





Abstract
Carbon fixation relies on Rubisco and 10 additional enzymes in the Calvin–Benson–Bassham cycle. Epimerization of xylulose-5-phosphate (Xu5P) into ribulose-5-phosphate (Ru5P) contributes to the regeneration of ribulose-1,5-bisphosphate, the substrate of Rubisco. Ribulose-5-phosphate-3-epimerase (RPE, EC 5.1.3.1) catalyzes the formation of Ru5P, but it can also operate in the pentose-phosphate pathway by catalyzing the reverse reaction. Here, we describe the structural and biochemical properties of the recombinant RPE isoform 1 from Chlamydomonas (Chlamydomonas reinhardtii) (CrRPE1). The enzyme is a homo-hexamer that contains a zinc ion in the active site and exposes a catalytic pocket on the top of an α8β8 triose isomerase-type barrel as observed in structurally solved RPE isoforms from both plant and non-plant sources. By optimizing and developing enzyme assays to monitor the reversible epimerization of Ru5P to Xu5P and vice versa, we determined the catalytic parameters that differ from those of other plant paralogs. Despite being identified as a putative target of multiple thiol-based redox modifications, CrRPE1 activity is not affected by both reductive and oxidative treatments, indicating that enzyme catalysis is insensitive to possible redox alterations of cysteine residues. We mapped phosphorylation sites on the crystal structure, and the specific location at the entrance of the catalytic cleft supports a phosphorylation-based regulatory mechanism. This work provides an accurate description of the structural features of CrRPE1 and an in-depth examination of its catalytic and regulatory properties highlighting the physiological relevance of this enzyme in the context of photosynthetic carbon fixation.
Introduction
The Calvin–Benson–Bassham cycle (CBBC) is the metabolic phase of photosynthesis that allows the endergonic conversion of carbon dioxide into carbohydrates (Emerson and Arnold 1932; Johnson 2016). Fixation of carbon dioxide occurs through the carboxylation of the acceptor ribulose-1,5-bisphosphate (RuBP) by the enzyme ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) (Benson et al. 1952). Subsequently, enzyme-driven regeneration of RuBP is crucial to sustain Rubisco catalysis and requires epimerization of xylulose-5-phosphate (Xu5P) into ribulose-5-phosphate (Ru5P) at its chiral carbon 3. Epimerization is catalyzed by ribulose-5-phosphate 3-epimerase (RPE, enzyme class 5.1.3.1), which is generally considered to be a metalloenzyme belonging to the ribulose phosphate binding superfamily (Chen et al. 1998). In parallel with RPE, the formation of Ru5P is also guaranteed by the isomerization of ribose-5-phosphate (R5P) catalyzed by ribose-5-phosphate isomerase (RPI). Following epimerase/isomerase activities, the regeneration of RuBP is ensured by the activity of phosphoribulokinase (PRK) which catalyzes the adenosine triphosphate (ATP)-dependent phosphorylation of Ru5P (Jakoby et al. 1956).
Chlamydomonas (Chlamydomonas reinhardtii), a model unicellular green alga, encodes two RPEs at nuclear genome loci Cre12.g511900.t1.2 (RPE1) and Cre02.g116450.t1.2 (RPE2) (Merchant et al. 2007). RPE1 and RPE2 proteins share 41% sequence identity but RPE2 (Uniprot A8I2N1) is predicted to be cytosolic while RPE1 (Uniprot A8IKW6) is predicted to be targeted to the chloroplast. Like in plants, the pentose-phosphate pathway (PPP) is duplicated in the chloroplast and the cytosol of Chlamydomonas (Johnson and Alric 2013). This suggests that RPE2 contributes to cytosolic PPP while RPE1 is active in both the chloroplast PPP and the CBBC. Recent proteomic studies employed Chlamydomonas as a model organism to identify protein targets undergoing post-translational modifications such as thiol-based redox modifications and serine-threonine phosphorylations. The chloroplast RPE1 isoform was identified as a putative target by redox proteomics probing S-glutathionylation and S-nitrosylation sites (Zaffagnini et al. 2012; Morisse et al. 2014), and thioredoxin-mediated disulfide/dithiol exchange (Pérez-Pérez et al. 2017). Likewise, Chlamydomonas (CrRPE1) was found to undergo multiple phosphorylations on three sites at residues Ser50, Thr220, and Ser239 (Wang et al. 2014). Nevertheless, the effective modulation of protein activity through post-translational modifications is still elusive. Over the last decades, several studies characterized RPE isoforms from different organisms including protozoan parasites, photosynthetic and non-photosynthetic bacteria, yeast, mammals, and land plants. The various RPEs showed rather different structural and functional features likely reflecting an adaptation to their specific physiological context. At the biochemical level, the sole enzymatic activity monitored so far is the conversion of Ru5P to Xu5P (i.e. the PPP-type activity) and the corresponding Michaelis–Menten constants (KM) for Ru5P determined for plant and non-plant RPEs revealed large variations, from high to low affinities (0.2–15 mM range) (Kiely et al. 1973; Akana et al. 2006). Similarly, turnover numbers (kcat) are highly variable, ranging from ∼0.1 to 104 s−1 (Teige et al. 1998; Chen et al. 1999). Focusing on plant enzymes, the recombinant form of spinach (Spinacia oleracea) (SoRPE) exhibited a kcat of 7,100 s−1 and a KM for Ru5P of 0.22 mM (Chen et al. 1998; Chen et al. 1999), whereas the catalytic parameters of RPE purified from spinach leaf chloroplasts were estimated at 0.138 s−1 and 0.25 mM for kcat and KM, respectively (Teige et al. 1998). These values reflected a similar affinity for the substrate while turnover numbers deviate by four orders of magnitude. Consequently, derived catalytic efficiencies (kcat/KM) strikingly differ from 3.23 × 107 M−1·s−1 for the recombinant enzyme to 5.53 × 102 M−1·s−1 for the native enzyme. To date, structures of 23 RPE orthologs have been experimentally determined and reported in the Protein Data Bank, among which four from Homo sapiens (Liang et al. 2011), one from Streptococcus pyogenes (Akana et al. 2006), one from Plasmodium falciparum (Caruthers et al. 2006), and one from Trypanosoma cruzi (Gonzalez et al. 2017). Three RPE structures were determined from photosynthetic organisms: one from the cyanobacterium Synechocystis sp. PCC 6803 (Wise et al. 2004), one cytosolic isoform from rice (Oryza sativa) (Jelakovic et al. 2003), and one chloroplast isoform from potato (Solanum tuberosum) solved at 2.3 Å resolution (Kopp et al. 1999). For all known RPEs from both plant and non-plant sources, the protein folds as an α8β8 barrel of the triose-phosphate isomerase (TIM-barrel) superfamily. The carboxy-end of the 8 parallel β-strands in the barrel defines a shallow surface onto which a metal ion is chelated by a conserved tetrad of 2 aspartates and 2 histidines. Xu5P and Ru5P substrate ligands are typically accommodated in the vicinity of the metal site where reversible epimerization is catalyzed. The identity of the metal was determined for RPE from Escherichia coli (Sobota and Imlay 2011) and attributed to iron II (Fe2+), while other structural data locate a zinc ion at this position (Jelakovic et al. 2003; Wise et al. 2004; Akana et al. 2006). The structural analysis of chloroplast RPE from potato revealed the absence of a metal ion, but instead, a molecule of water was accommodated in the active site center (Kopp et al. 1999). Overall, previous studies may lack accuracy in measuring enzyme reaction rates, metal allocation or its presence after purification of recombinant or native enzymes.
In the current study, we present a thorough examination of the structural features of RPE1 from CrRPE1 by means of small angle X-ray scattering (SAXS) analysis and size-exclusion chromatography coupled with immunoblot analysis of algal protein extracts. The hexameric structure was further confirmed by the resolution of the crystal structure of CrRPE1, determined at a resolution of 1.9 Å. Our model confirms the general features of the epimerase fold and catalytic site, with better resolution compared to the only other known chloroplastic isoform from potato.
To get insights into the kinetic properties of CrRPE1, we set up and optimized in vitro coupled-enzyme assays to monitor the enzymatic conversion of Ru5P to Xu5P (i.e. PPP-related activity), and the epimerization of Xu5P to Ru5P (i.e. CBBC-related activity). A biochemical comparison with the recombinant form of the spinach homolog was also conducted, showing that the 2 enzymes have similar kinetic features, in contrast to previous studies. Finally, we monitored the thiol-based redox sensitivity of CrRPE1 and the specific activities of phospho-mimicking mutants to assess possible regulatory mechanisms based on redox and phospho-based modifications, also mapping putative sites on the molecular structure.
This study presents the experimental high-resolution structure of Chlamydomonas RPE1, its kinetic properties in the 2 possible directions of epimerization, and possible regulatory mechanisms, highlighting the physiological relevance of this enzyme that plays a crucial role in carbon metabolism in a photosynthetic model organism.
Results
Quaternary structure of recombinant and native CrRPE1
Based on previous findings, chloroplast RPE from land plants assembles in a homo-hexameric structure (Kopp et al. 1999; Kopriva et al. 2000; Jelakovic et al. 2003), albeit an octameric form was initially assigned (Chen et al. 1998; Teige et al. 1998). The cytosolic counterpart, however, was incontrovertibly found as a dimer (Karmali et al. 1983; Kopriva et al. 2000). To assess the oligomerization state of CrRPE1, the enzyme was heterologously expressed, purified to homogeneity, and analyzed by size-exclusion chromatography (SEC, Supplementary Fig. S1A). We observed a single monodisperse peak with an apparent molecular weight of 134 kDa, which corresponds to roughly 5 times that of a 27 kDa subunit. The oligomeric nature of the protein along with a precise estimate of the number of subunits was further investigated by SAXS (Supplementary Fig. S1, B and C). The X-ray diffusion curve was analyzed by ATSAS PRIMUS-QT which derived a radius of gyration of 35.53 Å, and a molecular weight of 166,581 Da corresponding to 6 subunits per particle. Because SAXS is considered a more rigorous method to estimate protein molecular weights with respect to analytical chromatography, we concluded that CrRPE1 folds as a homo-hexamer.
To further inspect the oligomerization state of CrRPE, we analyzed the quaternary structure of the native enzyme extracted from Chlamydomonas cell cultures. The soluble protein fraction was submitted to analytical SEC (Supplementary Fig. S1D), and elution fractions were analyzed by western blot using primary polyclonal anti-CrRPE1 antibodies. Fractions containing detectable amounts of RPE corresponded to elution volumes comprised between 15.6 mL and 17.2 mL, with a maximum signal in the 16.0 to 16.4 mL fraction (Supplementary Fig. S1E). This elution volume matches that of the recombinant protein (16.25 mL, Supplementary Fig. S1A) that we observed on the same chromatographic system. We conclude that RPE1 extracted from cultivated algae has the same molecular weight as the pure recombinant protein, and that CrRPE1 assembles as a homo-hexamer in vivo.
CrRPE1 folds as an α8β8 TIM-barrel
To expand the current knowledge of the structural properties of photosynthetic RPE, so far limited to the RPE isoform of potato, we solved the three-dimensional structure of CrRPE1 by X-ray crystallography at a resolution of 1.9 Å. CrRPE1 crystallized in the monoclinic space group P 1 21 1 with 12 subunits that assemble in hexameric structures (Fig. 1). Crystal packing analysis thus confirmed the homo-hexameric organization of CrRPE1, in agreement with the quaternary structure assessed by SAXS analysis. All subunits are virtually identical with root mean square deviations (RMSDs) values in the 0.122 to 0.171 Å range. In the representative chain A, residues Thr31 to Pro260 of mature protein were modeled into continuous electron density. A structural alignment with potato RPE revealed RMSD values ranging from 0.276 Å to 0.347 Å, indicating a native folding substantially conserved between photosynthetic RPE from green algae and land plants.
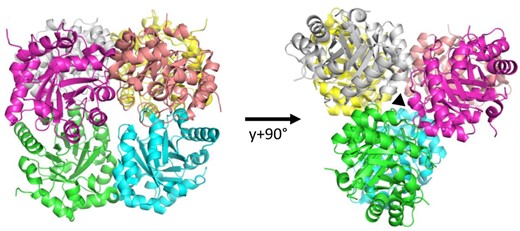
Crystal structure of ribulose-5-phosphate 3-epimerase isoform 1 from CrRPE1. Quaternary structure of CrRPE1. Subunits of the homo-hexamer are colored. Dimer pairs are colored in green-blue, magenta-salmon, and yellow-white. Trimerization axis is represented with a triangle on the right.
As previously observed in structurally solved RPEs (Meloni et al. 2023), CrRPE1 monomers adopt a canonical (β/α)8-barrel fold (TIM-barrel, CATH topology 3.20.20, SCOPe family c.1.2.2) with 8 parallel β-strands forming the central cylindrical β-sheet surrounded by 8 α-helices joined by loops (Fig. 2). An additional short amino-terminal α-helix caps the basal, amino-exposing side of the β-barrel. The overall content of secondary elements in the crystal structure (helices: 28.1%; strands: 17.4%; turns: 12.2%; other: 42.3%) reflects the TIM-barrel fold and this was further confirmed by in-solution analysis using circular dichroism (CD) spectroscopy (Supplementary Fig. S2). Structural alignment of EBI AlphaFold2 entry A8IKW6 and our independently determined crystal structure yield a RMSD of 0.241 Å. Both models are almost identical except for local loop discrepancies, side chains rotamers, and the additional information content of crystallography with regard to water molecules, bound ions, and quaternary packing. We conclude that CrRPE1 folds similarly to other TIM-barrel RPEs.
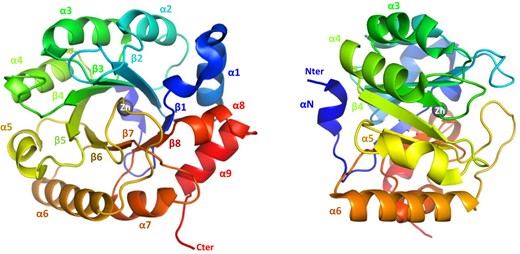
CrRPE1 folding. Main chain is traced in cartoon colored from blue (amino-terminus) to red (carboxy-terminus). Secondary structure elements are annotated. A zinc ion is placed in the center top of the β-barrel.
CrRPE1 packs as a trimer of dimers
Crystal packing analysis identified the nearest neighbors in and around the unit cell. EBI-PISA most probable assembly is composed of the 6 chains ABDFGK, burying a total surface of contact of 14,140 Å2. The computed free energy of dissociation is 20.3 kJ/mol. Three equivalent interfaces form sub-dimers between chains G-A, K-D, and F-B while 6 additional interfaces form between B-A, D-B, D-A, K-G, G-F, and K-F to trimerize the dimers into the full hexamer (Fig. 1). In the representative G-A dimer, the interface involves 10 hydrogen bonds: Leu56G-Ser54A, Gly57G-Ser54A, Gly87G-Thr85A, Arg79G-Glu110A (2 bonds), Ser54G-Leu56A, Ser54G-Gly57A, Thr85G-Gly87A, Glu110G-Arg79A (2 bonds), and 5 salt bridges: Arg79G-Glu110A (3 bridges), and the mirror equivalent residues pair Glu110G-Arg79A (2 bridges). The interface significance score is 1, implying it as essential for complex formation. Trimer interfaces such as that of subunits B-A only form with 6 hydrogen bonds, in residue pairs Ser133B-Gln132A (2 bonds), Ser161B-Glu166A, Phe80B-His137A, Asp181B-Arg140A, and the mirror pair Ser163B-Ser163A. EBI-PISA complex significance score is 0.328, attributing a reliable auxiliary role to these polar interactions in the stabilization of the full complex. The assembly sequence for hexameric CrRPE1 probably starts with the association of dimers, before they can contribute to several weak trimerization contacts that eventually gather the full hexamer altogether. This oligomeric organization was previously described for RPE isoforms from potato, Synechocystis, and Streptococcus pyogenes (Supplementary Fig. S3) (Kopp et al. 1999; Wise et al. 2004; Akana et al. 2006).
CrRPE1 is a metal-containing protein with a conserved catalytic pocket
RPEs from both plant and non-plant sources typically contain a metal ion which has a stabilizing effect toward the substrate (Liang et al. 2011). However, some RPE isoforms with available crystallographic structures do not contain any metal ion supporting that RPE folding does not require the presence of a specific metal within the active site. What remains to be established is whether enzymes that lack a metal ion are catalytically functional. Crystal structure analysis of CrRPE1 revealed that a spherical electron density is located close to the carboxy-exposing side of the barrel. A zinc ion was modeled into the density (Fig. 2) and the identity of the metal was further confirmed by anomalous scattering at the Zn K-edge (Supplementary Fig. S4A). This is in accordance with what was previously observed for cytosolic RPEs from rice (PDB entry: 1H1Z) and from Streptococcus pyogenes (PDB entry: 2FLI). The metal attribution is unknown for the structure of the chloroplast ortholog from potato (PDB: 1RPX). The canonical tetrad of residues involved in metal coordination is perfectly conserved in CrRPE1 and comprises His72, Asp74, His105, and Asp216 (CrRPE1 numbering) (Supplementary Fig. S5). His72 and Asp74 belong to β-strand 2, while His105 and Asp216 belong to β-strand 3 and β-strand 7, respectively. Besides stabilizing the metal ion, the 2 aspartate residues play a crucial role in the catalytic mechanism by exchanging protons with the epimerized carbon atom of the substrate (Chen et al. 1999).
