





Abstract
Desiccation is typically fatal, but a small number of land plants have evolved vegetative desiccation tolerance (VDT), allowing them to dry without dying through a process called anhydrobiosis. Advances in sequencing technologies have enabled the investigation of genomes for desiccation-tolerant plants over the past decade. However, a dedicated and integrated database for these valuable genomic resources has been lacking. Our prolonged interest in VDT plant genomes motivated us to create the “Drying without Dying” database, which contains a total of 16 VDT-related plant genomes (including 10 mosses) and incorporates 10 genomes that are closely related to VDT plants. The database features bioinformatic tools, such as blast and homologous cluster search, sequence retrieval, Gene Ontology term and metabolic pathway enrichment statistics, expression profiling, co-expression network extraction, and JBrowser exploration for each genome. To demonstrate its utility, we conducted tailored PFAM family statistical analyses, and we discovered that the drought-responsive ABA transporter AWPM-19 family is significantly tandemly duplicated in all bryophytes but rarely so in tracheophytes. Transcriptomic investigations also revealed that response patterns following desiccation diverged between bryophytes and angiosperms. Combined, the analyses provided genomic and transcriptomic evidence supporting a possible divergence and lineage-specific evolution of VDT in plants. The database can be accessed at http://desiccation.novogene.com. We expect this initial release of the “Drying without Dying” plant genome database will facilitate future discovery of VDT genetic resources.
Introduction
Desiccation tolerance (DT) is an ancient trait that can be observed across all lineages of land plants, primarily in reproductive tissues such as seeds and spores. However, a relatively small number of species can endure drying of vegetative tissues, reaching tissue water potentials of −100 MPa or lower without dying; a practical definition of DT (Alpert and Oliver 2002). Vegetative desiccation tolerance (VDT) has evolved in a relatively small number of vascular plants, whereas VDT species are more widely distributed in bryophytes (Wood 2007; Marks et al. 2021). Molecular mechanisms that are common to VDT have been identified (Oliver et al. 2020), and signatures of convergent evolution of VDT, such as massive tandem duplications of early light-induced protein (ELIP) genes in VDT plants, have been documented (VanBuren et al. 2019, 2023). Nevertheless, there is also evidence to support lineage- and species-specific VDT mechanisms (Xu et al. 2018; Oliver et al. 2020), indicating and reflecting the complex physiology and evolution underlying VDT.
Substantial advances have been made in our understanding of the molecular and physiological mechanisms of DT (Oliver et al. 2020) including the genomic underpinnings of VDT that has been advanced by the sequencing of several VDT plant genomes in the last decade. Within the VDT eudicots, the sequenced genomes all belong to the order Lamiales. The first sequenced VDT angiosperm genome was reported for the eudicot Dorcoceras hygrometricum (formerly Boea hygrometrica) of the Gesneriaceae family (Xiao et al. 2015). Other sequenced DT eudicots are Lindernia brevidens (Linderniaceae), together with a desiccation sensitive (VDS) closely related species, Lindernia subracemosa (VanBuren et al. 2018a). Most recently, the complex genome of the model VDT plant blue gem (Craterostigma plantagineum, also Linderniaceae) (Bartels 2005) was sequenced and revealed a massive expansion of ELIP genes (VanBuren et al. 2023).
Of the monocot VDT genomes, the majority are representative grasses. The genome of Oropetium thomaeum, with a small genome size (∼244 Mbp) and a compact genome structure compared to other grass genomes, was the first to be sequenced (VanBuren et al. 2015). Two more VDT grass genomes were both sequenced along with a closely related species in the same genus: the poikilochlorophyllous VDT (PDT) Eragrostis nindensis and its desiccation-sensitive (DS) relative teff (Eragrostis tef) (Pardo et al. 2020; VanBuren et al. 2020), and Sporobolus stapfianus together with its desiccation-sensitive (DS) sister species S. pyramidalis (Chavez Montes et al. 2022). In PDT species chlorophyll is degraded, and thylakoids are dismantled during desiccation which contrasts with homoiochlorophyllous VDT (HDT) plants where chlorophyll is retained, and the photosynthetic apparatus is well protected. PDT has only been found in monocots as currently surveyed (Marks et al. 2021). Another two sequenced VDT genomes belong to the monocot family Velloziaceae (Pandanales). One was Xerophyta schlechteri (previously Xerophyta viscosa), another poikilochlorophyllous VDT (PDT) species with a sequenced genome (Costa et al. 2017). The second VDT member of the Velloziaceae that has a sequenced genome is Acanthochlamys bracteata, which is also cold tolerant (Gao et al. 2021; Xu et al. 2022).
The first lycophyte genome sequenced was that of Selaginella moellendorffii, a DS species (Banks et al. 2011). Moreover, two Selaginella species, the flower of stone (S. lepidophylla) and S. tamariscina, which are VDT, have since been sequenced (Xu et al. 2018; VanBuren et al. 2018b). Additionally, the genome of Isoetes taiwanensis, a lycophyte that exhibits crassulacean acid metabolism, has been sequenced and has reported to be VDT (Huang et al. 2018; Wickell et al. 2021; Alejo-Jacuinde and Herrera-Estrella 2022). However, it is only the corm that survives desiccation as the leaves do not survive drying (Huang et al. 2018), so it is questionable that this species is indeed VDT. In liverworts, the first released and improved chromosomal-level genome was that of Marchantia polymorpha (Bowman et al. 2017; Diop et al. 2020). DT can be induced in the normally DS M. polymorpha by the application of exogenous abscisic acid. Although a genome is available for Marchantia paleacea (Radhakrishnan et al. 2020), it is unclear if M. paleacea is VDT. Nonetheless, we have included it in the database as VDT is a phenotype that exhibits a high degree of plasticity and is more frequent in bryophytes (Wood 2007; Stark 2017; Oliver et al. 2020; Marks et al. 2021) and it will also serve in comparative genomic analyses for future studies.
