) and Ifng. Blocking RORγt, however, only had a significant suppressive effect on Ifng in skin. Additionally, IL-23 overexpression caused significant upregulation of Tnf expression in all tissues but a reduction by PF-06747711 therapy was only seen in knee synovium and colon. S100a8 and S100a9, which form the heterodimer calprotectin, are two pro-inflammatory members of the S100-alarmin family that are classically associated with psoriasis [32]. As expected, a striking upregulation of both genes was detected in the IL-23 EEV model, which was completely blocked by RORγt inhibition (Fig. 6A).
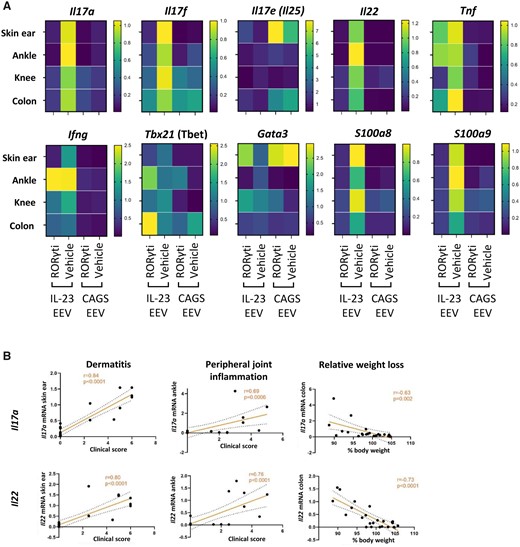
RORγt inhibition profoundly modulates IL-23-induced type 17 and alarmin gene signatures in PsA-related tissues. qRT-PCR analysis evaluating gene expression in disease relevant tissues on day 28 (IL-23 EEV n=8–9/group, CAGS EEV n =3/group). (A) Heatmaps showing mean fold change in gene expression compared to IL-23 EEV + vehicle. (B) Correlation of IL-17A and IL-22 gene expression (fold change) with clinical scores for dermatitis and peripheral joint inflammation and relative weight loss at endpoint (Pearson correlation coefficient)
Discussion
Despite the critical importance of the IL-23/IL-17 immune axis in PsA, conflicting results obtained with IL-23 and IL-17 blocking antibodies in various PsA disease domains point towards a complex regulation of cytokine networks that is not yet fully understood [2–4]. Targeting key downstream molecules in the type 17-signalling pathway may help elucidate some of these uncertainties and could lead to new therapeutic approaches. Therefore, we evaluated an oral RORγt inhibitor, PF-06747711, in the IL-23 EEV mouse model, an enhanced version of the minicircle model, with long-term systemic IL-23 overexpression and development of PsA-like symptoms [23]. PF-06747711 demonstrated significant therapeutic efficacy in all disease domains, including gut inflammation. This is in sharp contrast to the inefficacy of IL-17 targeting in treatment of human IBD, with even a risk of inducing flares [6, 7]. Conversely, IL-17 inhibitors ameliorate spinal inflammation, while anti-IL-23 therapy has failed to improve axial disease in clinical trials [8, 9], an unexpected finding given the multiple genetic associations of IL-23 and related genes with AS. In our study, RORγt inhibition did not significantly improve axial arthritis either. A caveat is that sustained IL-23 overexpression only led to mild sacroiliitis. In that context, it might be valuable to assess RORγt-antagonism in a murine model with more established axial disease. Sherlock et al. briefly described presence of axial enthesitis and sacroiliitis in the IL-23 minicircle model [28]. However, two recent studies could not confirm these findings, although only vertebrae and not sacroiliac joints were examined [20, 23]. Overall, the mild signs of sacroiliitis we observed upon IL-23 exposure in vivo, seem to confirm that IL-23 is not a main driver of axial disease. To formally assess the role of IL-23 in PsA patients with axial involvement, results of ongoing randomized clinical trials need to be awaited [26].