In CrRPE1, the zinc ion is positioned at the bottom of a deep cleft sided by loop A (Leu49-Phe53), loop B (Met76-Gly87), and loop C (Val180-Lys188) (Fig. 3A). Loops A, B, and C respectively project from β-strands 1, 2, and 6. Loops A and B contact each other by hydrophobic bonds between side chains of Leu49 and Thr85, and between side chains of Phe53 and Ile86. Loops B and C make multiple hydrophobic contacts involving the following pairs of residues: Val81-Pro182, Val81-Gly183, Pro82-Gly183, and Pro82-Phe184. The constrained positioning of the three loops restricts the path of an incoming substrate (Xu5P or Ru5P).
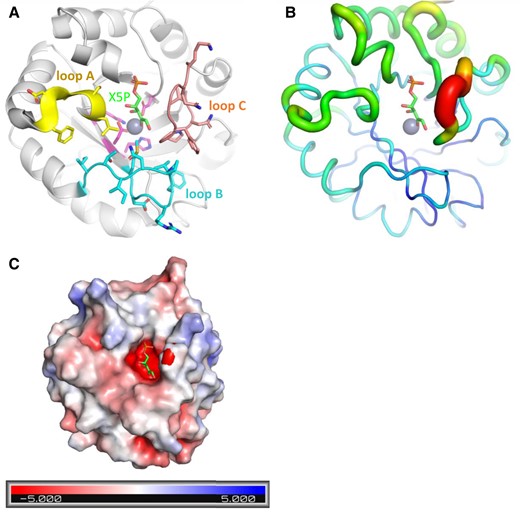
CrRPE1 active site model. A) Cartoon representation of CrRPE1 main chain. Zinc ion is represented as a gray sphere. Xu5P position was inferred by alignment of CrRPE1 structure with that of Homo sapiens RPE co-crystallized with Xu5P (3OVR (Liang et al. 2011)). Loops A, B, and C surrounding the active site are colored yellow, cyan, and teal with residue side chains represented in sticks. B) Local mobility of CrRPE1 is represented by crystallographic B-factors: thin and blue ribbon represents low B-factors, while large and red ribbon represents high B-factors. C) CrRPE1 electrostatic surface calculated by PyMOL APBS (Jurrus et al. 2018) is represented in a gradient from blue (electropositive) to red (electronegative).
Since attempts to crystallize protein in the presence of the substrate have proved unsuccessful, we predicted the substrate binding mode in the active site of CrRPE1 by aligning CrRPE1 crystal structure onto the complex of human RPE (HsRPE) and its substrate Xu5P (PDB entry: 3OVR) with an RMSD = 0.804 Å. Xu5P appears positioned in the space defined between the zinc ion and the 3 aforementioned loops, respecting the lock-and-key complementarity within the active site cleft (Fig. 3A). The conformation defined by CrRPE1 specific side chains hence fully accommodates the substrate dimension. The main structural difference between CrRPE1 and HsRPE complexed with Xu5P is the repositioning of the portion Phe184-Ph189 of the highly conserved loop C closer to the substrate. Xu5P binding probably induces a fit of the enzyme that closes around its substrate. Loop Phe184-Phe189 of our model displays the highest crystallographic B-factors of the whole protein, suggesting a functional mobility of these residues upon substrate and product accommodations (Fig. 3B). Loop between strand β6 and helix α6 was similarly considered important for Xu5P binding in chloroplast ortholog from potato (Kopp et al. 1999).
The substrate binding pocket presents an overall electronegative potential (Fig. 3C). The 5-phosphatidyl group of the modeled substrate is oriented in proximity to main chain amino groups of Gly217, Gly218, Gly238, and Ser239. Electrostatic interactions between electropositive or hydrogen bond donor amines and negatively charged or hydrogen bond acceptor phosphate favor the alignment of the substrate carbonyl and the hydroxyl group of the C3 substrate at 2.8 Å and 3.4 Å from the zinc ion, respectively. This conformation of the active site appears optimal to ensure efficient Xu5P to Ru5P interconversions.
Kinetic analysis of CrRPE1 in the epimerization of Ru5P to Xu5P
Since kinetic comparison of plant RPE orthologues reveals large discrepancies, we sought to determine the kinetic features of recombinant CrRPE1. To this end, we replicated and optimized protocols from previous studies (Davis et al. 1972; Kiely et al. 1973; Chen et al. 1999; Akana et al. 2006). The initial velocity (vi) of CrRPE1 ascribed to the PPP-related activity (i.e. Ru5P epimerization to Xu5P) was measured by coupling Ru5P epimerization to NADH oxidation via 3 reporter enzymes (CrTK, CrTPI, and α-GDH) (Supplementary Fig. S6). When CrRPE1 was omitted in the assay cuvette, we detected a slow and time-limited NADH consumption likely attributed to Xu5P contamination of Ru5P powder. Consequently, the further oxidation of NADH observed after adding CrRPE1 in the assay mixture indicates that the formation of Xu5P by CrRPE1 is strictly required to allow the continuous functioning of coupled enzymes (i.e. the CrTK-dependent formation of glyceraldehyde-3-phosphate and subsequent catalysis by CrTPI and α-GDH, Supplementary Fig. S6). After establishing the enzymatic assay, we analyzed the dependency of CrRPE1 activity on protein concentration and metal ion. As shown in Fig. 4A, we found that protein activity (ΔAbs340/min) displayed a linear relationship with increasing protein concentration in the 2.5 to 10 nM range, corresponding to a specific activity of 388 ± 30 μmol/min/mg (Supplementary Fig. S7A). Exposure of CrRPE1 to chelating agents (i.e. EDTA and TPEN) substantially decreased enzyme catalysis, while treatment with equimolar or higher (10- and 100-fold excess) concentrations of zinc chloride did not enhance CrRPE1 activity (Supplementary Figs. S4, B and C). The kinetic parameters were then determined using variable Ru5P concentrations and activity data were analyzed by non-linear regression using the Michaelis–Menten equation (Fig. 4B). Enzyme activities plotted versus Ru5P concentration displayed a typical hyperbolic response and CrRPE1 catalyzed the epimerization of Ru5P to Xu5P with a KM value of 1.52 ± 0.19 mM and a kcat of 273 ± 17 s−1. By comparing the kinetic properties of CrRPE with recombinant and native RPE from SoRPE, which is the only plastidial isoform to have been kinetically characterized to date, we noted that the affinity for Ru5P is markedly different as native and recombinant SoRPE showed a ∼6-fold lower Michaelis–Menten constant (0.22 to 0.25 mM) (Table 1). A striking diversity was also observed when comparing turnover numbers since CrRPE catalyzes the reaction with a value about 25-fold lower and 2,000-fold higher than those previously reported for the recombinant and native form of spinach RPE, respectively (Table 1) (Chen et al. 1998; Teige et al. 1998; Chen et al. 1999).
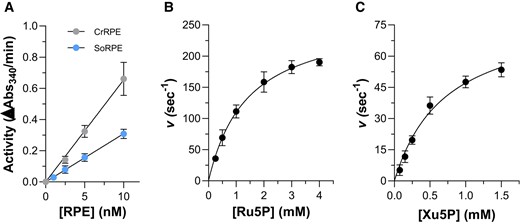
In vitro enzymatic activity of CrRPE1 and SoRPE. A) Linear dependence of PPP-related activity on CrRPE1 concentration expressed as ΔAbs340/min. The data are represented as mean ± SD. (n = 3). B) Variation of apparent turnover number (s−1) catalyzed by 5 nM CrRPE1 in the presence of varying Ru5P concentrations (0–4 mM). C) Variation of apparent turnover number (s−1) catalyzed by 5 nM CrRPE1 in the presence of varying Xu5P concentrations (0–1.5 mM). For panels B) and C), turnover represents moles of NADPH oxidized/s in the presence of 1 mol of CrRPE1. The data are represented as mean ± SD (n = 3). The best fit was obtained using the Michaelis–Menten equation.