The model moss Physcomitrium patens (previously known as Physcomitrella patens) was the first bryophyte genome to be reported, and although fragmented initially it was improved to a chromosomal-level assembly (Rensing et al. 2008; Lang et al. 2018). Similar to the liverwort M. polymorpha, priming or hardening is also required for VDT in P. patens (Koster et al. 2010). Other mosses that have been well studied for VDT mechanisms include tortula moss (Syntrichia ruralis), S. caninervis, and silvergreen bryum moss (Bryum argenteum), which are emerging as VDT models. The S. caninervis genome has been reported (Silva et al. 2021), and more Syntrichia genomes are being sequenced by Oliver and colleagues. We have also included the B. argenteum genome (Gao et al. 2023) in the database. The ceratodon moss (Ceratodon purpureus) and antifever fontinalis moss (Fontinalis antipyretica) are experimentally determined as VDT mosses (Wood 2007), and their genomes are available (Yu et al. 2020; Carey et al. 2021). The genome of the Antarctic pohlia moss (Pohlia nutans) has been sequenced and it is considered VDT, based on the conditions found in its extreme terrestrial habitats (Liu et al. 2022), and that several Pohlia species have been documented to be VDT (Wood 2007; Marks et al. 2021). The hypnalean moss Calohypnum plumiforme (previously Hypnum plumaeforme) was also reported to be VDT (Liu et al. 2022) with a genome available (Mao et al. 2020). However, VDT evaluations for another two sequenced hypnalean mosses, the seductive entodon moss (Entodon seductrix) and curveleaf hypnum moss (Hypnum curvifolium) (Yu et al. 2022), have not been reported.
Our objective for this study was to consolidate the genomic resources for VDT plants into a database that would serve as a resource for those in the field and to stimulate interest in this important topic. We have named the database “Drying without Dying” and it was constructed by integrating the genomic and transcriptomic resources for VDT plants that were currently available. With the increasing number of plant genomes being sequenced, the database will be updated with more VDT genomes and improved bioinformatic functions over time. In addition to the comprehensive annotation and basic bioinformatic tools provided in the database, we assessed its utility and effectiveness by analyzing tandemly duplicated gene families and their contributions to the expansion of corresponding PFAM families which revealed species- and lineage-specific “expansion by tandem duplication” patterns of several VDT-related families. Our findings suggest that CUPIN genes, which encode seed storage proteins, and cytochrome p450 (CYP) genes are among the most widely tandemly duplicated families across taxa. We discovered that the drought-responsive ABA transporter AWPM-19 (Yao et al. 2018) and chaperone DnaJ-C families are significantly tandemly duplicated in bryophyte genomes but rarely tandemly duplicated in tracheophytes. Along with transcriptomic analyses we have provided evidence to support a divergent and lineage-specific road map for the evolution of VDT. We hope that the release of this database will be useful to researchers in the field and stimulate further research on both VDT and seed DT alike.
Results and discussion
Collection and general characterizations of VDT genomes
To construct the database, we collected a total of 16 validated VDT genomes together with 10 closely related plant genomes as essential outgroups, as introduced above, including 3 eudicots, 7 monocots, 4 lycophytes, 2 liverworts, and 10 mosses. We included the 10 moss genomes in our database to not only facilitate VDT research but also enable phylogenetic analyses to incorporate more genes from nonvascular plants, which have long been underrepresented by the lone P. patens genome. Among the 26 genomes, only 12 were assembled to the chromosomal level (Fig. 1).
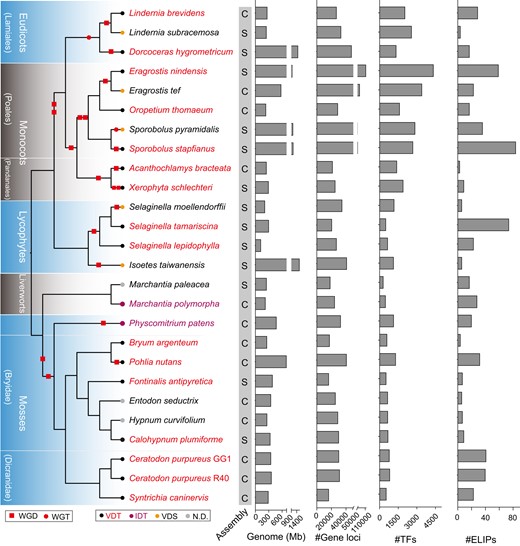
Summary of sequenced plant genomes from distinct clades with DT-related phenotypes. A consensus phylogeny was depicted along with reported whole-genome duplication (WGD) or whole-genome triplication (WGT) events. The genomes that have been assembled at scaffold or chromosome levels were denoted as “S” or “C” to the right of each species. Assembled genomes size, number of annotated genes, number of TF and ELIP genes were also revealed. The delineated sections to the left of the phylogenetic tree depict different plant lineages. Dorcoceras hygrometrium, previously B. hygrometrica; X. schlechteri, previously X. viscosa; P. patens, previously Physcomitrella patens. VDT, vegetative desiccation tolerant; IDT, VDT is inducible by special conditions such as exogenous ABA; VDS, vegetative desiccation sensitive; ND, not specifically determined; TF, transcription factor; ELIP, early light-inducible protein. The phylogenetic relationships were depicted as unscaled cladograms.