A potential explanation for the contrasting success of IL-17 inhibitors in AS could be IL-23 independent IL-17 production in spinal entheses [4]. Intriguingly, it was shown that resident spinal entheseal γδ-T cells are able to produce IL-17A in the absence of IL-23 [17]. Our serum and qPCR analyses clearly demonstrated an inhibitory effect of RORγt-blockade on respectively systemic and peripheral tissue IL-17A (and IL-17F) expression in the IL-23 EEV model. However, this does not exclude that secondary IL-23 (and potentially RORγt) independent IL-17 responses are responsible for the profound axial phenotype in humans, which warrants further investigation.
Accumulating evidence shows a prominent role for γδ-T cells in the pathogenesis of psoriasis, PsA and other spondyloarthritides [2, 4, 10, 11, 33, 34]. In line with this, γδ-T cells were indispensable in both the transdermal IL-23 injection and imiquimod models of psoriasis [10], next to the systemic IL-23 minicircle model of PsA [16], as TCRδ-/- mice were protected from disease. In the IL-23 EEV model, we observed a significant increase of splenic γδ-T cells, with a proportional decrease of conventional T cells. Interestingly, the systemic expansion of γδ-T cells was completely interrupted upon RORγt-antagonism. This is of particular interest as it was recently shown that peripheral γδ-T cells play a pivotal modulatory role in this model by recruiting neutrophils to skin and joint tissues where they induced local inflammation [16]. Consistently, we found a strong upregulation of alarmin S100a8 and S100a9 gene expression in PsA-related tissues, which was completely prevented by blocking RORγt. Of note, this was similarly detected in colon, where faecal calprotectin, the heterodimer formed by S100A8 and S100A9, represents an established biomarker for IBD in SpA patients [35].
The γδ-T cell dependent influx of neutrophils to the joint and skin was not surprising because γδ-T cells are major producers of IL-17 [2, 29], a well-known neutrophil recruiter. This was confirmed in our study, as γδ-T cells were the predominant IL-17A source in spleen and even more in joint-draining lymph nodes, over conventional T cells. Likewise, the percentage of IL-17A+ cells was ∼10-fold higher in γδ-T cells than in conventional T cells. Their paramount IL-17 secreting capacity underscores the significance of our observation that γδ-T cell expansion was selectively suppressed by RORγt inhibition. However, it should be noted that in terms of absolute numbers, conventional T cells are the more abundant population and Th17 cells were clearly induced upon IL-23 overexpression, indicating they might also play a significant role in SpA pathology [18, 20]. Therefore, it was particularly interesting that blocking RORγt abolished IL-17 and IL-22 secretion by both γδ-T cells and conventional Th17 cells, respectively consistent with our previous work using human peripheral blood cells (IL-17) [11] and with findings in the CIA model (IL-22) [36]. In contrast, human IL-22+ γδ-T cells seemed selectively spared in vitro [11]. This differential effect on IL-22 could reflect discrepancies in human vs murine γδ-T cell responses or be attributed to the different origin of isolated cells. Serum cytokine measurement and gene expression analysis of skin, joint and gut tissue showed suppression of both IL-17A and IL-22 by PF-06747711 as well, which could be beneficial as a pro-inflammatory role has been ascribed to IL-22 in arthritis [37, 38] and psoriasis [39]. Although IL-22 protects intestinal barrier integrity [40], we observed an improvement of colitis upon RORγt-antagonism, despite reduction of IL-22. In fact, IL-22 can also induce inflammatory responses in the gut [41, 42]. Moreover, both IL-17A and IL-22 are strongly involved in bone remodelling. We confirmed the induction of bone resorption and surface erosions by IL-23 [18, 23], using µCT scans. As expected by the strong local suppression of type 17 responses, RORγt-blockade efficiently restored all bone parameters.