Kinetic parameters of RPE
| Source organism | KMRu5P (mM) | kcat (s−1) | kcat/KMRu5P(M−1 s−1) | References |
|---|---|---|---|---|
| Chlamydomonas reinhardtii | 1.52 | 273 | 1.80*105 | This work |
| Spinacia oleracea | ||||
| chloroplast extract | 0.25 | 0.1382 | 553 | Teige et al. (1998) |
| recombinant protein | 0.22 | 7,100 | 3.23*107 | Chen et al. (1999) |
| Spinacia oleracea | ||||
| recombinant protein | 1.56 | 105 | 6.73*104 | This work |
| Source organism | KMRu5P (mM) | kcat (s−1) | kcat/KMRu5P(M−1 s−1) | References |
|---|---|---|---|---|
| Chlamydomonas reinhardtii | 1.52 | 273 | 1.80*105 | This work |
| Spinacia oleracea | ||||
| chloroplast extract | 0.25 | 0.1382 | 553 | Teige et al. (1998) |
| recombinant protein | 0.22 | 7,100 | 3.23*107 | Chen et al. (1999) |
| Spinacia oleracea | ||||
| recombinant protein | 1.56 | 105 | 6.73*104 | This work |
Catalytic parameters of RPE from Chlamydomonas (Chlamydomonas reinhardtii) and spinach (Spinacia oleracea).
Kinetic parameters of RPE
| Source organism | KMRu5P (mM) | kcat (s−1) | kcat/KMRu5P(M−1 s−1) | References |
|---|---|---|---|---|
| Chlamydomonas reinhardtii | 1.52 | 273 | 1.80*105 | This work |
| Spinacia oleracea | ||||
| chloroplast extract | 0.25 | 0.1382 | 553 | Teige et al. (1998) |
| recombinant protein | 0.22 | 7,100 | 3.23*107 | Chen et al. (1999) |
| Spinacia oleracea | ||||
| recombinant protein | 1.56 | 105 | 6.73*104 | This work |
| Source organism | KMRu5P (mM) | kcat (s−1) | kcat/KMRu5P(M−1 s−1) | References |
|---|---|---|---|---|
| Chlamydomonas reinhardtii | 1.52 | 273 | 1.80*105 | This work |
| Spinacia oleracea | ||||
| chloroplast extract | 0.25 | 0.1382 | 553 | Teige et al. (1998) |
| recombinant protein | 0.22 | 7,100 | 3.23*107 | Chen et al. (1999) |
| Spinacia oleracea | ||||
| recombinant protein | 1.56 | 105 | 6.73*104 | This work |
Catalytic parameters of RPE from Chlamydomonas (Chlamydomonas reinhardtii) and spinach (Spinacia oleracea).
In view of the values reported here and in previous studies, any comparative conclusion on the catalytic features is risky since the catalytic constants for spinach native and recombinant RPE isoforms are extremely discordant. One possible explanation was provided by Chen and coauthors who considered the presence of the reducing agent dithiothreitol (DTT), which was used during the purification procedure of the native form, as a destabilizing agent that alters the functionality of the spinach protein (Chen et al. 1998). To further inspect the kinetic diversity with SoRPE, we expressed and purified the spinach enzyme and determined its biochemical properties (Fig. 4A). The recombinant SoRPE catalyzed the epimerization of Ru5P to Xu5P with a specific activity ∼2-fold lower compared to CrRPE (Supplementary Fig. S7A) and exhibited a similar KM with respect to CrRPE1 (1.56 ± 0.17 mM) and 2.5-fold lower kcat value (105.4 ± 13.5 s−1) (Supplementary Fig. S7B). Taken together, our results on spinach enzyme are in contrast to previous studies differing for both substrate affinity and catalytic proficiency (i.e. specific activity or turnover numbers) (Table 1). In this regard, several reasons can be proposed that may depend on multiple factors: (i) the functional and redox state of the RPE enzyme, (ii) the source and specificity of the coupled enzymes, and (iii) the purity and integrity of the chemicals used. However, other possible explanations could justify these differences, including the amount of catalytic metal in purified recombinant proteins, and for this reason, we do not intend to question further this striking discrepancy.
Kinetic properties of CrRPE1 catalyzing the conversion of Xu5P to Ru5P
While PPP-related RPE activity has been widely employed to dissect the catalytic properties of RPE enzymes, there is a lack of knowledge about the biochemical features of RPE related to the conversion of Xu5P into Ru5P (i.e. CBBC-related activity). To assess the catalytic properties related to the catalytic capacity to use Xu5P as substrate, we employed the enzymatic assay typically used to monitor the activity of PRK (Gurrieri et al. 2019). The enzyme catalyzes the ATP-dependent phosphorylation of Ru5P, and in the assay we developed, the substrate of PRK is provided by CrRPE1 through epimerization of Xu5P. We observed that the CrRPE1 amount in the assay cuvette was the only limiting factor of the assay. Consistently, we found that protein activity (ΔAbs340/min) displayed a linear relationship with increasing protein concentration in the 2.5 to 20 nM range. The derived specific activity was 116 ± 7 μmol/min/mg, which is ∼3.3-fold lower compared to the PPP-related activity (Supplementary Fig. S7A).
Kinetic constants were then determined by using variable Xu5P concentrations (0 to 1.5 mM) and activity data were analyzed by non-linear regression analysis using the Michaelis–Menten equation (Fig. 4C). As observed for the opposite reaction, enzyme activities plotted versus Xu5P concentration displayed a typical hyperbolic response and CrRPE1 catalyzed the epimerization of Xu5P to Ru5P with a KM value of 0.716 ± 0.09 mM and a kcat of 80.7 ± 7.9 s−1. Comparing these kinetic data with those obtained from the reverse reaction, CrRPE1 shows a higher affinity for Xu5P compared to Ru5P but displays a lower capacity to convert Xu5P into Ru5P with respect to the reverse reaction. Consequently, derived catalytic efficiencies (kcat/KM) slightly differ from 1.13 × 105 M−1/s for the activity employing Xu5P as a substrate to 1.80 × 105 M−1/s for the epimerization of Ru5P to Xu5P.
CrRPE1 displays limited intrinsic instability and redox sensitivity to reducing agents
The stability of a given protein under different cellular conditions (e.g. low temperature or reducing conditions) is a crucial parameter because of the direct connection between structural integrity and functionality. In a previous study, Chen and colleagues showed that the chloroplast SoRPE (recombinant form) was highly unstable when incubated at 4 °C (Chen et al. 1998). Moreover, the activity of the recombinant enzyme decreased after exposure to reducing agents such as 2-mercaptoethanol. In relation to this evidence, we investigated the response of CrRPE1 activity to incubation at low temperature (4 °C) and treatments with 2 chemical reducing agents, namely 2-mercaptoethanol and DTT. CrRPE1 activity was not affected by incubation at low temperature (∼99% activity compared to control), whereas a loss of activity was observed following exposure with 2-mercaptoethanol (∼40% inhibition after 2 h incubation). Intriguingly, no significant alteration of the enzymatic activity was detected upon treatment with DTT. Although DTT and 2-mercaptoethanol share similar reactivity toward protein cysteines, the redox mechanism underlying the inhibitory effect of the latter is unclear. In this regard, it is worth noting that a reductive-based redox mechanism would imply a pre-existing oxidized state of the protein, which would be subsequently modified by reducing treatments. However, this is in contrast with the observation that in the crystal structure all cysteine thiols are found in a reduced state (Fig. 5A). Therefore, we can assume that the observed inactivation mediated by 2-mercaptoethanol eludes a cysteine-based redox-type mechanism but rather derives from other properties of the molecule likely affecting protein folding and/or metal coordination.
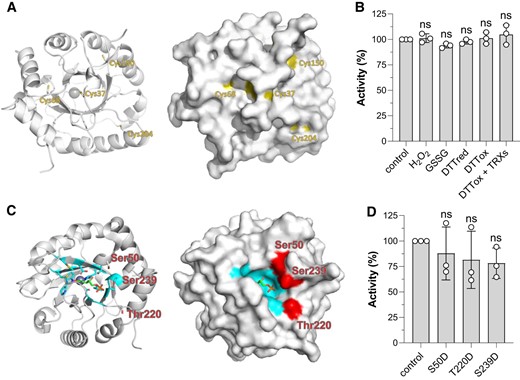
Post-translational modification sites of CrRPE1. A) Cartoon (left) and surface (right) representation of the protein main chain with potential cysteine redox sites highlighted in yellow. B) Determination of redox sensitivity of CrRPE1 after treatment with oxidizing and reducing compounds (see “Material and Methods” for further details). Control activity was set to 100% and used to calculate the percentage of residual activity of CrRPE1 after incubation (60 min) in the presence of reducing or oxidizing conditions. The data are represented as mean ± SD (n = 3). Statistical analysis was performed using one-way ANOVA followed by Tukey's post-hoc test for multiple comparisons. P values reported on graphs as follows: *P ≤ 0.05, **P ≤ 0.01, ns: not significant. C) Cartoon (left) and surface (right) representation of the protein main chain with phosphorylation target amino acids side chains highlighted in red. Xu5P substrate was modeled by alignment with structure 3OVR as in Fig. 3. Residues at 4 Å from Xu5P are colored in cyan. D) Determination of specific activities of phospho-mimicking CrRPE mutants. Purified recombinant CrRPE1 mutants bearing negatively-charged aspartate residues replacing Ser50, Thr220, or Ser239 were assayed monitoring the PPP-related activity (Ru5P to Xu5P conversion). Activity of WT CrRPE1 was set to 100% and used to calculate the percentage of activity of CrRPE1 mutants. The mutant data are represented as mean ± SD (n = 3). Statistical analysis was performed using one-way ANOVA followed by Tukey's post-hoc test for multiple comparisons. P values reported on graphs as follows: *P ≤ 0.05, **P ≤ 0.01, ns: not significant.
Redox response of CrRPE1 to oxidative treatments
CrRPE1 was identified as a putative target of thioredoxin implying that it might contain cysteine residues involved in the formation of one or more disulfide bonds (Pérez-Pérez et al. 2017). Besides, CrRPE1 was also found to undergo S-nitrosylation (Morisse et al. 2014) and S-glutathionylation (Zaffagnini et al. 2012). Our crystal structure of CrRPE1 positions the 4 cysteines with relative surface accessibility of 0.180 for Cys37, 0.076 for Cys150, and 0.000 for both Cys68 and Cys204 (Fig. 5A). The thiol of Cys37 is the most accessible to the solvent and therefore it is likely able to react with oxidizing molecules, even though minor conformational changes may allow the other cysteines to partially or fully expose their side chains to the solvent. To determine the number of accessible cysteine thiols, we employed the Ellman's reagent (5,5-dithio-bis-(2-nitrobenzoic acid)) and measured one accessible thiol group in vitro (1.0 ± 0.2), suggesting that only one cysteine is likely competent for redox exchange.
We then examined the redox sensitivity of CrRPE1 after exposure with oxidized DTT alone or in combination with pure recombinant chloroplast thioredoxins from Chlamydomonas. As shown in Fig. 5B, no significant alteration of protein activity was observed indicating that dithiol/disulfide interchanges do not constitute a regulatory mechanism of CrRPE1 activity. The catalytic response of CrRPE1 to oxidative modifications was also assessed in the presence of oxidized glutathione (GSSG), and hydrogen peroxide (H2O2) alone or in combination with reduced glutathione (GSH). Again, we did not observe any significant variation of CrRPE1 activity with respect to control conditions (Fig. 5B). Taken together, these results led us to the conclusion that CrRPE1 catalysis is insensitive to possible variations in the redox state of cysteine residues, and this is consistent with the distal positions of the cysteines from the active site (Fig. 5A).
Post-translational phosphorylation sites
A phosphoproteomic study identified 3 phosphorylation sites on CrRPE1, namely Ser50, Thr220, and Ser239 (Wang et al. 2014), which map at the vicinity of the active site, at respectively, 11 Å, 17 Å, and 13 Å of the zinc ion. In our crystal structure, the three residues exposed to solvent their unmodified hydroxyl side chains in a 14 Å arch that aligns at the entrance of the active site (Fig. 5C). Possible recognition and modification by a kinase are allowed by this solvent exposure. RPE substrates (i.e. Xu5P and Ru5P) are expected to be exchanged with solvent following a path gated by Ser50, Thr220, and Ser239. Phosphorylation of 1 or more of these sites may hence hinder the formation of the Michaelis–Menten complex, causing a reduction of the enzyme activity.
To assess this hypothesis, we designed and prepared 3 mutated proteins in which serine or threonine were substituted by aspartates to tentatively mimic with β-carboxylates the introduction of phosphate charges. However, the activity of phospho-mimicking single mutants was not significantly reduced as compared to wild-type (WT) (Fig. 5D) either because phosphorylation introduces more charges and larger steric hindrance than the carboxylate mimics, or because more than one site needs to be modified to affect the enzymatic activity.
Discussion
In this study, we describe the structure of RPE1 from the model microalga Chlamydomonas (Chlamydomonas reinhardtii) determined by X-ray crystallography at a resolution of 1.9 Å. Recombinant CrRPE1 purifies as a homo-hexamer built from the trimerization of dimers as revealed by in-solution analysis and resolution of the 3D structure. We also unveiled that native RPE extracted from algae cultures elutes at the same apparent molecular mass as the purified recombinant protein. Hexamerization is not observed in the crystal structure of non-plant RPE (Liang et al. 2011) but is reported for chloroplastic RPE from potato (Solanum tuberosum) (Kopp et al. 1999). Our structural model reveals that the catalytic pocket contains a zinc ion and lies close to both the dimerization and the trimerization interfaces. We propose that the formation of oligomers of RPE supports allosteric cross-talk between subunit active sites, possibly by restricting the mobility of loops around the catalytic pocket. Interestingly, this could contribute to the variation of the enzymatic parameters reported for photosynthetic and non-photosynthetic RPEs. The 3D structure computes B-factors that quantify the local mobility of the protein in the crystal. We observed 4 elements of high mobility surrounding the active site (Fig. 3B), that are good candidates for such an allosteric regulation. The determination of RPE high-resolution structures in the presence of the substrate or less constrained environments, e.g. by nuclear magnetic resonance or single-particle cryogenic electron microscopy, would provide more information on the dynamic around the active site.
In the chloroplast of Viridiplantae, RPE participates to either PPP or CBBC catalyzing the configuration exchange between Ru5P and Xu5P. CrRPE1, which is localized in the chloroplast stroma, likely performs both epimerization reactions given that both pathways co-exist in the same cellular compartment. Catalytic interconversion between Xu5P and Ru5P consists in an acid–base catalysis involving proton abstraction and donation from and to the C3 atom of the substrate and is proposed to occur through the formation of a 2–3-ene-diolate intermediate (Chen et al. 1999; Akana et al. 2006). Active site zinc ion along with conserved methionine residues (Met76, Met107, and Met178) participate in the stabilization of this high energy intermediate interacting with carbon 2 and carbon 3 hydroxyl oxygens and carbon 2 oxyanion. Asp74 and Asp216 successively complete the metal coordination sphere and can exchange protons with the reaction intermediates. Depending on the bound substrate, the reaction proceeds with its deprotonation being carried out by 1 of the 2 catalytic Asp (i.e. Ru5P to Xu5P conversion) or with the taking of a proton from the second catalytic Asp. Based on the kinetic analysis of CrRPE1 here reported, the algal enzyme has similar affinities for Xu5P and Ru5P with a slightly higher preference for the former. Consequently, the kinetic direction of the reaction does not depend on the kinetic properties of the enzyme but rather on the intracellular concentration of Xu5P/Ru5P owing to the similar tendency of the two different substrates to bind.
While the enzymatic mechanism is clearly established for several RPE orthologs, it is not yet known whether the enzyme is subject to any regulatory mechanisms. In the present study, we mapped the sites of post-translational modifications that may contribute to the modulation of RPE activity according to the environmental conditions or the energetic demand of the cell. Recent proteomic studies on Chlamydomonas cell extracts revealed that RPE has 3 sites of phosphorylation (Ser50, Thr220, and Ser239) (Wang et al. 2014; McConnell et al. 2018). Notwithstanding their distant position in the sequence, all 3 residues are located in a small area in close proximity to the catalytic pocket. We propose that insertion of 1, 2, or 3 bulky negatively-charged phosphates might interfere with the binding of the Xu5P/Ru5P substrate and Ru5P/Xu5P product release, thus slowing down RPE catalysis. Phosphorylation sites (Ser50, Thr182, and Ser239) are conserved in other RPE isoforms from photosynthetic organisms and also in the human enzyme, suggesting that phosphorylation might constitute a conserved regulation mode of RPE activity. Future studies are required to shed light on the possible modulation of photosynthetic-related metabolism as other CBBC enzymes were identified as putative targets of phosphorylation (Wang et al. 2014; McConnell et al. 2018). Cysteine redox status is coupled with photosystems illumination through the ferredoxin-thioredoxin reduction pathway that was demonstrated to activate several CBBC enzymes by reducing regulatory disulfide bonds (for review see Michelet et al. (2013)). In addition, accessible and reactive protein thiols could act as redox sensors of oxidative stress conditions by reacting with reactive oxygen/nitrogen species resulting primarily in S-nitrosylation or S-glutathionylation of proteins (Zaffagnini et al. 2019). Considering that CrRPE1 was identified as a putative target of multiple thiol-based redox modifications (Zaffagnini et al. 2012; Morisse et al. 2014; Pérez-Pérez et al. 2017), we evaluated whether CrRPE1 enters these thiol-switching mechanisms due to the redox alteration of its cysteine residues, knowing that only Cys37 is accessible to the solvent. Various attempts made in vitro, however, revealed no change in CrRPE1 activity indicating that the redox state of CrRPE1 cysteine(s) is not altered by redox treatments or, alternatively, that cysteine oxidation alone is not sufficient to affect protein catalysis. This is consistent with the position of CrRPE1 cysteines, which are all located far from the catalytic pocket. Overall, this result is reminiscent of the redox sensitivity of triose-phosphate isomerase, another CBBC enzyme. This enzyme was identified as a putative redox target, but no alteration in protein activity was detected upon exposure to various oxidizing treatments (Zaffagnini et al. 2014). While being functionally resistant to oxidative modifications, we can hypothesize that alteration of the redox state of CrRPE1 cysteine thiols might induce local conformational changes allowing the enzyme to interact with partner proteins. Such interactions would act as a regulatory mechanism as observed for PRK and glyceraldehyde-3-phosphate dehydrogenase forming an inactive supramolecular complex bridged by CP12 (Gurrieri et al. 2021), or alternatively, they might be functional in the formation of larger enzymatic assemblies to channel CBBC metabolites and increase metabolic efficiency. In this regard, plant RPE was recently proposed to engage in such an activating complex with RPI (Kuken et al. 2018) and to colocalize with CBBC enzymes in a liquid partition of the stroma (Wang et al. 2022; Wang et al. 2023). Further research on native RPE isolated from algae extracts is needed to evaluate the complexity and dynamics of the RPE proteome and redox-dependent protein-protein interactions.
The current knowledge on kinetic features of plant RPE makes it rather complicated to accurately estimate how the enzyme truly functions in the physiological context of the stroma, which could in turn result into important physiological implications likely to be linked to the substrate-to-product conversion in the Michaelis–Menten complex. Nonetheless, if we consider in vitro catalytic properties of CrRPE1 related to photosynthetic activity (i.e. CBBC-related activity), we can estimate a kinetic modeling in the physiological context of a photosynthetic cell under light conditions. In Chlamydomonas, RPE1 concentration was quantified at 3.3 µM (Hammel et al. 2020). Considering that (i) RPE1 is the sole isoform present in the chloroplast stroma with a concentration of 3.3 µM, (ii) its substrate Xu5P is available at 10 to 50 µM (Mettler et al. 2014), and (iii) RPE functions with a KM of 0.716 mM and a kcat of 81 s−1, we expect an in vivo Xu5P to Ru5P flow ranging from 3.7 to 17 µM/s. These values are comparable to or lower than those calculated for Rubisco of 18.9 µM/s (kcat = 5.8 s−1, KM = 0.029 mM (Tcherkez et al. 2006), [RuBP] = 200 µM (Mettler et al. 2014), RbcS stromal concentration of 183 µM (Hammel et al. 2020)).
Relying on in vitro kinetic constants coupled with metabolome and proteome values of substrates and enzymes concentrations, it appears likely that any increase in RPE protein abundance and optimization of its function may result in improved regeneration efficiency of RuBP and thus increased Rubisco activity as suggested by recent models (Raines 2022) and reported in Arabidopsis rpe mutants (Li et al. 2022). If this was confirmed by further analyses, synthetic biology engineering toolkits available in Chlamydomonas and other microalga (Li et al. 2016; Shin et al. 2016; Crozet et al. 2018) could overcome the RPE limitation by either changing protein quantities or altering kinetic features (i.e. substrate affinity and turnover number) to improve catalytic efficiency. Modifying a synthetic CBBC from engineering principles will support the understanding of its physio-chemical properties and open important opportunities to improve photosynthetic efficiency and likely crops yield.
Materials and methods
Cloning and protein preparation
Chlamydomonas (Chlamydomonas reinhardtii) open reading frame of gene Cre02.g116450 of the UniProt entry A8IKW6 was searched for chloroplast transit peptide with ChloroP (Emanuelsson et al. 1999), PredAlgo (Tardif et al. 2012), and multiple sequence alignments (Supplementary Fig. S5). Nucleotide sequence encoding residues 28 to 265 of the predicted mature protein was PCR-amplified from template AV634644 (HC036a02) of Chlamydomonas expressed sequence tag database (Kazusa). PCR product was digested by NcoI and BamHI and ligated into pET-3d vector in fusion with an in frame N-terminal hexa-histidine tag, yielding plasmid pET-3d-His6-CrRPE1. The plasmid was used to transform Escherichia coli BL21 Rosetta2 (DE3) expression strain (Merck, Darmstadt, Germany), grown to exponential phase in 1 L lysogeny broth medium supplemented with 100 µg/mL ampicillin. Expression was induced by addition of 0.2 mM isopropyl-β-D-thiogalactopyranoside for 16 h at 30 °C. Cells were harvested by centrifugation, resuspended in 30 mM Tris-HCl, pH 7.9 (buffer A) and lysed by sonication. Clarified lysate was loaded on 3 mL Ni-NTA resin, washed with buffer A supplemented with 30 mM imidazole and step-eluted with buffer A supplemented with a final imidazole concentration of 250 mM. Eluate was desalted on PD-10 column pre-equilibrated with buffer A and concentrated by ultrafiltration to 5–10 mg/mL. The molecular mass and purity of recombinant protein were examined by SDS-PAGE and the resulting homogeneous protein solutions were stored at −20 °C. Protein concentration was determined spectrophotometrically using a molar extinction coefficient at 280 nm of 14,105 mM−1/cm and a molar mass of 26,772.9 Da.
Analytical size-exclusion chromatography
Recombinant CrRPE1 was injected on a Superose 6 Increase 10/300 GL column (GE Healthcare, Chicago, IL, USA) and isocratically eluted in 50 mM Tris-HCl (pH 7.5) and 150 mM KCl at a 0.5 mL/min flow rate. The column was calibrated with standard globular proteins (Bio-Rad, Hercules, USA), namely bovine thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), and horse myoglobin (17 kDa).
Small angle X-ray scattering
An aliquot of 50 µL of pure CrRPE1 concentrated at 9.5 mg/mL was injected on BioSEC-3 300 size-exclusion chromatography column (Agilent Technologies, Santa Clara, USA) equilibrated in 20 mM Tris-HCl (pH 7.9) and 100 mM NaCl, in line with the SAXS exposure capillary at the synchrotron beamline SWING (SOLEIL, Saint Aubin, France). Collected diffusion images were analyzed on the application Foxtrot 3.3.4 (Xenocs, Sassenage, France) and the ATSAS 2.8.3 suite (Petoukhov et al. 2012; Franke et al. 2017). Primus-QT calculated a radius of gyration of 35.49 ± 0.03 Å and estimated SAXS CrRPE1 molecular weight at 173,354 Da (Qp), 169,124 Da (MoW), 158,056 Da (Vc), and 165,791 Da (size and shape). Oligomeric state was finally computed as mean SAXS molecular weight of 166,581 Da, divided by monomeric recombinant CrRPE1 sequence molecular weight of 26,773 Da and resulting in an estimated number of ∼6 subunits per oligomer.
Crystallization, structure determination, and model refinement
Sitting drops of 100 nL of pure CrRPE1 concentrated at 9.5 mg/mL were mixed with 100 nL of each of the 384 precipitant conditions of the joint center for structural genomics sparse-matrix screen for crystallization (Qiagen, Hilden, Germany) (Lesley and Wilson 2005). Condition 53 of screen III (160 mM calcium acetate; 80 mM sodium cacodylate pH 6.5; 20% v/v glycerol; 14.4% w/v polyethylene glycol 8,000) yielded macled rods of 100 µM length. Crystals were flash-frozen in cryo-loops and tested for diffraction at ESRF beamline ID30A-3 (Grenoble, France). A collection of 7,200 frames of 0.05° tilt each were collected for a complete dataset of space group P21 (Table 2). Indexation, integration, and scaling were performed with XDS (Kabsch 2010) on the beamline software EDNA (Incardona et al. 2009). Phases were determined by molecular replacement with PHASER-MR (McCoy et al. 2007) from protomers of CrRPE1 as modeled by homology with PHYRE2 prediction algorithm (Kelley et al. 2015). Twelve subunits were searched for, to respect a solvent content of 48.8% in the asymmetric unit. Initial density map and structural model were interpreted by iterative cycles of automated building with AUTOBUILD (Terwilliger et al. 2008), manual building with COOT (Emsley et al. 2010), and refinement with PHENIX.REFINE (Afonine et al. 2012) until geometry (Ramachandran 97.48% allowed, 2.49% favorable) and statistics (Rwork = 0.1880; Rfree = 0.2124) were judged acceptable (Table 2). All other crystallographic utilities were found in the PHENIX package (Adams et al. 2010). Images of the crystallographic model were traced with PyMOL (Schrödinger, LLC, New York, NY) version 2.0.6. Relative solvent exposure of cysteine residues was calculated with ASAview (Ahmad et al. 2004) and scaled from 0 (no exposure) to 1 (full exposure). Interface analysis was conducted by PISA (EBI) (Krissinel and Henrick 2007). Crystallographic data are registered at the Protein Data Bank under accession code 7B1W.
Crystallographic data collection and model refinement statistics
| CrRPE1 7b1w | |
|---|---|
| Wavelength (Å) | 0.9679 |
| Resolution range (Å) | 49.29–1.935 (2.005–1.935) |
| Space group | P 1 21 1 |
| Unit cell (Å, °) | 77.293 297.322 77.747 90 116.301 90 |
| Total reflections | 165,0136 (151,270) |
| Unique reflections | 229,414 (21,621) |
| Multiplicity | 7.2 (7.0) |
| Completeness (%) | 98.65 (93.36) |
| Mean I/sigma(I) | 9.79 (1.40) |
| Wilson B-factor | 30.34 |
| R-merge | 0.1273 (1.049) |
| R-meas | 0.1372 (1.131) |
| R-pim | 0.05063 (0.4178) |
| CC1/2 | 0.997 (0.616) |
| CC* | 0.999 (0.873) |
| Reflections used in refinement | 229,338 (21617) |
| Reflections used for R-free | 2,409 (227) |
| R-work | 0.1880 (0.2890) |
| R-free | 0.2124 (0.3261) |
| CC (work) | 0.966 (0.744) |
| CC (free) | 0.961 (0.633) |
| Number of non-hydrogen atoms | 22,618 |
| macromolecules | 20,867 |
| ligands | 12 |
| solvent | 1,739 |
| Protein residues | 2,772 |
| RMS (bonds) | 0.008 |
| RMS (angles) | 0.90 |
| Ramachandran favored (%) | 97.48 |
| Ramachandran allowed (%) | 2.49 |
| Ramachandran outliers (%) | 0.04 |
| Rotamer outliers (%) | 0.52 |
| Clash score | 6.26 |
| Average B-factor | 34.73 |
| macromolecules | 34.24 |
| ligands | 34.16 |
| solvent | 40.61 |
| CrRPE1 7b1w | |
|---|---|
| Wavelength (Å) | 0.9679 |
| Resolution range (Å) | 49.29–1.935 (2.005–1.935) |
| Space group | P 1 21 1 |
| Unit cell (Å, °) | 77.293 297.322 77.747 90 116.301 90 |
| Total reflections | 165,0136 (151,270) |
| Unique reflections | 229,414 (21,621) |
| Multiplicity | 7.2 (7.0) |
| Completeness (%) | 98.65 (93.36) |
| Mean I/sigma(I) | 9.79 (1.40) |
| Wilson B-factor | 30.34 |
| R-merge | 0.1273 (1.049) |
| R-meas | 0.1372 (1.131) |
| R-pim | 0.05063 (0.4178) |
| CC1/2 | 0.997 (0.616) |
| CC* | 0.999 (0.873) |
| Reflections used in refinement | 229,338 (21617) |
| Reflections used for R-free | 2,409 (227) |
| R-work | 0.1880 (0.2890) |
| R-free | 0.2124 (0.3261) |
| CC (work) | 0.966 (0.744) |
| CC (free) | 0.961 (0.633) |
| Number of non-hydrogen atoms | 22,618 |
| macromolecules | 20,867 |
| ligands | 12 |
| solvent | 1,739 |
| Protein residues | 2,772 |
| RMS (bonds) | 0.008 |
| RMS (angles) | 0.90 |
| Ramachandran favored (%) | 97.48 |
| Ramachandran allowed (%) | 2.49 |
| Ramachandran outliers (%) | 0.04 |
| Rotamer outliers (%) | 0.52 |
| Clash score | 6.26 |
| Average B-factor | 34.73 |
| macromolecules | 34.24 |
| ligands | 34.16 |
| solvent | 40.61 |
Crystallographic data collection and model refinement statistics
| CrRPE1 7b1w | |
|---|---|
| Wavelength (Å) | 0.9679 |
| Resolution range (Å) | 49.29–1.935 (2.005–1.935) |
| Space group | P 1 21 1 |
| Unit cell (Å, °) | 77.293 297.322 77.747 90 116.301 90 |
| Total reflections | 165,0136 (151,270) |
| Unique reflections | 229,414 (21,621) |
| Multiplicity | 7.2 (7.0) |
| Completeness (%) | 98.65 (93.36) |
| Mean I/sigma(I) | 9.79 (1.40) |
| Wilson B-factor | 30.34 |
| R-merge | 0.1273 (1.049) |
| R-meas | 0.1372 (1.131) |
| R-pim | 0.05063 (0.4178) |
| CC1/2 | 0.997 (0.616) |
| CC* | 0.999 (0.873) |
| Reflections used in refinement | 229,338 (21617) |
| Reflections used for R-free | 2,409 (227) |
| R-work | 0.1880 (0.2890) |
| R-free | 0.2124 (0.3261) |
| CC (work) | 0.966 (0.744) |
| CC (free) | 0.961 (0.633) |
| Number of non-hydrogen atoms | 22,618 |
| macromolecules | 20,867 |
| ligands | 12 |
| solvent | 1,739 |
| Protein residues | 2,772 |
| RMS (bonds) | 0.008 |
| RMS (angles) | 0.90 |
| Ramachandran favored (%) | 97.48 |
| Ramachandran allowed (%) | 2.49 |
| Ramachandran outliers (%) | 0.04 |
| Rotamer outliers (%) | 0.52 |
| Clash score | 6.26 |
| Average B-factor | 34.73 |
| macromolecules | 34.24 |
| ligands | 34.16 |
| solvent | 40.61 |
| CrRPE1 7b1w | |
|---|---|
| Wavelength (Å) | 0.9679 |
| Resolution range (Å) | 49.29–1.935 (2.005–1.935) |
| Space group | P 1 21 1 |
| Unit cell (Å, °) | 77.293 297.322 77.747 90 116.301 90 |
| Total reflections | 165,0136 (151,270) |
| Unique reflections | 229,414 (21,621) |
| Multiplicity | 7.2 (7.0) |
| Completeness (%) | 98.65 (93.36) |
| Mean I/sigma(I) | 9.79 (1.40) |
| Wilson B-factor | 30.34 |
| R-merge | 0.1273 (1.049) |
| R-meas | 0.1372 (1.131) |
| R-pim | 0.05063 (0.4178) |
| CC1/2 | 0.997 (0.616) |
| CC* | 0.999 (0.873) |
| Reflections used in refinement | 229,338 (21617) |
| Reflections used for R-free | 2,409 (227) |
| R-work | 0.1880 (0.2890) |
| R-free | 0.2124 (0.3261) |
| CC (work) | 0.966 (0.744) |
| CC (free) | 0.961 (0.633) |
| Number of non-hydrogen atoms | 22,618 |
| macromolecules | 20,867 |
| ligands | 12 |
| solvent | 1,739 |
| Protein residues | 2,772 |
| RMS (bonds) | 0.008 |
| RMS (angles) | 0.90 |
| Ramachandran favored (%) | 97.48 |
| Ramachandran allowed (%) | 2.49 |
| Ramachandran outliers (%) | 0.04 |
| Rotamer outliers (%) | 0.52 |
| Clash score | 6.26 |
| Average B-factor | 34.73 |
| macromolecules | 34.24 |
| ligands | 34.16 |
| solvent | 40.61 |
Circular dichroism spectroscopy
Samples of CrRPE1 (10.7 μM) were prepared in buffer A and quantified by spectrophotometric analysis at 280 nm in a 1 cm cell (ε280 = 13,980 M−1/cm based on primary structure) (Pace et al. 1995). Far-UV CD spectra (250–195 nM) were measured at room temperature on a J-810 spectropolarimeter (Jasco, Japan), using a QS-quartz cell with 0.5 mM optical pathlength (Hellma Analytics, Germany), a 2 nM spectral bandwidth, a 20 nM/min scanning speed, a 4 s data integration time, a 0.2 nM data interval, and an accumulation cycle of 3 scans per spectrum. The resulting CD spectra were blank-corrected and converted to molar units per residue (Δεres, in M−1/cm).
In vitro reporter assays of CrRPE1 activity
The epimerization of Ru5P into Xu5P (i.e. PPP-related activity) was measured using a 4-step coupled assay as previously described (Nowitzki et al. 1995) (Supplementary Fig. S6) with minor modifications. As reporter enzymes, we used the recombinant transketolase and triose-phosphate isomerase from Chlamydomonas (CrTK and CrTPI, respectively) (Zaffagnini et al. 2014; Pasquini et al. 2017), and the rabbit muscle α-glycerophosphate dehydrogenase (α-GDH; Sigma-Aldrich, Saint Louis, USA). Prior to the activity assays, CrRPE1 was separated by size-exclusion chromatography and eluted fractions corresponding to the hexameric form were pooled and desalted in buffer A. The catalytic activity was assayed spectrophotometrically at 25 °C in a reaction mixture containing 50 mM Tris-HCl (pH 7.9), 15 mM MgCl2, 0.1 mM thiamin pyrophosphate, 0.1% (w/v) bovine serum albumin, 1 µM CrTK, 4 nM CrTPI, 2 units/mL α-GDH, 2 mM ribose-5-phosphate (R5P), 0.25-4 mM Ru5P, and 0.2 mM NADH. The reaction was initiated by the addition of CrRPE1 and the enzymatic activity was measured by following the decrease in the absorption at 340 nM using a Cary60 UV/Vis spectrophotometer (Agilent Technologies). Previous studies highlighted interferences in the assay derived from traces of Xu5P in the Ru5P powder and from traces of epimerase activity in the auxiliary enzymes from commercial sources (Wood 1979). To establish whether NADH oxidation was effectively dependent upon CrRPE1 catalysis (i.e. Xu5P formation and subsequent reactions catalyzed by CrTK, CrTPI, and α-GDH; Supplementary Fig. S6), we measured the consumption of NADH in the absence of CrRPE1 and found a limited NADH oxidation corresponding to 5% of the initial NADH (∼10 µM) that we ascribed to Xu5P contamination in the Ru5P solution.
To measure the CBBC-related activity, namely the conversion of Xu5P to Ru5P, we employed a recently developed 4-step coupled assay involving three reporter enzymes: the recombinant form of phosphoribulokinase from Chlamydomonas (CrPRK, (Gurrieri et al. 2019)), and commercial pyruvate kinase (PK) and lactate dehydrogenase (LDH) from Saccharomyces cerevisae (Sigma-Aldrich, Saint Louis, USA) (Supplementary Fig. S6). The catalytic activity was measured at 25 °C in a reaction mixture containing 50 mM Tris-HCl (pH 7.9), 10 mM MgCl2, 2 mM ATP, 2.5 mM phosphoenolpyruvate, 5 units/mL PK, 6 units/mL LDH, 6 units/mL CrPRK, 0.075 to 1.5 mM Xu5P, and 0.2 mM NADH. The reaction was initiated by the addition of CrRPE1 and monitored at 340 nM using a Cary60 UV/Vis spectrophotometer (Agilent Technologies). CrRPE1-dependent enzymatic activities were determined after subtracting background activities measured in the absence of CrRPE1.
Protein stability and redox sensitivity
The stability of CrRPE1 was assessed by incubating the enzyme (5 μM) at 4 °C or 25 °C in buffer A. At the indicated times, aliquots (1–2 μL) were withdrawn to carry out activity measurements as described above. The redox sensitivity of CrRPE1 (5 μM) was evaluated by treating the enzyme with reducing agents (10 mM 2-mercaptoethanol or 10 mM DTT) or oxidizing molecules such as 2 mM oxidized GSSG, or 2 mM hydrogen peroxide (H2O2), or 10 mM oxidized DTT in the absence/presence of recombinant chloroplast thioredoxins f2, m, x, y, and z from Chlamydomonas (5 µM each) (Lemaire et al. 2018; Marchand et al. 2019). All incubations were carried out at 25 °C in buffer A. Control experiments were performed by incubating the enzyme in the presence of buffer alone. At the indicated times, aliquots (1–2 μL) were withdrawn to carry out activity measurements as described above.
Metal stability and requirement
The stability of the zinc ion in CrRPE1 and the possible activation by exogenous zinc ion were assessed by incubating the protein (10 μM) in the presence of chelating agents (10 mM EDTA or 10 mM TPEN) or zinc chloride (1, 0.1, and 0.01 mM). All incubations were carried out for 60 min at 25 °C in buffer A. Control experiments were performed by incubating the enzyme in the presence of buffer alone. At the indicated times, aliquots (1–2 μL) were withdrawn to carry out activity measurements as described above.
Extraction of algal proteins
A single colony of wild-type Chlamydomonas reinhardtii strain D66 was grown in 50 mL TAP medium (Gorman and Levine 1965) under continuous 120 rpm agitation and illumination at an intensity of 36 µE/s/m2. At 20*106 cells/mL, the culture was harvested by 5 min centrifugation at 3,000 g and stored at −20 °C. Cell pellet (600 mg) was thawed and resuspended in 12 mL of 20 mM Tris-HCl supplemented with 100 mM NaCl (pH 7.9), and lysed by passage through CellD (Constant systems, Daventry, UK) at 30 kPSI. Soluble fraction was separated by 10 min centrifugation at 30,000 g at 4 °C and filtered through 0.2 µm membrane. The resulting suspension (300 µL) was injected on a Superose 6 Increase 10/300 GL column and eluted isocratically at 0.3 mL/min in 20 mM Tris-HCl supplemented with 150 mM KCl (pH 7.5). Elution fractions (400 µL) were collected, starting from column dead volume 7.2 mL (fraction 18) and up to column total volume 28.0 mL (fraction 70), snap-frozen in liquid nitrogen, and stored at −20 °C for western blot detection using rabbit polyclonal antibodies raised against recombinant CrRPE1 (Covalab, Bron, France).
Western blot
Size-exclusion chromatography of Chlamydomonas soluble protein extract fractions 19 to 44 were mixed with reducing Laemmli loading buffer, boiled, and resolved by 200 V electrophoresis through SDS-PAGE. Proteins were subsequently transferred to Protran 0.2 µm nitrocellulose membrane (GE Healthcare, Chicago, USA). Efficiency of protein transfer was checked by Ponceau staining. Primary polyclonal anti-CrRPE1 antibody was incubated overnight at a dilution of 1:5,000. Secondary anti-rabbit IgG coupled to peroxidase (Sigma-Aldrich reference A9169, Saint Louis, USA) was incubated 2 h at a dilution of 1:10,000 and revealed by ECL Prime colorimetric assay (GE Healthcare, Chicago, USA).
Replicates and statistical analyses
All the results reported are representative of at least 3 independent biological replicates and expressed as mean ± SD. Statistical analysis was performed using one-way ANOVA followed by Tukey's post-hoc test for multiple comparisons. P values reported on graphs as follows: *P ≤ 0.05, **P ≤ 0.01.
Accession numbers
Sequence data from this article can be found in the EMBL data library under accession number PNW74744.1.
Acknowledgments
We acknowledge Marion Hamon and Dr. Christophe Marchand at the mass spectrometry platform of the Institut de Biologie Physico-Chimique (FRC 550 CNRS) for technical support. The Institut de Biologie Physico-Chimique provided access to crystallization platform. We acknowledge the European Synchrotron Radiation Facility (Grenoble, France) for provision of synchrotron radiation facilities at beamline ID30A-3 MASSIF-3 and SOLEIL (Gif-sur-Yvette, France) for provision of synchrotron radiation facilities on beamlines SWING, Proxima-1, and Proxima-2a. M.M. and M.Z. sincerely thank Dr. C. de Witte for valuable scientific discussions.
Author contributions
M.Z. and J.H. designed the research; M.M., S.Fa., D.T., L.G., S.Fe., M.Z., and J.H. performed the research; M.M., S.Fa., D.T., L.G., P.T., S.Fe., S.D.L., M.Z., and J.H. analyzed the data; and M.M., S.Fa., D.T., L.G., P.T., S.Fe., S.D.L., M.Z., and J.H. wrote the manuscript.
Supplementary data
The following materials are available in the online version of this article.
Supplementary Figure S1. In vitro quaternary structure analysis of CrRPE1.
Supplementary Figure S2. Recombinant CrRPE1 far-UV circular dichroism spectrum.
Supplementary Figure S3. Comparison of RPE quaternary structures.
Supplementary Figure S4. Analysis and identification of zinc ion in CrRPE1.
Supplementary Figure S5. Multiple sequence alignment of RPE from photosynthetic eukaryotes.
Supplementary Figure S6. Schematic view of the enzymatic reporter assays used in this study.
Supplementary Figure S7. In vitro specific activity of plant RPE.
Funding
This work was funded by CNRS, Sorbonne Université, and Agence Nationale de la Recherche grants LABEX DYNAMO (11-LABX-0011), CALVINDESIGN (ANR-17-CE05-001), and CALVINTERACT (ANR-19-CE11-0009).
Dive Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
References
Author notes
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://dbpia.nl.go.kr/plphys/pages/General-Instructions) is: Julien Henri ([email protected]).
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}