Based on reported genome duplication events for these genomes (Fig. 1), it appears that VDT in angiosperms has frequently evolved from “recent” polyploids that were postulated to be more resilient to the extreme environmental stresses that occur during ancient mass extinction events (Lohaus and Van de Peer 2016). Doubled genomes generated extra gene dosage and provided the genetic materials necessary for functional innovations. In lycophytes, an extra round of genome duplication in the DS S. moellendorffii was observed (Wang et al. 2020), but there is no report that provides evidence for a genome duplication event in liverworts. In mosses, an ancient genome duplication event was assessed to have occurred upon the early diversification of Bryopsida, which led to the significant retainment of stress-responsive genes after the paleo-polyploidy event (Gao et al. 2022). Overall, the evolutionary linkage between polyploidy and VDT seems weak. We have summarized the genome size and number of gene loci in each genome, which revealed a high level of variability in these genomes (Fig. 1). Transcription factors in these genomes were also annotated, with angiosperms, in general, possessing conspicuously larger transcription regulatory reservoirs than bryophytes and lycophytes.
The tandem proliferation of ELIP genes has been linked to VDT in plants (VanBuren et al. 2019). We observed conspicuously stronger expansions of ELIP members in VDT plants than in their close relatives in the same genus (Fig. 1), such as L. brevidens vs L. subracemosa, E. nindensis vs E. tef, S. stapfianus vs S. pyramidalis, and S. tamariscina vs S. moellendorffii. The DS species E. tef and S. pyramidalis have also accumulated larger numbers of ELIPs than VDT species such as D. hygrometricum, Xerophyta schlecteri, and O. thomaeum, but this might reflect their high-light habitats or their extensive polyploidy. However, the strong expansion of ELIPs were not observed in documented VDT species such as Acanthochlamys bracteate, C. plumiforme, and B. argenteum, further suggesting the existence of intricate lineage- and species-specific VDT mechanisms.
Construction and functional overview of the “Drying without Dying” database
The information and bioinformatic tools provided in the database are intuitive and easy to use, and we included examples of how each tool can be used (Fig. 2). Each gene in each genome can be accessed by the user to obtain basic functional descriptions, as well as the ability to visualize gene structure and chromosomal location via the JBrowser hyperlink. The Gene Ontology (GO) terms and KEGG assignments for each gene are linked to the respective database for further investigation, and nucleotide and amino acid sequences in FASTA format are readily available. Users can perform blast searches for homologs, and the target gene information is linked in the database, and alignment results can be graphically visualized and downloaded. In addition to blast searches, we have incorporated orthofinder (Emms and Kelly 2019) clustering results in the database for easy access to homologs through the “Homolog Search” tool. The number of homologs in each genome, sequence alignments, and sequences of homologous genes can be conveniently inspected and downloaded. We have also included “gene sequence extraction” and “sequence fetch” tools for quick access to protein-coding sequences and genomic DNA sequences in a target region, such as untranslated, promoter, and intergenic regions.
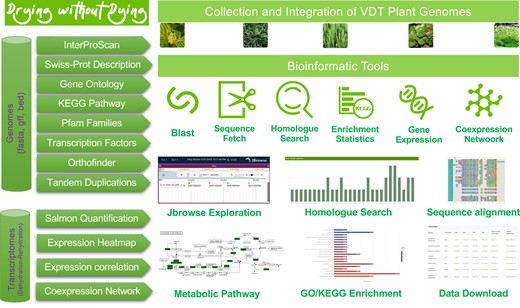
Data sources, bioinformatic integration, database structure and functions of the “Drying without Dying” plant genome database. All genomic and transcriptomic data, including functional annotations using multiple databases and homologous clustering results, together with the bioinformatic tools were incorporated into MongoDB to construct the database. The original data can be accessed from the download section of the website (http://desiccation.novogene.com).
In terms of transcriptomics, our focus was primarily on the changes in transcript abundance during dehydration and rehydration. To this end, we have made it easy to obtain abundance profiles by providing a list of genes of interest for a given species. This generates a table of transcripts per million (TPM) values and a preliminary heatmap. The abundance data file and heatmap can be customized by selecting preferred time points and color schemes. Additionally, we have incorporated co-expression relationships of gene pairs within a species, which can be easily queried by providing a list of interested genes. The output is a three-column table with gene pairs and corresponding Pearson correlation coefficient (PCC) values above a specific threshold, which can be imported to network visualization tools such as Cytoscape (Saito et al. 2012) and Gephi (Bastian et al. 2009). We have also implemented the GO and KEGG pathway enrichment tools in the database by conducting widely employed hypergeometric tests. Enrichment analyses can be achieved by providing a list of genes of interest, for example, a group of genes with significantly differential transcript abundances at different hydration stages. The statistical enrichment results can be visualized using column or point charts, and the gene count and P-value thresholds are adjustable. All detailed statistical results can be exported to a table and further scrutinized, and the corresponding GO terms and metabolic pathways can be conveniently linked out for further detailed exploration.
Statistical analyses of tandem gene duplications
Gene duplications provide genetic material for functional innovations, which can facilitate adaptations to environmental stresses, particularly lineage-specific tandem duplications (Hanada et al. 2008). Tandemly duplicated genes (TDGs) in VDT species, such as O. thomaeum (VanBuren et al. 2015), S. lepidophylla (VanBuren et al. 2018b), X. schlechteri (Costa et al. 2017), and L. brevidens (VanBuren et al. 2018a), are enriched in functional categories related to dehydration. The discovery of an association between massively tandemly duplicated ELIPs and VDT also prompted us to further analyze the TDGs in all 26 genomes (Fig. 3A). Our integrated analyses showed that grasses have conspicuously more TDGs (Fig. 3A) and duplication clusters (Fig. 3B) which occupy higher proportions of respective genomes (Fig. 3, C and D) than other lineages, particularly for the two Eragrostis species. While for moss genomes, tandem duplications had less variations in the terms of both TDGs and tandem clusters, and take up a smaller proportion of the genomes. It should be noted that TDG analyses might be hindered by fragmented genome assemblies (e.g. F. antipyretica). In the “Drying without Dying” database, we deposited the list of tandem duplication clusters for each genome in the “Download” section.
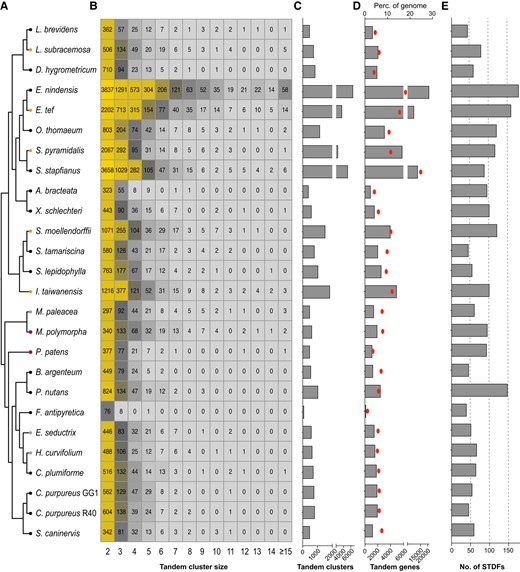
Tandem gene duplication profiles of the investigated plant genomes. A) The species phylogeny was consistent with that depicted in Fig. 1. B) Numbers of tandem duplication clusters containing different numbers of genes were depicted in the heatmap. Tandem duplication clusters containing more than 14 genes were combined. C) The numbers of tandem duplication clusters in each genome, tandemly duplicated homologous genes within a maximum distance of 10 genes were merged into one tandem cluster. D) The numbers of TDGs were shown as bar charts, and their corresponding proportions of the whole genomes are depicted as red dots. E) The numbers of significantly tandemly duplicated PFAM families (STDFs) within each of the investigated genomes were depicted as bar charts. STDFs are identified using a hypergeometric test with a FDR threshold of ≤ 0.05. The phylogenetic relationships were depicted as unscaled cladograms.
Considering the distinct genome evolutionary background observed across taxa (Figs. 1 and 3), we conducted rigorous over-representative analyses of TDGs to assess their contribution to corresponding PFAM families. We probed significantly tandemly duplicated families (STDFs) in each of the genomes (Fig. 3E, Supplementary Table S1) and subsequently analyzed a manually collected subset of families across these taxa. We generated a heatmap depicting the phylogenetic distributions for STDFs (Fig. 4A, Supplementary Table S1), which revealed some intriguing patterns. For instance, genes containing the cupin domain (PF00190) were significantly tandemly duplicated across land plants (Fig. 4A), with bicupins (i.e. 11S and 7S seed storage proteins) known to be related to DT (Dunwell et al. 2000). This wide phylogenetic distribution of significant tandem duplications of cupin genes may indicate the importance and contributions of seed-related DT genes to terrestrial adaptations in embryophytes. Similarly, genes encoding p450 (PF00067) enzymes were also found to be tandemly expanded across land plants. Notably, two STDFs encoding cell wall proteins exhibit intriguing expansion patterns across land plants; with expansin (PF01357, Expansin_C) not significantly expanded in eudicots and Dirigent (PF03018) not significantly expanded in the Bryidae. We discovered other lineage-specific STDFs, including the caleosin in monocots, LEA-1 in three VDT monocots, LEA-5 in bryophytes and lycophytes, the chaperone DnaJ in P. patens and Bryidae, and the energy metabolism-related ATPase (PF14363, AAA_assoc) in three VDT lycophytes and two VDT monocots. However, we did not see significant tandem duplications of other families of the LEA group, such as LEA-2, 3, 4, 6, and SMP families.
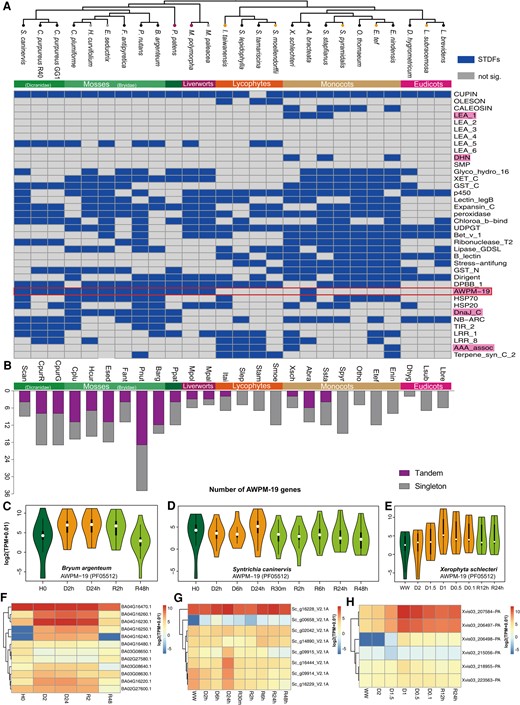
Analysis of STDFs in each of the genomes demonstrates the AWPM-19 family is widely expanded in bryophytes. A) A heatmap illustrates the exemplar STDFs in each of the plant genomes. Blue color indicates that tandem duplications have significantly (hypergeometric test, FDR ≤ 0.05) contributed to the expansion of the PFAM family in corresponding plant genomes, and the AWPM-19 family emerged as a widely STDF in bryophytes but rarely tandemly duplicated in tracheophytes. B) The number of AWPM-19 family members identified in each of the plant genomes. C to E) Overall transcript abundance changes in the two model VDT mosses S. caninervis (8 genes) and B. argenteum (12 genes) and X. schlechteri (6 genes). Dark green, gold, and light green colors indicate hydrated, desiccated, and rehydrated samples, respectively. The white dot represents the median, the thick gray bar in the center represents the interquartile range, the thin gray line represents the rest of the distribution. F to H) Heatmaps show detailed expression profiles of AWPM-19 genes in the three VDT plants.
Prolonged interest in DT in bryophytes prompted us to focus on the AWPM-19 family. AWPM-19 is an ABA transporter that was experimentally validated to mediate ABA influx and to respond to drought in rice (Oryza sativa) (Yao et al. 2018). Through integrated analyses, we discovered that the AWPM-19 gene family was significantly tandemly duplicated in all bryophyte genomes and in only one angiosperm, A. bracteata, a VDT monocot. This phylogenetic distribution pattern (Fig. 4A), coupled with the prevalence of VDT in bryophytes, suggested that AWPM-19 may have played a critical role in the VDT of nonvascular plants. The important role of AWPM-19 in VDT might be further supported by the fact that VDT in P. patens (Khandelwal et al. 2010; Koster et al. 2010), X. schlechteri seedlings (Costa et al. 2017) as well as several other bryophytes (Marks et al. 2021) can be induced by applying exogenous ABA. Further analyses of member distributions across taxa revealed a prominent expansion of AWPM-19 in bryophytes, especially in mosses (Fig. 4B). We also analyzed the expression patterns of AWPM-19 genes in the two VDT moss models, B. argenteum (Gao et al. 2017) and S. caninervis (Yang et al. 2023), which indicated that transcript abundance of AWPM-19 was higher in desiccated tissues, especially in the dried state (Fig. 4, C and D). Dehydration and rehydration transcriptomes in A. bracteata are not yet available but we examined the dehydration transcriptomes of its close relative X. schlechteri, which also exhibited tandem duplications of AWPM-19. We found a substantial transcript abundance increase during dehydration in X. schlechteri (Fig. 4E). Additionally, detailed expression profiles of individual AWPM-19 genes revealed specific members that might play major roles in the dehydration response (Fig. 4, F to H) and could serve as important target genes for future molecular experiments aimed at investigating VDT mechanisms in bryophytes.
Application of the “Drying without Dying” database to investigate VDT
Employment of the genomic and transcriptomic datasets, along with the bioinformatic tools with graphic interface embedded in the database, enabled an exploration of a possible divergence in the evolutionary and functional characteristics of VDT plants. For example, as a group of secondary metabolites, flavonoids (including flavonols, anthocyanins, and proanthocyanins) demonstrate diversified biosynthesis pathways across distinct plant lineages (Saito et al. 2013; Saigo et al. 2020), displaying essential physiological roles in reproductive organs and responses to environmental factors such as light (Ma et al. 2021). To investigate flavonoid metabolism in VDT plants, we explored the presence and absence variation (PAV) of genes encoding enzymes that participate in the biosynthesis of flavonoids (Fig. 5). The analyses was conducted using the “Homolog search” function (http://desiccation.novogene.com/homologousgenesearch) with known Arabidopsis (Arabidopsis thaliana) enzyme genes used as baits (Saito et al. 2013). When searching homologs of CINNAMIC ACID 4-HYDROXYLASE (C4H; At2g30490), we observed that C4H is widely distributed across taxa and is present in all 28 plant genomes within the database (Supplementary Fig. S1), suggesting the conservation of the phenylpropanoid metabolic pathway across land plants. However, in a search of the homologs of the A. thaliana DIHYDROFLAVONOL REDUCASE (DFR, At5g42800), homologs were only found in the 12 angiosperm genomes (Supplementary Fig. S2), suggesting the relatively shallow origin of this gene. Similarly, presence–absence patterns were also observed in several anthocyanin biosynthetic genes such as anthocyanidin synthase (ANS), UDP-dependent glycosyltransferase (UDT78D), flavone 7-O-glucosyltransferase (F7GlcT), flavonol 7-O-rhamnosyltransferase (F7RhaT), anthocyanin 5-O-glucosyltransferase (A5GlcT) (Fig. 5). Overall, integrated PAV analyses of the biosynthetic genes of flavonoids provided an exemplar footprint illustrating divergence in evolution of genomes and the metabolic characteristics between angiosperms and nonseed plants.
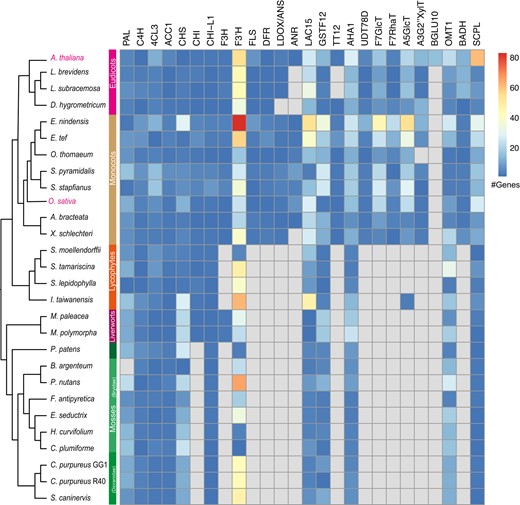
Summarized presence and absence analyses of genes participating the flavonoid biosynthesis based on the “Homolog Search” function implemented in the “Drying without Dying” database. The summarized analyses demonstrated wide-spread absence of canonical genes encoding enzyme that are essential for the anthocyanin biosynthesis in bryophytes and lycophytes. Functions of key enzymes are listed below. PAL, phenylalanine ammonia-lyase; C4H, cinnamic acid 4-hydroxylase; 4CL3, 4-coumaric acid: CoA ligase; ACC1, acetyl-CoA carboxylase; CHS, chalcone synthase; CHI, chalcone isomerase; F3H, flavanone 3-hydroxylase; F3′H, flavonoid 3′-hydroxylase; FLS, flavonol synthase; DFR, dihydroflavonol reductase; LDOX/ANS, leucoanthocyanidin dioxygenase/anthocyanidin synthase; ANR, anthocyanidin reductase; LAC15, laccase; GSTF12, glutathione S-transferase; TT12, multi-drug and toxic efflux (MATE) transporter; AHA10, P-type H+-ATPase; UGT, UDP-dependent glycosyltransferases; BGLU10, anthocyanin 3-O-6″-O-coumaroylglucoside: glucosyltransferase; GlcT, glucosyltransferase; RhaT, rhamnosyltransferase; OMT1, methyltransferase; A5GIcMaIT, anthocyanin malonyltransferase (BADH); A3GlcCouT, anthocyanin coumaroyltransferase (BADH); SCPL, anthocyanin sinapoyltransferase. The phylogenetic relationships were depicted as unscaled cladograms.
To further explore the possible evolutionary and functional divergences within VDT plants, we utilized the dehydration transcriptomes within the database to generate and explore a set of transcripts with extreme changes in abundance during desiccation (i.e. well-watered vs desiccated samples). Only transcripts with fold changes of above 10 and TMP > 1 were included in this exemplar analyses (Supplementary Tables S2 to S4). Overall, the number of transcripts that exhibited the high-level abundance changes were fewer in nonseed plants than in angiosperms, which supports the hypotheses of “unaltered messenger RNA pools” as described for DT bryophytes (Oliver and Bewley 1984). A GO enrichment analysis was conducted to assess these transcripts using the “GO/KEGG Enrichment” tool (http://desiccation.novogene.com/tools/enrichment) included in the “Drying without Dying” database (Fig. 6). We identified significantly more GO terms for transcripts with decreased abundance than for those whose abundance increased (Fig. 6, Supplementary Fig. S3), consistent with the physiologically dormant status of desiccated tissues. Notably, statistically enriched GO terms rarely overlapped across the individual VDT plants in the database (Fig. 6, Supplementary Fig. S3), indicating perhaps lineage- or species-specific variations in the desiccation responses. This is likely a reflection of the independent evolution of VDT across angiosperm resurrection plant lineages (Gaff and Oliver 2013) and the plasticity exhibited by this trait in the bryophytes (Stark 2017). For transcripts that increased in abundance, the conspicuously-related GO terms such as “response to desiccation”, “response to water”, “response to abscisic acid”, and “embryo development ending in seed dormancy” are enriched in monocots S. stapfianus, X. schelectri, and E. nindensis. The “monolayer-surround lipid storage body” was enriched for “desiccation-induced” transcripts in three VDT angiosperms (S. stapfianus, E. nindensis, and L. brevidens), implying possible signatures of convergent responses to desiccation.
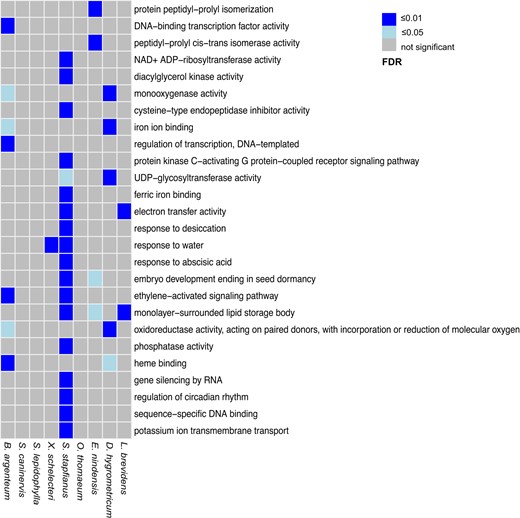
Summary of enriched GO terms for transcripts that demonstrate remarkable abundance increase upon desiccation in VDT species. GO enrichment analyses of gene sets were conducted using the “KEGG/GO Enrichment” function implemented in the “Drying without Dying” database.
Statistically enriched GO terms for transcripts that decreased in abundance are more numerous in angiosperms (Supplementary Fig. S3), in particular those transcripts encoded by photosynthesis-related genes; the terms “photosynthesis” and “photosystem II” are significantly repressed in five VDT angiosperms. In the moss S. caninervis, “ATP hydrolysis activities” and “regulation of transcription, DNA templated” are significantly repressed upon desiccation, coinciding with the quiescent status of energy metabolism and transcription activities in fully desiccated gametophytes. Unlike the numerous transcripts that represent the commonly repressed biological processes in angiosperms only a relatively small number of the highly abundant transcripts are detected in nonseed plants. This resulted in the lack of significantly repressed GO terms in the analysis, further implying “unaltered messenger RNA pools” in desiccated bryophytes. These transcriptomic observations, focusing on well-watered and fully desiccated samples, also supports our hypothesis for diverged evolution of VDT during the evolution of bryophytes and angiosperms.
Conclusions and perspectives
We have established a dedicated plant genome database, “Drying without Dying,” specifically focused on VDT, which can be accessed freely at http://desiccation.novogene.com. The VDT genomes in our database can be downloaded for local analysis or utilized directly using the online bioinformatic tools available. We conducted well-tailored and integrated enrichment analyses of TDGs based on PFAM family annotations, in a statistically rigorous framework that minimizes the influence of the distinct evolutionary baggage accumulated in different phylogenetic lineages. Our analysis revealed insights into the genomic adaptations of land plants, emphasizing the rewiring roles of modern-day seed-related DT genes in both terrestrial adaptation and VDT. Our focus on VDT in bryophytes uncovered the lineage-specific tandem duplication of AWPM-19 as a potentially important evolutionary signature underlying the widespread VDT in bryophytes. Exemplar functional analyses of a small set of genes with extreme abundance changes upon desiccation also supported the “unaltered messenger RNA pools” hypothesis in bryophytes and diverged evolution of VDT between bryophytes and angiosperms at the transcript abundance responsive level. We also suggest that incorporating the “Drying without Dying” genomic resources with DS plant genomes from databases such as Phytozome, and the use of other annotation labels, such as KEGG orthologue and InterPro accessions, may reveal additional interesting phylogenetic patterns of STDFs. Finally, our analyses of both genomes (e.g. widely tandem duplications of AWPM-19 in bryophytes) and transcriptomes (e.g. divergent responses in angiosperms and “unaltered messenger pool in bryophytes”) support the divergent aspects of the evolution of VDT in land plants.
Materials and methods
All the genomes and transcriptomes used in this study were obtained from published reports (http://desiccation.novogene.com/about), with the exception of the silvergreen bryum moss (B. argenteum) genome, which is currently under peer review (Gao et al. 2023). To ensure clarity and facilitate further annotation and comparisons of genomic features, we retained only one representative mRNA for each gene locus. We annotated conserved PFAM families or domains (Mistry et al. 2021) and gene ontology terms using InterProScan v5.52–86.0 with the “-f TSV -goterms -iprlookup -appl Pfam” options for each proteome (Jones et al. 2014). Transcription factor genes were annotated using the prediction function in the PlantTFDB v5.0 database (Tian et al. 2019). KEGG pathway information was annotated using KofamKOALA v1.3.0 (Aramaki et al. 2019) by running the “exec_annotation” module with “–format mapper -e 0.01” parameters.
We used the python package jcvi v1.2.7 (https://github.com/tanghaibao/jcvi) to screen for tandemly and proximally duplicated genes in each plant genome. Homologous genes with a default minimal coverage of 50% of coding sequences (−percent_overlap = 50) and a maximum distance of 10 genes (−tandem_Nmax = 10) were merged into one tandem cluster. Homologous gene pairs separated by 10 or fewer genes have been widely recognized as proximally duplicated genes (Qiao et al. 2019). To simplify the terminology, we referred to all tandem (directly adjacent to each other on the chromosome) and proximal (separated by other nonhomologous genes) genes as TDGs. We conducted PFAM family enrichment analyses of TDGs against the whole genome annotation for each plant genome based on PFAM annotations obtained earlier. The objective was to determine the statistical significance of the contributions made by TDGs for each PFAM family. Specifically, we used the “enricher” function in the clusterProfiler R package (Yu et al. 2012) to conduct the PFAM enrichment analyses for TDGs. The P-values were adjusted using the false discovery rate (FDR) method (pAdjustMethod = “fdr”), and the range of enriched family size was set to [5,1000] (minGSSize = 5, maxGSSize = 1000). PFAM families containing TDGs with FDR ≤ 0.05 were considered STDFs, indicating that tandem duplications had significantly contributed to the expansion of the corresponding family. For each plant genome, we obtained a list of PFAM families which had significantly expanded through tandem gene duplications, and the statistical results for each species are listed in Supplementary Table S1. We subsequently focused on the validated VDT plant genomes depicted in Fig. 1 and manually selected a list of STDFs that could be found in a large number of VDT genomes or in a specific phylogenetic lineage. Finally, we generated a heatmap using the “pheatmap” v1.0.12 package in the R v4.2.2 environment to illustrate the phylogenetic distributions of STDFs.
For transcriptome analysis, we obtained the transcriptomic reads associated with hydration, desiccation, and rehydration processes from the NCBI-SRA database (Supplementary Table S3). The extracted sequencing reads were cleaned with fastp v0.23.2 (Chen et al. 2018) software with default parameters. We estimated the expression levels using salmon v1.9.0 (Patro et al. 2017) against the respective gene sets annotated from the genomes of each species, gene sequences were first indexed using the “salmon index” function. Subsequently, TPM values were estimated to indicate transcript abundance levels using the “salmon quant” module with the option “-numBootstraps 100” and “salmon quantmerge” was employed to generate an expression matrix table for each species (Supplementary Table S2). Transcripts with high-level abundance changes (FC > 10 and TPM > 1) were then filtered based on the average TPM values by comparing well-watered and desiccated samples in each VDT species. Expression values of genes could be queried from http://desiccation.novogene.com/tools/geneexpression. For dehydration and rehydration transcriptomes in each species, we calculated co-expressions using the time-ordered gene co-expression network (TO-GCN) package, which has been updated to support single time-series transcriptomic data (Chang et al. 2019). We used the “cutoff” command to determine a dataset-specific threshold of the PCC value, and estimated PCC values of gene pairs using the “GCN” command. Gene pairs with PCC values greater than the threshold were considered co-expressed and the co-expressions could be queried at http://desiccation.novogene.com/tools/Coexpression.
The genomic and transcriptomic data, along with the bioinformatic tools, have been organized in a MongoDB database and stored on a dedicated Linux server running the CentOS operating system on a high-performance cloud computer cluster. The web interface was designed using the React.js framework in JavaScript, and Python was used to build query and bioinformatic functions for data search and visualization of results. All genome annotations and homologous clustering results were organized in a relational database structured in several tables, including a primary table with multifaceted gene annotations from all VDT-related genomes, such as SwissProt descriptions, KEGG pathways, GO terms, InterProScan classifications, PFAM families, and transcription factors. The website has been tested for compatibility in widely used browsers such as Google Chrome, Microsoft Edge, and Safari on MacOS. The website is also optimized for cell phone screen displays. The genome sequences and associated annotations can be accessed via the “Download” section of the database (http://desiccation.novogene.com/download).
Accession numbers
Sequence data employed in this article can be found in the GenBank/EMBL data libraries under accession numbers listed in the Supplementary Table S3.
Acknowledgments
We appreciate the generous contribution of genomic data, and critical and insightful comments provided by Professor Robert VanBuren (Michigan State University).
Author contributions
B.G. conceived the project, processed the genomic and transcriptomic data, constructed the database, performed the bioinformatic analyses, and wrote the manuscript. X.L., Y.Liang., M.C., Y.Liu. assisted in the collection of genomic and transcriptomic data and participated in the construction of the database. J.W. and H.L. assisted in and provided suggestions for the website user interface design. J.Z. and Y.Z. provided insightful suggestions for the manuscript and the constructions of the database. M.O. and D.Z. supported and supervised the whole project. All authors read and approved the manuscript.
Supplementary data
The following materials are available in the online version of this article.
Supplementary Figure S1. A screenshot displaying the “Homolog Search” results by searching an Arabidopsis C4H gene (At2g30490) as bait.
Supplementary Figure S2. A screenshot displaying the “Homolog Search” results by searching an Arabidopsis DFR (dihydroflavonol reductase) gene (At5g42800) as bait.
Supplementary Figure S3. Summary of enriched GO terms for transcripts that demonstrate remarkable abundance decrease upon desiccation in VDT species.
Supplementary Table S1. Summary of PFAM over-representative statistical results of tandem duplicated genes in each of the plant genomes.
Supplementary Table S2. Summary of expression values (TPM) in each of the VDT plants upon dehydration and rehydration.
Supplementary Table S3. Accession information of dehydration and rehydration RNA-Seq libraries incorporated in the “Drying without Dying” database.
Supplementary Table S4. Number of transcripts that demonstrate remarkable abundance changes upon extreme desiccation in VDT species.
Funding
This project was supported by the National Natural Science Foundation of China (32100256 to B.G.), the Ministry of Science and Technology of China (Third Xinjiang Scientific Expedition Program) (2021xjkk0500 and 2022xjkk1500), the National High-Level Young Talent Programs (2022000005 and 2022000243 to B.G.), Strategic Biological Resources Capacity Building Project, CAS (KFJ-BRP-017-72), and the U.S. National Science Foundation Dimensions of Biodiversity Program Award (1638972 to M.J.O.).
Data availability
The data underlying this article are available in the article and in its online supplementary material.
Dive Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
References
Author notes
Yuanming Zhang, Melvin J. Oliver and Daoyuan Zhang Senior authors.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://dbpia.nl.go.kr/plphys/pages/General-Instructions) is Daoyuan Zhang.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}