IL-17A is typically the most studied member of the IL-17 family, although IL-17F also contributes to PsA and psoriasis pathology and simultaneous targeting of both cytokines is therefore thought to provide better disease control [2, 43, 44]. RORγt-blockade resulted in distinct downregulation of both Il17f and Il17a in PsA-affected tissues, illustrating a therapeutic advantage over IL-17A inhibitors. Il17e on the other hand, was significantly downregulated in skin and colon after IL-23 overexpression, but not in the joint. It is known that IL-17E can act as anti-inflammatory by suppressing Th17 function [31, 45] while promoting type 2 responses, which is in line with decreased Gata3 expression in skin upon IL-23 overexposure. Interestingly, PF-06747711 could restore both Il17e and Gata3 expression. However, some reports mention pathogenic effects of IL-17E in the skin [31], contrasting our results and necessitating further investigation to understand the role of IL-17E in PsA and other inflammatory diseases. In contrast to type 2 responses, type 1 genes were upregulated in the IL-23 EEV model. RORγt-antagonism reduced Ifng expression in skin, but otherwise type 1 genes were not significantly altered. IL-23 gene transfer additionally induced Tnf expression, which was restored by PF-06747711 in synovium and colon. However, the contribution of TNF-α in this model might be marginal as it has been shown that TNF-α inhibition does not prevent disease in IL-23 overexpressing mice [28]. One limitation of our work includes that we did not look at the local cellular source of IL-17 and other upregulated signature cytokines in skin, joint and gut tissue upon IL-23 overexpression. However, Leys et al. demonstrated that T cells were the main source of IL-17A in the skin of mice upon IL-23 stimulation [46] and another study identified dermal γδ-T cells as the dominant IL-17 producers [10]. Nonetheless, we performed immune profiling of spleen and joint-draining lymph nodes, where γδ-T cells were clearly the prominent producers of IL-17A and IL-22. In addition, IL-17-producing iNKT cells were also strongly suppressed upon RORγt-blockade, suggesting that other IL-17 producing cells such as MAIT cells or ILCs may be influenced by RORγt targeting as well [19, 20, 22].
Strikingly, we noticed a duality in clinical outcome upon RORγt-blockade, with good and partial responders. The latter especially displayed a diminished suppression of arthritis while dermatitis was still strongly reduced, which is of particular interest because this mimics the divergent skin-joint response in human PsA [47]. Partial responders showed identical suppression of IL-17A but remarkably had more IL-17A-IFN-γ+ conventional T cells, which could imply an immunological escape with residual inflammation. Such a mechanism could provide a rationale for targeted treatment approaches, which as proof-of-concept showed promising results in PsA [48].
In conclusion, modulating RORγt transcriptional activity demonstrated clinical efficacy in a preclinical PsA model with protection towards intestinal inflammation, as opposed to IL-17 inhibitors in human clinical studies. Mechanistically, a broader action with suppression of both IL-17A, IL-17F and IL-22 was obtained. γδ-T cells, master producers of these cytokines, were effectively targeted, next to Th17 cells.
Supplementary material
Supplementary material is available at Rheumatology online.
Data availability
The data underlying this article are available upon reasonable request to the corresponding author.
Funding
This work was supported by the Research Foundation-Flanders (Fonds voor Wetenschappelijk Onderzoek Vlaanderen) (FWO1199117N/); Pfizer (WI231622) and the special research fund of the Ghent University (BOF-UGent) (BOF.EXP.2017.0007) to the UGCT Centre of Expertise.
Disclosure statement: C.A., M.H., P.S., S.S. and G.B. are/were employees of Pfizer Inc. The authors declare no other potential conflicts of interests.
Acknowledgements
We would like to thank Dr Anton De Spiegeleer, Karlijn Debusschere and Gillian Blancke for their technical support. We would like to thank Ivan Josipovic for the help he provided for the µCT scans. We would like to acknowledge the following colleagues from Pfizer Inc: Sarah Osgood, Stacey Becker, Christopher Holliman and Andre Negahban for their technical work in determining the exposure values of RORγt antagonist and Lisa Schopf and Cara Williams for critical reading of the manuscript.
References
Gracey E, Hromadova D, Lim M et al. TYK2 inhibition reduces type 3 immunity and modifies disease progression in murine spondyloarthritis. J Clin Invest 2020;
Author notes
Present address: Novartis Institutes of Biomedical Research, Cambridge, MA, USA
D.E. and K.V. contributed equally.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments