





Abstract
Class IV homeodomain leucine-zipper transcription factors (HD-Zip IV TFs) are key regulators of epidermal differentiation that are characterized by a DNA-binding HD in conjunction with a lipid-binding domain termed steroidogenic acute regulatory-related lipid transfer (START). Previous work established that the START domain of GLABRA2 (GL2), a HD-Zip IV member from Arabidopsis (Arabidopsis thaliana), is required for TF activity. Here, we addressed the functions and possible interactions of START and the HD in DNA binding, dimerization, and protein turnover. Deletion analysis of the HD and missense mutations of a conserved lysine (K146) resulted in phenotypic defects in leaf trichomes, root hairs, and seed mucilage, similar to those observed for START domain mutants, despite nuclear localization of the respective proteins. In vitro and in vivo experiments demonstrated that while HD mutations impair binding to target DNA, the START domain is dispensable for DNA binding. Vice versa, protein interaction assays revealed impaired GL2 dimerization for multiple alleles of START mutants, but not HD mutants. Using in vivo cycloheximide chase experiments, we provided evidence for the role of START, but not HD, in maintaining protein stability. This work advances our mechanistic understanding of HD-Zip TFs as multidomain regulators of epidermal development in plants.
Introduction
Class IV homeodomain leucine-zipper transcription factors (HD-Zip IV TFs) are critical regulators of epidermal and sub-epidermal differentiation in plants (Nakamura et al., 2006). In Arabidopsis (Arabidopsis thaliana), there are 16 steroidogenic acute regulatory (StAR)-related lipid transfer (START) domain-containing TFs belonging to the HD-Zip IV family. The founding member, GLABRA2 (GL2), functions in a complex regulatory network to dictate epidermal cell fate. Genetic studies initially showed that GL2 promotes trichome differentiation in shoots (Rerie et al., 1994) and mucilage biosynthesis in seeds (Western et al., 2001, 2004), while it suppresses hair formation in nonhair root cells (Di Cristina et al., 1996; Masucci et al., 1996). More recent mutant analyses revealed that GL2 regulates the accumulation of seed oil (Shen et al., 2006; Shi et al., 2012) and anthocyanin biosynthesis (Wang et al., 2015). In addition to GL2, the functionally redundant family members A. thaliana MERISTEM LAYER1 (ATML1) and PROTODERMAL FACTOR2 (PDF2) have been extensively studied for their developmental role in specification of the epidermis in the embryo (Abe et al., 2003; Ogawa et al., 2015; Iida and Takada, 2021). HD-Zip IV proteins are comprised of four defined domains including a DNA-binding HD, a leucine-zipper domain termed zipper loop zipper (ZLZ), a START domain, and a START-associated domain (SAD or STAD) (Schena and Davis, 1994; Di Cristina et al., 1996; Ponting and Aravind, 1999; Riechmann et al., 2000; Schrick et al., 2004).
The HD is a DNA-binding domain of approximately 60 amino acids that is highly conserved in the eukaryotes (Carrasco et al., 1984; McGinnis et al., 1984; Gehring et al., 1990; Bürglin, 1994; Derelle et al., 2007). It was discovered in Drosophila genes, whose mutation or overexpression cause homeotic transformations affecting the body pattern (Garber et al., 1983; McGinnis et al., 1984; Scott and Weiner, 1984). The 3D structure of the HD shows a flexible amino-terminus and three α-helices, the second and third of which form a helix–turn–helix motif (Gehring et al., 1990; Kissinger et al., 1990; Liu et al., 1990; Otting et al., 1990; Gehring, 1994). DNA-binding specificity occurs through major groove contacts via the third helix as well as minor groove contacts via the amino-terminal region (Schofield, 1987; Mann et al., 2009; Burglin and Affolter, 2016). The earliest reported homeobox gene from plants was maize (Zea mays) Knotted-1 (KN-1), whose gain-of-function phenotype is marked by uncontrolled cell divisions in lateral veins, forming outpocketings or knots (Vollbrecht et al., 1991). Since the discovery of Kn-1, numerous plant homeobox genes have been identified and implicated in a wide variety of developmental processes. In Arabidopsis, there are 110 predicted HD proteins that are subdivided into 14 classes including HD-Zip TFs (Classes I–IV) (Mukherjee et al., 2009).
HD-Zip III and IV TFs contain a START domain that occurs in the middle of the protein, C-terminal to the HD-Zip. Initially discovered in mammalian StAR cholesterol-binding transporters (Kallen et al., 1998), START domains are modules of approximately 210 amino acids (Ponting and Aravind, 1999) conserved across plants and animals as well as several protists, bacteria, and archaea (Iyer et al., 2001). The domain structure comprises of an α/β helix-grip fold that forms a binding pocket (Tsujishita and Hurley, 2000; Thorsell et al., 2011). Human START domain-containing proteins bind ligands such as sterols, phospholipids, ceramides, bile acids, and fatty acids (Kallen et al., 1998; Tsujishita and Hurley, 2000; Roderick et al., 2002; Kudo et al., 2008; Létourneau et al., 2012; Tillman et al., 2020). In mammals, START domains function in lipid/sterol transport or metabolism and several act in intracellular signaling pathways (Clark and Stocco, 1995; Lin et al., 1995; Watari et al., 1997; Stocco, 2001; Soccio and Breslow, 2003).
Studies in plants indicate similar roles for START domains in lipid transfer and/or lipid sensing. In the liverwort Marchantia polymorpha, a START domain-containing protein (STAR2) was recently implicated in the incorporation of ER-derived C20 fatty acids into chloroplast glycolipids (Hirashima et al., 2021). Our previous work showed that the START domain is essential for TF activity of HD-Zip IV member GL2 (Schrick et al., 2014). Several mechanisms have been proposed to explain how the ligand-bound START domain can stimulate TF activity, including conformational changes, stabilization of protein levels, and/or promotion of protein–protein interactions (Schrick et al., 2014). However, lack of detailed information on how START modulates TF activity poses a major challenge.
The goal of this study was to understand how the START domain modulates HD-Zip IV TF activity either independently or in synergy with the HD. We generated HD mutations in wild-type and START domain-deleted backgrounds and studied their effect on TF function. HD mutants lacking the predicted DNA-binding α-helices or containing missense mutations in a conserved lysine imparted defective phenotypes in trichomes, roots, and seeds, similar to those of START mutants. While HD mutations abolished DNA binding, multiple mutations affecting the START domain retained DNA-binding affinity in vitro and in vivo. Gel shift experiments led to the surprising discovery that HD-Zip IV proteins lacking the START domain can bind DNA as monomers. Using interaction studies in yeast and Nicotiana benthamiana, we confirmed dimerization defects from mutations affecting START but not HD. Finally, cycloheximide chase experiments showed that protein levels drop rapidly in several START domain mutants but not in HD mutants, consistent with a unique role of START in maintaining protein stability.
Results
HD and START domain are highly conserved across the HD-Zip IV TFs
The HD is evolutionarily conserved across all major eukaryotic lineages (Carrasco et al., 1984; McGinnis et al., 1984; Derelle et al., 2007; Mukherjee et al., 2009). We found that the crystal structure of HOMEOBOX PROTEIN ENGRAILED-2 (Gg-En-2) from chicken exhibits predicted structural similarity with the HD from HD-Zip IV members. Aligning HDs from all 16 HD-Zip IV TFs (GL2, PDF2, ANL2, ATML1, and HDG1-12) with reference to Gg-En-2 revealed highly conserved α-helices as well as amino acid residues (Supplemental Figure S1A). The lysine at position 45 of the HD displays 100% conservation in HD-Zip IV members (Figure 1A; Supplemental Figure S1A). This conserved lysine selectively recognizes the 3′-CC dinucleotide adjacent to the TAAT core sequence (Amendt et al., 1998; Cazorla et al., 2000; Lebel et al., 2001) in the vertebrate Bicoid-like TFs PITX2 and PITX3 (Semina et al., 1996, 1997; Gage and Camper, 1997; Smidt et al., 1997).

HD mutants exhibit loss-of-function phenotype in leaves, seeds, and roots. A, Schematic of wild-type GL2 and mutant gl2ΔHD and gl2ΔSTART proteins containing HD, ZLZ, START, and STAD. Amino acid lengths of proteins are indicated (right). Alignment of GL2 and PDF2 HD domains generated by Geneious 7.0.6 (Biomatters). Identical and similar amino acid residues are indicated by black and gray shading, respectively. See Supplemental Figure S1A. The various HD deletions are shown below the alignment. Lysine targeted for missense mutations (K-E or K-T) is indicated by box and asterisk. B and B′, Wild-type GL2 rosettes display trichomes covering leaf surfaces. In contrast, mutants (gl2ΔST, gl2ΔHD46, gl2ΔHD20, gl2ΔHD11, gl2K146E, gl2ΔHD20;ΔST, gl2K146T;ΔST, and gl2K146E;ΔST) exhibit trichome differentiation defects. gl2K146T displays an intermediate phenotype. Bar = 1 mm. C and (C′, Left: Ruthenium red staining of seeds to assay mucilage production. Wild-type GL2 seeds display a normal mucilage layer, while the various gl2 mutants exhibit defective mucilage production. gl2K146T displays a partial phenotype. Bar = 100 μm. Right: Root hair phenotypes of 4- to 5-day-old seedlings. In contrast to wild-type GL2 and the partial phenotype of gl2K146T, all other mutants exhibit excessive root hair formation. Bar = 1 mm. D, Total number and branching pattern of trichomes on the first leaves. Gray bars indicate trichomes with two to five branches in plants expressing wild-type EYFP-tagged GL2. Mutant plants of the genotypes gl2-5, and the other gl2 mutants exhibit unbranched trichomes, indicated by red bars. gl2K146T plants display partial trichome differentiation defects. Values are mean ± sd for n = 20 plants. Letters denote significant differences from ordinary one-way ANOVA (P < 0.05) using Tukey’s multiple comparisons test. E, Numbers of root hairs on primary roots of 4- to 5-day-old seedlings. Genotypes correspond to those in (D). All the gl2 mutants display increases in the number of root hairs. The gl2K146T seedlings exhibit an intermediate phenotype. Values are mean ± sd for n = 6–10 seedlings. Letters denote significant differences from one-way ANOVA (P < 0.05) using Tukey’s multiple comparisons test.
The conserved features of the START domain among HD-Zip IV members were studied using a structure-based sequence alignment with STARD5, a mammalian START protein with known crystal structure (Soccio et al., 2002; Rodriguez-Agudo et al., 2005; Thorsell et al., 2011). Despite low sequence identity, the alignment revealed consensus α-helices and β-strands, as well as conserved amino acids implicated in ligand binding (Supplemental Figure S1B).
To study the role of the HD in HD-Zip IV TFs, we generated several constructs with HD mutations in both wild-type GL2 (and/or PDF2) and START-deleted (ΔST) backgrounds (Figure 1A). The gl2ΔST mutant lacks 235 residues encoding the START domain (Schrick et al., 2014). We constructed HD deletion mutants that remove the consensus α-helices in GL2 (ΔHD11, ΔHD20, and ΔHD46) and missense mutants (K-E, K-T) targeting a highly conserved lysine (K146 in GL2; K107 in PDF2) (Figure 1A). This positively charged lysine was replaced with a negatively charged glutamic acid or with a neutral polar residue, threonine. Transgenic lines were constructed in the gl2-5 null mutant background with wild-type GL2 or mutant gl2 expressed under the native GL2 promoter with the enhanced yellow fluorescent protein (EYFP) translationally fused at the amino-terminus. For each genotype, we screened more than 20 independent T1 transformants to monitor nuclear expression of EYFP and phenotypic complementation of gl2-5 (Supplemental Table S1), and chose representative homozygous T3 lines for further characterization.
Mutations affecting the HD of GL2 exhibit defects in leaf, root, and seed epidermis
Deletion of the START domain in GL2 was previously shown to cause visible defects in differentiation of the epidermis in the leaf (trichomes), root (nonhair cells), and seed (seed coat mucilage) similar to gl2-5 null mutant phenotypes (Schrick et al., 2014). We examined the phenotypic effects of mutations in the HD in presence or absence of START domain deletion (Figure 1). Unlike wild-type plants, rosette leaves from HD mutants lacking consensus α-helices (gl2ΔHD11, gl2ΔHD20, gl2ΔHD46) or containing a K-E substitution in the conserved lysine (gl2K146E) displayed trichome defects and were indistinguishable from ΔST or gl2-5 null mutants (Figure 1, B and B′). Another missense mutation in the same lysine (K146T) resulted in partial rescue (Figure 1B′). Importantly, three lines harboring both HD and START domain mutations (gl2ΔHD20;ΔST, gl2K146E;ΔST, and gl2K146T;ΔST) were also defective in trichome differentiation similar to ΔST mutants (Figure 2A′). Quantification of trichomes on the first leaves revealed a reduction in total number as well as the abnormal presence of unbranched trichome in these mutants (Figure 1D).

Nuclear localization of wild-type EYFP-tagged GL2 and mutant gl2 TFs. A and A′, Imaging of leaf trichomes under white light (green, chlorophyll). Bar = 100 μm. B and B′, Matching fluorescence images (red, chlorophyll) indicate nuclear localization of EYFP-tagged GL2, gl2ΔST, gl2ΔHD46, gl2ΔHD20, gl2ΔHD11, gl2K146T, gl2K146E, gl2ΔHD20;ΔST, gl2K146T;ΔST, and gl2K146E;ΔST proteins in trichome cells. Arrows mark the brightest nuclear signal in each image. Bar = 100 μm. C and C′, Confocal laser scanning microscopy of developing seeds reveals nuclear expression of EYFP-tagged wild-type and mutant proteins in seed coat cells. Bar = 100 μm. D and D′, Magnified images from (C) and (C′). Bar = 50 μm.
Apart from trichome defects, mutation of HD resulted in excessive root hair formation characteristic of gl2-5 null mutants, irrespective of accompanying START domain mutation (Figure 1, C, C′, and E). To assess the effect of mutations on differentiation of the mucilage secretory cells, imbibed seeds were stained with ruthenium red, a dye that binds to pectinaceous polysaccharides. Reduced seed coat mucilage was observed in HD mutants gl2ΔHD11, gl2ΔHD20, gl2ΔHD46, and gl2K146E, while the K146T mutation resulted in a partial mucilage defect (Figure 1, C and C′) consistent with partial phenotypes observed in trichomes and roots (Figure 1, D and E). HD and START domain double mutants (gl2ΔHD20;ΔST, gl2K146E;ΔST, and gl2K146T;ΔST) were also defective in seed mucilage similar to ΔST mutants (Figures 1, C and C′). These results demonstrate that both the HD and START domain are critical for epidermal differentiation in multiple cell types, and that mutation in one domain is not able to suppress the loss-of-function phenotype in the other domain.
HD and START domain mutants retain nuclear localization of GL2
GL2 is expressed in developing trichomes of the leaf epidermis (Rerie et al., 1994; Szymanski et al., 1998), nonhair cell files in the root epidermis (Di Cristina et al., 1996; Masucci et al., 1996) and hypocotyl (Hung et al., 1998), and seed coat cells (Western et al., 2004). As a TF, GL2 is presumably translocated to the nucleus where it binds DNA to regulate gene expression. Using EYFP fusions to GL2, it was previously shown that the START domain is not necessary for nuclear localization (Schrick et al., 2014). Here we monitored the expression of EYFP:GL2 transgenes harboring various combinations of HD and START domain mutations in leaf trichomes and developing seeds. Using live imaging of trichomes, we observed nuclear localization of the EYFP-tagged proteins in all the HD mutants similar to that for wild-type GL2 and gl2ΔST (Figure 2, A, A′, B, and B′). Nuclear localization remained intact in trichomes of mutants for both the HD and START domains (gl2ΔHD20;ΔST, gl2K146E;ΔST, and gl2K146T;ΔST) (Figure 2, A′ and B′). Confocal laser scanning microscopy of developing seeds indicated nuclear localization of HD and START mutant proteins in the seed coat cells (Figure 2, C, C′, D, and D′). The results demonstrate that both HD and START domain are dispensable for translocation of GL2 to the nucleus.
START domain missense mutants exhibit loss of function despite nuclear localization of GL2
We next tested whether missense mutations in the START domain result in phenotypic defects similar to those seen in the START deletion mutant, gl2ΔST (Figure 3A). Transgenic lines carrying EYFP:gl2 constructs were established according to the strategy described above for the HD mutants and gl2ΔST. Phenotypic analysis showed that three START missense mutants (gl2E375G;R392M, gl2Amo, and gl2L480P) and a mutant deleted for the six terminal amino acids of the HD and ZLZ domain (gl2ΔHD6;ΔZLZ), display leaf trichome defects and seed mucilage defects (Figure 3, B, C, and C′), as well as excess root hair formation (Supplemental Figure S2A) similar to the gl2-5 null mutant. The mutations in gl2E375G;R392M disrupt a salt bridge that is thought to be critical for ligand binding (Roostaee et al., 2009). In gl2Amo, the R386L and E387L missense mutations, which remove charged residues, are followed by an 11-residue duplication in a predicted ligand-contact region (Figure 3A; Supplemental Figure S1B). In gl2L480P, the mutation affects a lipid contact site in the C-terminal α-helix of the START domain (Roostaee et al., 2009; Alpy and Tomasetto, 2014; Wojciechowska, 2021).
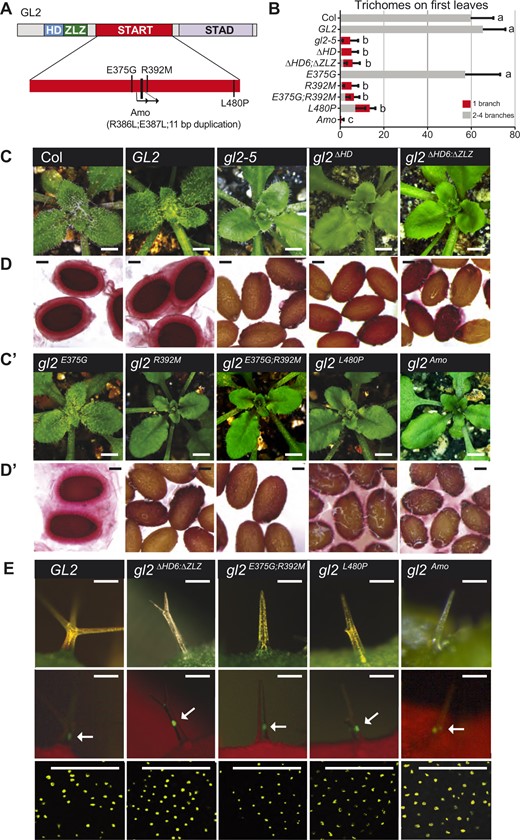
START domain missense mutants exhibit loss-of-function phenotypes despite nuclear localization of the corresponding EYFP-tagged proteins. A, Schematic showing the functional domains of GL2. Positions of mutations within the START domain are indicated (see Supplemental Figure S1B). B, Total number and branching pattern of trichomes on the first leaves. Gray bars indicate normal trichomes in plants expressing Col wild-type, and EYFP-tagged GL2 and gl2E375G. Mutant gl2-5 plants and those expressing EYFP-tagged gl2ΔHD, gl2ΔHD6;ΔZLZ, gl2R392M, gl2E375G;R392M, gl2L480P, and gl2Amo exhibit unbranched trichomes (red bars). Values are mean ± sd for n = 20 plants. Letters denote significant differences from one-way ANOVA P < 0.05) using Tukey’s multiple comparisons test. C and C′, Wild-type Col, EYFP:GL2, and EYFP:gl2E375G expressing rosettes display trichomes covering leaf surfaces. In contrast, gl2-5 mutants as well as the EYFP-tagged HD mutants (gl2ΔHD, gl2ΔHD6;ΔZLZ) and START domain mutants (gl2R392M, gl2E375G;R392M, gl2L480P, and gl2Amo) exhibit trichome differentiation defects. Bar = 1 mm. D and D′, Ruthenium red staining of seeds to assay mucilage production. Wild-type Col, EYFP:GL2, and EYFP:gl2E375G seeds display a normal mucilage layer, while gl2-5 mutants as well as the EYFP-tagged HD mutants (gl2ΔHD, gl2ΔHD6;ΔZLZ) and START mutants (gl2R392M, gl2E375G;R392M, gl2L480P, and gl2Amo) exhibit defective mucilage production. Bar = 100 μm. E, Nuclear localization of EYFP-tagged GL2 and mutant gl2 TFs. Top: Imaging of leaf trichomes under white light (green, chlorophyll). Bar = 100 μm. Middle panel: Matching fluorescence images (red, chlorophyll) indicate nuclear localization of EYFP-tagged proteins in trichomes. Arrows mark the brightest nuclear signal in each image. Bar = 100 μm. Bottom: Confocal laser scanning microscopy of developing seeds reveals nuclear expression of EYFP-tagged GL2 and mutant proteins in seed coat cells. Bar = 50 μm.
We identified another missense allele, W279R, that maps to the N-terminus of the START domain, which also results in loss-of-function (Supplemental Figure S3). In gl2W279R, a highly conserved tryptophan is replaced with a charged residue within a predicted hydrophobic region of the binding pocket (Supplemental Figures S1, B and S3, A). In contrast, missense mutations in the predicted N-terminal α-helix of START (gl2A258P, gl2M271I, gl2I289N, and gl2E294Q) do not significantly impair function (Supplemental Figure S3, A–E).
Despite loss-of-function phenotypes, the four missense START alleles (gl2E375G;R392M, gl2Amo, gl2L480P, and gl2W279R) and the gl2ΔHD6;ΔZLZ mutant retained nuclear localization of the corresponding EYFP:tagged proteins (Figure 3; Supplemental Figures S2, B and S3, F). These results provide additional evidence that the HD and START domain are not required for nuclear location of GL2.
HD, but not the START domain, is required for DNA binding in vitro
HD-Zip IV TFs regulate the expression of various downstream target genes by binding to cis-elements in the respective promoter regions (Nakamura et al., 2006; Khosla et al., 2014). We performed electrophoretic mobility shift assays (EMSA) to examine whether DNA binding is regulated by the HD alone or by HD in combination with the START domain. For the DNA probe, we used the L1-Box element (TAAATGTA) implicated in GL2-DNA binding that is found upstream of several GL2 targets (Ohashi et al., 2003; Tominaga-Wada et al., 2009; Khosla et al., 2014). We produced recombinant Halo-tagged proteins using wheat germ in vitro transcription–translation. Despite repeated attempts, we were unable to obtain full-length recombinant GL2 protein that was functional in EMSA.
Given the high degree of sequence similarity between HD-Zip IV proteins (Supplemental Figure S1), we selected PDF2 and mutant versions analogous to our gl2 HD and START mutants (pdf2K107E, pdf2K107T, pdf2ΔST, pdf2K107E;ΔST, and pdf2K107T;ΔST) for DNA-binding studies. ATML1, which functions redundantly with PDF2, is reported to bind the octamer TAAATG(C/T)A (Abe et al., 2001). Thus, we performed EMSAs with a Cy3-labeled probe containing three tandem repeats of TAAATGTA. Both wild-type PDF2 and pdf2ΔST caused a shift in probe mobility as an indication of DNA binding (Figure 4A). In contrast, the two missense mutations K107E and K107T resulted in defective mobility shift independently of the START domain. K107E completely abolished binding to the target sequence as indicated by a lack of mobility shift, while K107T resulted in partial DNA binding for both wild-type PDF2 (pdf2K107T) and pdf2ΔST (pdf2 K107T;ΔST) (Figure 4A). Western blotting confirmed uniform expression of the recombinant proteins used in our EMSA experiments (Figure 4B).
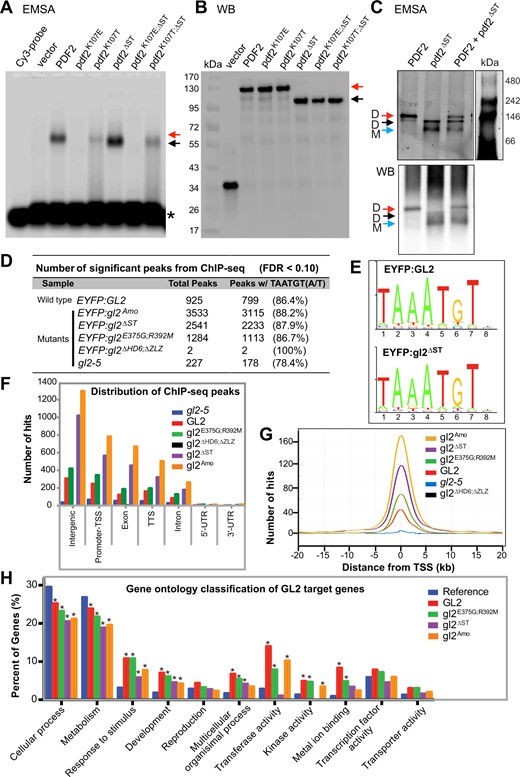
HD is required for DNA-binding activity of PDF2 and GL2, while START domain is dispensable for DNA binding. A, HD is required for in vitro DNA binding of PDF2 and pdf2ΔST proteins. EMSA reveal band shifts for Halo:PDF2 (red arrow) and Halo:pdf2ΔST(black arrow), but not for proteins harboring HD mutation K107E. The K107T mutation results in partial DNA–protein complex formation indicated by a mixed population of bound and smeared molecules. Migration of the Cy3 probe containing the L1-box TAAATGTA is indicated (asterisk). Representative experiment from three independent experiments. B, Western blot with anti-Halo Ab detects Halo-tagged proteins assayed for DNA binding in (A), representative of three independent experiments. C, The pdf2ΔST protein binds DNA as a monomer. EMSA coupled with polyacrylamide gel electrophoresis reveals that Halo:PDF2 binds the Cy3-labeled L1-box probe as a dimer (∼230 kDa) (red arrow) while Halo:pdf2ΔST binds DNA as a monomer (∼90 kDa) (blue arrow) and weakly as a dimer (∼180 kDa) (black arrow). Western blot (WB) (bottom) of the polyacrylamide gel with anti-Halo Ab detected Halo-tagged PDF2, and pdf2ΔST at the expected migration positions matching that of the EMSA (top). Representative blot of two replicates. See Supplemental Figure S4. D–H, Chip-seq data reveals that wild-type GL2 and mutants with compromised START domain function (gl2ΔST, gl2Amo, and gl2E375G;R392M) are proficient in DNA binding in vivo. In contrast, DNA binding was abolished in the gl2ΔHD6;ΔZLZ mutant. D, Number of significant peaks (false discovery rate, FDR < 0.1) from ChIP-seq data indicate that START domain mutants, but not the HD-ZLZ mutant, bind DNA target sites in vivo. Second column shows number and percentage of Chip-seq peaks containing one or more binding motifs with at least 80% sequence identity to DNA sequence TAAATGT(A/T). E, Sequence logo representing the information content of the GL2 consensus DNA-binding motif identified in 86.4% of the wild-type GL2 and 87.9% of the mutant gl2ΔST ChIP-seq peak regions, respectively (see Supplemental Figure S5A). F, Genomic distribution of ChIP-seq binding events for wild-type GL2 and mutant versions are indicated (FDR < 0.1). TTS, transcription termination site; UTR, untranslated region (see Supplemental Table S2). G, ChIP-Seq hit distribution from gene TSS. The distribution of significant ChIP-seq peaks (FDR < 0.1) upstream and downstream from the TSS is shown for wild-type GL2 and mutants (see Supplemental Figure S5B). H, Functional classification of target genes from ChIP-seq data indicate over-represented GO terms from target genes whose promoters were bound by wild-type GL2 and mutants gl2ΔST, gl2Amo, and gl2E375G;R392M. Genes were classified based on molecular function and biological process, and a given gene may be represented in more than one category. Enrichment analysis was performed using AgriGO (Du et al., 2010). Asterisks indicate significant differences from the Arabidopsis genome reference (blue) (Fisher’s exact test, P < 0.01).
Combining EMSA with native polyacrylamide gel electrophoresis, we observed that the wild-type PDF2 protein binds the DNA as a dimer. In contrast, the pdf2ΔST mutant binds the DNA target sequence as a monomer (Figure 4C; Supplemental Figure S4). The abnormal monomeric form was also seen in comparing the DNA-binding behavior of the mutant atml1ΔST protein to wild-type ATML1 dimer (Supplemental Figure S4).
The START domain is dispensable for DNA binding in vivo
We performed chromatin immunoprecipitation sequencing (ChIP-seq) assays with seedlings expressing EYFP-tagged wild-type GL2 and mutant proteins to test whether the START domain is dispensable for DNA binding in vivo. The experiments included three START domain mutants (gl2ΔST, gl2E375G;R392M, and gl2Amo) as well as the HD-ZLZ mutant (gl2ΔHD6;ΔZLZ), all of which display loss-of-function phenotypes similar to the gl2-5 null mutant, but whose proteins are nuclear-localized (Figures 1–3; Supplemental Figure S2; Supplemental Data Sets 1 and 2).
Data analysis revealed DNA-binding proficiency for wild-type GL2 and all three START domain mutants whereas little or no binding was detected for gl2ΔHD6;ΔZLZ and the gl2-5 null mutant control (Figure 4D; Supplemental Table S2). The consensus DNA-binding motif TAAATGT(A/T) was found in >85% of ChIP-seq peaks for wild-type GL2 and the START mutants (Figure 4, D and E; Supplemental Figure S5A). Genomic distributions of binding sites were similar for wild-type GL2 and the START domain mutants. Most of the ChIP-seq hits were mapped to promoter or intergenic regions, consistent with the role of GL2 as a transcriptional regulator (Figure 4F; Supplemental Table S2). The relative positions of binding sites to transcription start sites (TSS) were similar for the wild-type GL2 and START domain mutants (Figure 4G; Supplemental Figure S5B).
Gene ontology (GO) classification revealed overlapping functional classes of target genes bound by wild-type GL2 and the START domain mutants (Figure 4H). In the category “biological processes,” there was a similar enrichment for genes involved in cellular processes (GO:0009987; P = 4.8e-31) and metabolism (GO:0008152; P = 2.4e-42). In the category of “molecular function,” genes encoding transferase activity (GO:0016740; P = 8e-09), kinase activity (GO:0016301, P = 6.1e-07), and metal ion binding (GO:0046872; P = 4.5e-05) appeared as the most enriched genes for wild-type GL2 and two of the START mutants (gl2Amo and gl2E375G;R392M). Although the gene targets bound by the gl2ΔST protein were similarly enriched for metal ion binding, they were not enriched for transferase activity and kinase activity (Figure 4H). A list of target genes with ChiP-seq peaks in their respective promoter regions is presented in Supplemental Data Set 2. The MYB23/At5g40330 gene, which was previously identified as a direct target of GL2 (Khosla et al. 2014) was found to contain ChIP-seq peaks in wild-type GL2 as well as gl2ΔST, gl2E375G;R392M, and gl2Amo samples (Supplemental Data Set 2). Overall, the data indicate that START domain mutants retain DNA-binding activity in a manner that is comparable to wild-type.
The START domain is required for GL2 homodimerization, while the HD is dispensable for this activity
Previous studies demonstrated the important role of the leucine zipper for homo- and hetero-dimerization of HD-Zip TFs (Landschulz et al., 1988; Sessa et al., 1993). Our EMSA experiments indicated that the START domain is critical for homodimerization and heterodimerization of PDF2 and ATML1 when bound to target DNA (Figure 4C; Supplemental Figure S4). We tested GL2 in yeast two-hybrid (Y2H) assays to further address the role of START in homodimerization of HD-Zip IV TFs. Unlike several other HD-Zip family members, GL2 does not show bait autoactivation in the Y2H assay (Supplemental Figures S6, A and C). Y2H experiments confirmed that GL2 undergoes homodimerization but we did not detect heterodimerization of GL2 with other HD-Zip IV members nor was heterodimerization detected with two HD-Zip III members that were tested (Supplemental Figure S6B).
Next we assayed self-interaction between different segments of GL2. Only the full-length GL2 protein and the segment encoding START + STAD displayed homodimerization. The START domain alone did not exhibit homodimerization, while the STAD domain alone could not be tested because of its bait autoactivation (Figure 5A). Consistent with the importance of the START domain for self-interaction, deletion of START (gl2ΔST) and two loss-of-function missense mutants (gl2R392M and gl2L480P) abolished homodimerization. The gl2E375G mutant, whose phenotype is similar to wild-type (Figure 2) supported dimerization (Figure 5B).
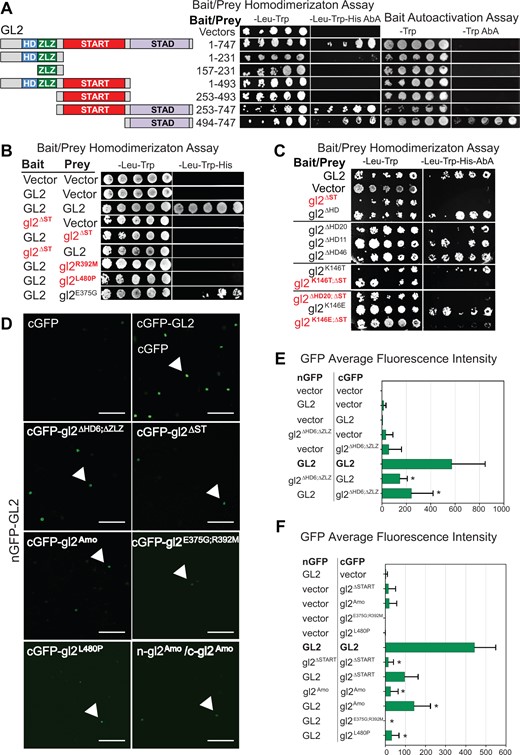
START domain is required for homodimerization of GL2. A, Schematic representation of full-length GL2 and segments for Y2H assays (left). GL2 homodimerization of full-length protein (1–747) and START–STAD fragment (253–747) was observed (middle). The STAD fragment exhibits bait autoactivation (right). Homodimerization was assayed on –Leu–Trp–His Aureobasidin A (AbA) while bait autoactivation was assayed on –Trp AbA media. See Supplemental Figure S7A. B, Deletion or missense mutations in the START domain disrupt homodimer formation in Y2H assays. GL2, gl2ΔST, and the indicated missense mutants were cloned into bait and prey vectors, co-expressed in yeast strain Y2HGold and assayed on the indicated growth media. Empty prey and bait vectors served as negative controls. C, Y2H assays show that bait and prey fusion proteins interact to homodimerize only when START domain is present. Homodimerization was assayed on selective media as in (A). HD mutants were proficient in homodimerization. START domain deletion resulted in loss of homodimerization in absence or presence of HD mutations (red). Wild-type GL2 bait and prey served as positive controls while empty bait and prey vectors represent negative controls (see Supplemental Figures S6, C–E and S7B). D–F, BiFC assays indicate homodimerization of wild-type GL2 but not HD-ZLZ mutant nor START domain mutants. D, Nicotiana benthamiana leaf abaxial epidermal cells were transiently transformed with the indicated constructs and silencing suppressor p19. Interaction of full-length GL2 tagged with split GFP at the N-terminus (nGFP-GL2) with vector with the split GFP at C-terminus (cGFP), full-length GL2 and various gl2 mutants. Detection of gl2Amo self-interaction is also shown. Nuclear expression is indicated by arrows. Scale bar = 50 µm (see Supplemental Figure S8). E and F, Quantitative analysis of BiFC assays in (D). Mean pixel intensities from 10 images are shown. Error bars indicate standard deviations for two independent transformants in two trials. Asterisks indicate statistically significant differences (*P < 0.0001 by t test).
We examined whether the HD plays a role in homodimerization. All of the HD mutants tested (gl2ΔHD, gl2ΔHD20, gl2ΔHD11, gl2ΔHD46, gl2K146T, and gl2K146E) exhibited homodimerization but not bait autoactivation similar to wild-type GL2 (Figure 5C; Supplemental Figure S6, C–E). In contrast, double mutants with deletion of START (gl2K146T;ΔST, gl2ΔHD20;ΔST, and gl2K146E;ΔST) abolished homodimerization (Figure 5C). These results show that GL2 dimerization is unaffected by HD mutations but is impaired by deletion of the START domain. Western blotting confirmed the expression of the various wild-type and mutant fusion proteins in yeast (Supplemental Figure S7).
Bimolecular fluorescence complementation (BiFC) assays in N. benthamiana (Figure 5, D–F) verified our GL2 homodimerization results in yeast. Controls expressing either the N- or C-terminal half of GFP showed little or no fluorescence (Supplemental Figure S8). In contrast, strong fluorescent signals were observed in nuclei for samples expressing both halves of GFP fused to wild-type GL2, indicating homodimerization. Consistent with the reported role of the ZLZ domain in dimerization, the fluorescence intensity was significantly reduced when wild-type GL2 was tested for interaction with gl2ΔHD6;ΔZLZ (Figure 5E). Reduced fluorescence intensity was similarly seen when wild-type GL2 was tested for interaction with four START domain mutant proteins (gl2ΔST, gl2Amo, gl2E375G;R392M, and gl2L480P), as well as for self-dimerization for both of the START mutants we tested (gl2ΔST and gl2Amo) (Figure 5F). Taken together, the interaction assays in yeast and N. benthamiana demonstrate the critical importance of START for homodimerization of GL2, whereas the HD domain is not required for this activity.
The START domain, but not HD, regulates GL2 protein stability
Little is known about how the level of HD-Zip TFs is regulated posttranslationally. It was reported that mutation of a putative lipid contact site in the START domain of ATML1 completely disrupts expression of the fluorescently tagged protein in lateral roots (Nagata et al., 2021). In our study, we observed that some EYFP-tagged gl2 proteins harboring mutations in either the HD or START, particularly gl2W279R, appeared weakly expressed in comparison to the wild-type protein (Figure 3; Supplemental Figure S3F).
To determine whether the HD and/or START domain play a role in regulating GL2 levels, we monitored protein half-life by treating Arabidopsis seedlings with cycloheximide, an inhibitor of protein synthesis. Proteins were extracted from seedlings at various intervals over a 10 or 24 h time course followed by immunodetection and quantification. We analyzed wild-type GL2, gl2ΔHD20 and gl2ΔST along with three of the START missense mutants: gl2W279R, gl2L480P, and gl2Amo. As revealed by Western blot and protein quantification, wild-type GL2 and gl2ΔHD20 proteins remained stable over 24 h, whereas protein levels of various START domain mutants (gl2ΔST, gl2L480P, gl2W279R, and gl2Amo) dropped rapidly (Figure 6, A and B; Supplemental Figure S9). The gl2ΔST and gl2W279R mutant proteins were particularly unstable, having barely detectable protein levels at the 2-h time point (Supplemental Figure S9). In case of gl2ΔST, a degradation product of ∼65 kDa also exhibited a diminished protein half-life (Supplemental Figure S9). We additionally examined the stability of proteins that harbored mutations in both the HD and START, but the low levels of expression made it difficult to interpret their half-lives in cycloheximide assays (Supplemental Figure S10). Taken together, our results provide strong evidence for the role of START in maintaining protein stability of GL2.
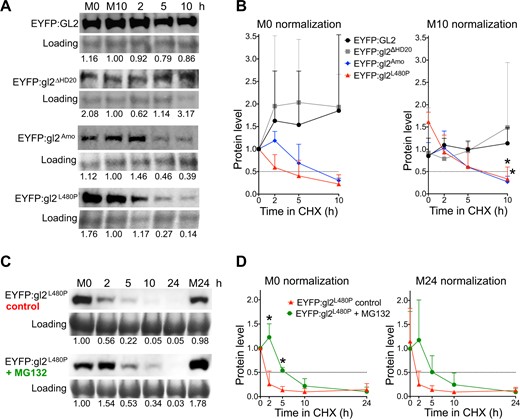
Cycloheximide experiments reveal that the START domain, but not HD, is critical for protein stability of GL2. A and B, HD mutant gl2ΔHD20 behaves similarly to wild-type GL2, whereas START domain mutants gl2Amo and gl2L480P exhibit decreases in protein half-life. C and D, Treatment with MG132 results in increased protein stability of START domain mutant gl2L480P. A and C, Levels of EYFP-tagged proteins were monitored by Western blotting. Immunodetection with anti-GFP Ab was followed by Coomassie blue staining for loading controls. Mock treatments with DMSO at 0 (M0) and 10 (M10) or 24 (M24) h are indicated. Blots for each genotype are representative of three independent experiments. A, Five-day-old seedlings expressing wild-type GL2 or mutant gl2 proteins were treated with cycloheximide (400 μM) for a 10-h time course. Values were normalized to loading controls and M10 (see Supplemental Figures S9 and S10). B, Five-day-old seedlings expressing EYFP-tagged gl2L480P were treated simultaneously with cycloheximide (400 μM) and DMSO (control) or proteasome inhibitor MG132 (50 μM) for a 24 h time course. Values were normalized to loading controls and M0. B and D, Protein quantification is shown for three experiments including that depicted in (A) and (C), respectively. Values were normalized to M0 (left) or M10 (or M24) (right) controls. Protein half-life (0.5) is indicated by intersection with dotted lines. Error bars indicate sd for means from three experiments. Two-tailed t test with unequal variance: *P ≤ 0.05.
To better understand the degradation pathway used to control protein levels, additional experiments were performed with one of the START mutants (L480P) in presence or absence of proteasome inhibitor MG132. Upon addition of MG132, the gl2L480P protein remained stable for ∼5–10 h during CHX treatment whereas the control treatment resulted in disappearance of the protein as early as 2 h (Figure 6, C and D). The data suggest the possible involvement of the Ubiquitin/26S proteasome system in the turnover of GL2 when the START domain is nonfunctional.
Discussion
Domain organization in HD-Zip IV TFs and nonoverlapping roles of HD and START
The combination of a DNA-binding HD with a lipid-sensing START domain in a single protein is plant-specific (Schrick et al., 2004) and likely represents a novel mode of action in a multidomain TF. At the outset of this study, we envisioned that the START domain regulates the activity of HD-Zip IV TFs through the DNA binding domain. Our overall goal was to investigate the possible association or overlap in the roles of the HD and START domains. In addition to assaying phenotypic effects of genetic mutations in planta, we considered several aspects of TF function, including subcellular localization, DNA binding, dimerization, and protein stability. We found that mutations affecting HD activity result in loss-of-function phenotypes, as illustrated by trichome differentiation defects, reduced seed mucilage and ectopic root hair formation similar to the null mutant gl2-5, ΔST, and multiple START missense mutants (Figures 1 and 3; Supplemental Figure S3). Both HD and the START domain are critical for epidermal differentiation, but the molecular mechanisms underlying their loss-of-function phenotypes are different. While mutations in the HD abolish DNA binding of the TF, the loss of START domain function results in dimerization defects and unstable protein.
Conserved role of HD in DNA binding
A missense mutation in the HD, changing a conserved lysine to a glutamic acid (K107E), completely disrupted the DNA binding activity of PDF2 (Figure 4A). The analogous K-E substitution in the vertebrate HD TFs PITX2 and PITX3 also abolishes DNA binding (Saadi et al., 2001, 2003; Sakazume et al., 2007). In contrast, we found that a K-T substitution at the same position (K107T) exerts a partial defect (Figure 4A). The effect of either of these two missense mutations on DNA binding was unaltered by the deletion of the START domain, consistent with our model that the HD functions in DNA binding independently of the START domain.
Our results indicate that START domain mutants are proficient in DNA binding under both in vitro and in vivo conditions (Figure 4). Several studies report that HD-Zip IV TFs bind DNA elements as dimers (Sessa et al., 1993; Palena et al., 1999; Tron et al., 2001). Surprisingly, our EMSA data suggested that START-deleted proteins bind the DNA as monomers (Figure 4C; Supplemental Figure S4). While the START domain is not required for DNA binding, we confirmed the importance of the START domain for dimerization using self-interaction/homodimerization assays in yeast (Figure 5, B and C; Supplemental Figure S6), and BiFC experiments in planta (Figure 5, D–F; Supplemental Figure S8). Thus, the START domain behaves differently from the ZLZ domain, which seems to be required for dimerization as well as DNA binding (Di Cristina et al., 1996).
The START domain is critical for protein stability
Our cycloheximide assays revealed that wild-type GL2 protein is relatively stable, while mutations in the START domain dramatically reduce protein half-life (Figure 6, A and B; Supplemental Figure S9). It is noteworthy that gl2L480P, gl2W279R, and gl2Amo harbor missense mutations in highly conserved amino acid residues predicted to be important for ligand binding (Supplemental Figure S1B). Thus, our findings support the hypothesis that ligand binding through the START domain stabilizes GL2 protein levels. Stabilization of protein levels might aid in activation of the protein, promotion of protein–protein interactions, or correct protein folding. It is noteworthy that deletion of the START domain results in very unstable protein (Supplemental Figure S9), but the gl2ΔST protein binds to the promoters of target genes in planta (Figure 4, D–H; Supplemental Figure S5), arguing against a general defect in protein folding. In eukaryotes, protein degradation acts as an important regulatory process that mediates optimum responses of cellular pathways to environmental cues. Our experiments with MG132 point to possible degradation of GL2 via the 26S proteasome (Figure 6, C and D). However, proteasome inhibition is known to cause a global inhibition of translation resulting in proteome-wide effects (Larance et al., 2013). Thus, MG132 treatment might induce other pathways affecting protein stability, such as autophagy (Carlsson and Simonsen, 2015; Kang et al., 2018). Further investigation will be required to characterize the precise mechanism of GL2 protein turnover including possible posttranslational modifications.
Model for a role of HD and START domains in promoting HD-Zip IV TF activity
We propose a model in which the function of the HD is to recruit the HD-Zip IV TF to the DNA target site (Figure 7). In this way, the multidomain TF is correctly positioned on the DNA to activate or repress target genes. If HD function is missing, the HD-Zip IV protein is stably expressed and is capable of dimerization via its START domain. However, if the START domain is missing or nonfunctional due to mutation, the protein fails to dimerize and becomes unstable. Since START is required for both dimerization and protein stability, it is possible that these two functions are tightly interlinked. Thus far, we have not identified a mutant or condition in which these two processes are uncoupled. Therefore, START-domain-mediated dimerization likely contributes to stabilization of the TF.
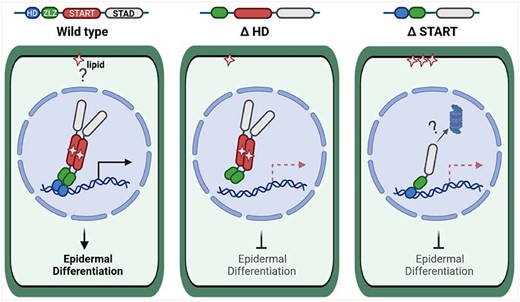
Model of HD and START domain roles in HD-Zip IV TFs. Wild-type HD-Zip IV TFs, as exemplified by GL2 (left), bind a lipid ligand that is possibly retrieved from a membrane compartment. The protein dimerizes and binds to DNA elements in the promoters of target genes, resulting in activation of transcription or repression of gene expression. For simplicity, only activation of gene expression is shown (horizontal arrow). Downstream target genes orchestrate cell differentiation of the plant epidermis. Deletion or mutation of the HD (K146E in GL2) (middle) abolish DNA binding of the TF, but dimerization and nuclear localization remain intact. Although the protein is found in the nucleus, epidermal defects in the corresponding mutants indicate that it is nonfunctional. Deletion of or missense mutations in the START domain (right) result in nonfunctional protein that is compromised in lipid binding. Nonetheless, the mutant protein is transported to the nucleus and binds DNA elements. Dimerization of the protein is affected, resulting in monomeric protein that is unstable, possibly due to degradation via a proteasome-dependent pathway. This figure was created with BioRender.com.
The sequence of events leading to transcriptional activity might involve several steps starting with ligand binding of the HD-Zip IV TF through START. Ligand binding could stabilize dimerization which in turn allows the TF to bind the DNA via its HD. It is noteworthy that in the absence of the START domain, the monomeric form of the TF binds to its DNA target site via its HD (Figure 4C; Supplemental Figure S4). In our model, unliganded monomers lacking the START domain are unstable, resulting in premature degradation.
Other possible domain interactions in HD-Zip IV TFs
While our study was focused on the HD and the START domain, several other domains or motifs might also exert regulatory roles either independently or in conjunction with these functional domains. It is intriguing that while the START domain is important for homodimerization of GL2, it was unable to form dimers on its own and appears to require cooperative action with STAD (Figure 5A), a C-terminal domain of unknown function. Another candidate is the ZLZ domain, a plant-specific leucine zipper that contains approximately 10–20 amino acid loop with two highly conserved cysteines (Schrick et al., 2004). One study suggested that these cysteines are involved in redox control of dimerization as well as the regulation of DNA binding (Tron et al., 2002). Our results demonstrate that both the HD and START domain are dispensable for nuclear localization of the GL2 TF in developing seeds and trichomes (Figures 2 and 3). An uncharacterized motif that maps elsewhere is likely responsible for nuclear localization of HD-Zip IV TFs. In previous studies, several plant HD-Zip I proteins were found to contain a nuclear localization signal either at their N- or C-terminus (Arce et al., 2011; Cao et al., 2016; Shao et al., 2018).
Taken together, our study reports distinct roles of the HD and START domain, both of which contribute to the overall activity of HD-Zip IV TFs. These TFs are critical for orchestrating plant epidermal development, and they additionally play vital roles in responses to biotic and abiotic stress. Our results not only advance our current understanding of these developmentally important TFs but also lay the groundwork for engineering of plant-derived HD and START domains as modular units in synthetic biology applications. Future studies will explore how HD-Zip IV protein levels are tightly regulated during epidermal differentiation. Because these TFs also contain a specialized leucine-zipper domain (ZLZ) and STAD, a plant-specific domain of unknown function, a promising avenue of future investigation will be to examine whether these domains work in association with HD and/or START to modulate various TF activities.
Materials and methods
Plant material, growth conditions, and Arabidopsis transformation
Wild-type Arabidopsis (A. thaliana) ecotype Columbia (Col) and the gl2 null allele gl2-5 (Khosla et al., 2014) were used for this study. Seeds were stratified at 4°C for 3 days and grown on soil containing PRO-MIX PGX, vermiculite, and perlite (Hummert International) in a ratio of 4:2:1 at 23°C under continuous light. The wild-type proGL2:EYFP:GL2 and mutant proGL2:EYFP:gl2 constructs were transformed into gl2-5 plants using Agrobacterium tumefaciens GV3101 (pMP90) mediated floral dip (Clough and Bent, 1998). T1 progeny were screened on 20 μg mL−1 hygromycin B. Independent T1 transformants (21–55) were characterized for each genotype and 3:1 segregation for EYFP expression was confirmed in T2 progeny. Representative T3 homozygous lines with stable EYFP expression were confirmed by polymerase chain reaction (PCR) genotyping and were used for further analysis.
HD and START domain structural alignments
The HD from GL2 was found to exhibit structural similarity with the solution structure of Gallus gallus HOMEOBOX PROTEIN ENGRAILED-2 (Gg-En-2) (PDB ID = 3ZOB) using the SWISS-MODEL homology modeling server (Arnold et al., 2006; Benkert et al., 2011; Biasini et al., 2014). A structural homolog suitable for HD-Zip IV-derived START domain modeling was identified as the crystal structure of human STARD5 (PDB ID = 2R55) using I-TASSER (Yang et al., 2015). Sequences from all 16 HD-Zip IV TFs from A. thaliana (Nakamura et al., 2006) were aligned to corresponding structural homologs in Clustal O (Madeira et al., 2019). Consensus α-helices and β-sheets were visualized using ESPript version 3.0 (Robert and Gouet, 2014).
Plant constructs
The GL2 cDNA was cloned into pENTR/D-TOPO using the pENTR/D-TOPO Cloning Kit (Invitrogen, Carlsbad, CA, USA). Deletion and missense mutations gl2ΔST, gl2ΔHD46, gl2ΔHD20, gl2ΔHD11, gl2K146T, gl2K146E, gl2A258P, gl2M271I, gl2W279R, gl2I289N, and gl2E294Q were generated using the Q5 Site-Directed Mutagenesis Kit (E0554S, New England Biolabs, Ipswich, MA, USA) following manufacturer’s protocol with primers listed in Supplemental Table S3. The gl2ΔST construct was used to generate gl2ΔHD20;ΔST, gl2K146E;ΔST, and gl2K146T;ΔST double mutants. After sequence confirmation, the wild-type and mutant pENTR/D-TOPO plasmids were transferred into the SR54 (ProGL2:EYFP:GL2) binary vector (Schrick et al., 2014). Inserts were amplified with gene specific primers using Q5 High Fidelity DNA polymerase (M0491, New England Biolabs), followed by purification of amplicons using the Nucleospin Gel and PCR Clean-up (Machery-Nagel) and Dpn1 digestion. The vector and insert DNA fragments were assembled in a ratio of 1:5 using NEBuilder HiFi DNA Assembly (E5520S, New England Biolabs). The gl2L480P, gl2E375G, gl2R392M, gl2E375G;R392M, and gl2Amo mutants were generated by one-step PCR-based site-directed mutagenesis (Scott et al., 2002) using PfuUltra II Fusion HS DNA polymerase (Agilent Technologies, Santa Clara, CA, USA). The Phusion Site-Directed Mutagenesis Kit (Thermofisher Scientific, Waltham, MA, USA) was used to generate the gl2ΔHD and gl2ΔHD6;ΔZLZ deletion mutants. Oligonucleotides for generation of mutants are listed in Supplemental Table S3. After verification by sequencing, the GL2 deletion constructs were restriction digested with KpnI and SalI and ligated into the binary vector SR54 (proGL2:EYFP:GL2) at the corresponding sites.
Phenotypic assays and microscopy of Arabidopsis
Arabidopsis lines were assayed for leaf trichomes, root hairs, and seed coat mucilage. Trichomes were quantified on first leaves from 20 individual plants. Leaf samples were placed on 0.8% w/v agar/water plates and trichomes were counted manually upon viewing with a stereomicroscope. For root hair density analysis, seeds were vapor sterilized and sown on 0.5× MS media (Murashige and Skoog, 1962), followed by stratification at 4°C for 5 days. Seedlings were germinated in vertical orientation at 23°C under continuous light. Primary roots were imaged with a stereomicroscope 2–5 days after germination. For seed mucilage analysis, seeds were stained with ruthenium red (R2751, Sigma-Aldrich, St Louis, MO, USA) following an established protocol (McFarlane, 2014) with slight modification. Briefly, 20–30 seeds per line were placed in a 24-well plate followed by addition of 800- μL 50-mM EDTA, and 2-h incubation with gentle shaking at room temperature. Seeds were washed to remove the EDTA and 800 μL 0.01% w/v ruthenium red was added followed by 2-h incubation with gentle shaking. Seeds were washed with water (pH 7.5) and mounted on glass slides for imaging with a Leica DMIL LED compound microscope. Rosettes, roots, and trichomes were imaged with a Leica M165 FC stereomicroscope. Live imaging was performed on trichomes and developing seeds to detect EYFP expression using the stereomicroscope and Zeiss LSM 880 confocal microscope, respectively. Argon laser excitation of EYFP was at 488 nm (10% Intensity), and a 519–607-nm band pass filter captured emission (Gain = 700). To image EYFP-tagged proteins in primary roots, 4-day-old seedlings were stained with propidium iodide (10 μg mL−1) to visualize cell boundaries. Seedlings were rinsed with distilled water and mounted in water under a coverslip. Images were collected using Zeiss LSM-5 Pascal confocal microscope. The EYFP excitation was performed using a 488-nm Argon laser (30% intensity), collecting emission with a 510–540-nm band pass filter (Gain = 800). Propidium iodide fluorescence was detected using a 543 nm laser (10% intensity) for excitation and a 587–625-nm band pass filter (Gain = 800) for emission. Images were processed using Adobe Photoshop C4.
In vitro transcription and translation
The full-length PDF2 and pdf2ΔST cDNAs were transferred from pENTR/D-TOPO into destination vector pIX-HALO (CD3-1742, ABRC) using Gateway LR Clonase II Enzyme mix (ThermoFisher Scientific). The K107E and K107T mutations were generated using the Q5 Site-Directed Mutagenesis Kit (E0552S, New England Biolabs) with primers listed in Supplemental Table S3 in pENTR/D-TOPO followed by transfer to pIX-HALO. The Halo fusion proteins were produced using 1.2-µg plasmid DNA of the respective pIX-HALO construct in a volume of 15 µL using the TNT SP6 High-Yield Wheat Germ Protein Expression System (L3260, Promega, Madison, WI, USA). Protein expression was confirmed by Western blot with Anti-Halo Tag monoclonal antibody (1:2,000) (G9211, Promega).
EMSA
Cy3-labeled and unlabeled dsDNA probes were generated with oligonucleotides listed in Supplemental Table S3. Annealing was performed with 25-µM oligonucleotides in 100-mM Tris–Cl (pH 7.5), 1-M NaCl, and 10-mM EDTA at 95°C for 2 min, followed by 57°C for 5 min, 57–37°C transition over 90 min and 37°C for 2 min. EMSA reactions (20 µL) were prepared as previously described (Evens et al., 2017) with the following modifications: 6 µL of in vitro translated product was preincubated with binding buffer at 28°C for 10 min. Binding reactions were initiated by adding 200 nM of Cy3-labeled dsDNA probe, followed by a 20 min incubation. After electrophoresis at 4°C in a 0.6% w/v agarose gel (1X TBE, pH 8.3) at 150 V for 1 h, or a 7.5% w/v polyacrylamide gel (456–1025, Bio-Rad, Hercules, CA, USA) at 100 V for 4 h, protein–DNA complexes were analyzed with a Typhoon Trio Imager (GE Healthcare, Chicago, IL, USA) using the 580-nm emission filter for BP30Cy3, 600 PMT voltage, 532-nm green laser and high sensitivity.
ChIP-seq assay
Seeds from EYFP-tagged GL2, gl2ΔST, gl2ΔHD6;ΔZLZ, gl2Amo, and gl2R392M;E375G and gl2-5 lines were surface sterilized and grown on 0.8% w/v agar containing 1× MS (Murashige and Skoog, 1962) and 1% w/v sucrose. Seeds were stratified for 3–5 days at 4°C and then transferred to 23°C and continuous light for 10 days. Seedlings were harvested and crosslinked with 1% v/v formaldehyde for 10 min in a vacuum chamber. Glycine was added to a final concentration of 0.125 M, and the reaction was terminated by incubation for 5 min under vacuum. Chromatin was fragmented using a Covaris 220 focused ultrasonicator (Duty Cycle: 10%; intensity: 5; Cycles per Burst: 200) to produce <500-bp fragments. ChIP experiment was performed as previously described (Khosla et al., 2014). Immunoprecipitated DNA was sequenced using Illumina sequencing technology. ChIP DNA was processed, prepared with six-fold multiplexed Tru-Seq ChiP-seq libraries, and sequenced using short-read Illumina technology at the Kansas University Medical Center Genome Sequencing Facility. Libraries were sequenced (50 cycles) using the TruSeq Single Read Clustering Kit version 3 and TruSeq SBS-HS v3 sequencing chemistry with index read.
Bioinformatic analysis of ChIP-seq data
ChIP-sequencing was performed by Illumina HiSeq 2500 (Illumina, San Diego, CA, USA) at a 50 base single read resolution. Read quality was assessed using FastQC (Andrews, 2010). Aligned read count statistics were obtained using the “flagstat” utility of the SAMtools suite (Li et al., 2009). Sequences were mapped to the A. thaliana genome (TAIR10.24) using Bowtie2 (Langmead and Salzberg, 2012) (version 2.2.3; parameters: -q –phred33 –end-to-end –sensitive -p 16 -t). The mapping rates varied from a high of 97% to a low of 3%. Due to the low mapping rates of some replicates, the replicate aligned ChIP samples were merged (using “view” utility of the SAMtools suite; Li et al., 2009) for greater coverage. The overall number of mapped reads for the merged ChIP samples varied from 20 to 70 million reads per sample (40 million reads per sample on average) except for the negative control sample, gl2-5, which did not express EYFP-tagged protein, and had only 5 million mapped reads (see Supplemental Data Set 1). The merged ChIP samples were analyzed along with their corresponding input samples for significant peaks using MACS (Zhang et al., 2008) (version 1.4.2; parameters: -f BAM -g 0.135e9). Peaks calls passing a false discovery rate cutoff of 0.1 were considered for downstream analysis.
Target GO analysis
AgriGO (http://bioinfo.cau.edu.cn/agriGO/index.php) was employed to extract functional annotations for genes whose promoter or intergenic regions were bound by the TFs (Du et al., 2010). Singular enrichment analysis was applied to determine GO term enrichment in each sample relative to the A. thaliana TAIR9 reference. Analysis was performed using Fisher’s exact test (P < 0.01) with Bonferroni correction.
Y2H assay
For Figure 5A, Bait and Prey constructs were prepared by restriction enzyme cloning with EcoRI and BamHI inserts into the pGBKT7 and pGADT7 plasmids (Clontech, Mountain View, CA, USA). Bait constructs were transformed into the haploid yeast strain Y2HGold (MATa, trp1-901, leu2-3,112, ura3-52, his3-200, gal4Δ, gal80Δ, LYS2::GAL1UAS-Gal1TATA-His3, GAL2UAS-GAL2TATA-Ade2, URA3::MEL1UAS-Mel1TATA AUR1-C, MEL1), and prey constructs were transformed into Y187 (MATα ura3-52 his3-200 ade2-101 trp1-901 leu2-3,112 gal4Δ gal80Δ, met-URA3::GAL1UAS-Gal1TATA-LacZ, MEL1), using the LiAc method (Gietz and Woods, 2002), followed by selection on -Trp and -Leu, respectively. As a negative control, empty vectors were co-transformed with the desired constructs. Mating experiments were performed following Matchmaker Gold Y2H System (Clontech). Diploids were replica plated to media that selects for reporter gene activity of HIS3, AUR1-C, and MEL1. To test for autoactivation of bait plasmids Y2HGold/pDEST32 were plated in -Trp media and replica plated to media that selects for activity of the reporter genes HIS3 and AUR1-C, respectively. All pairwise combinations were investigated for protein-protein interaction by growing mated cultures to a density of optical density (OD)600 = 1.0. Four serial dilutions at 4x each were prepared from the normalized cultures and spotted onto permissive and selective media for detection of interaction using a 48-pin multiplex plating tool (“Frogger,” Dankar, Inc.). Plates were incubated at 30°C for 3–5 days, followed by imaging. Bait protein expression was confirmed by immunoblotting with Hela-c-Myc (9E10) polyclonal antibody (Gift from Stella Lee, 1:10 dilution).
For Figure 5, B and C and Supplemental Figure S6, C–E, full-length GL2, PDF2, ATML1 cDNAs and the various deletion and missense mutations were transferred from pENTR/D-TOPO to pDEST32 bait and pDEST22 prey vectors (Proquest Two-Hybrid System, Invitrogen) using Gateway LR Clonase II Enzyme mix (ThermoFisher Scientific). For Figure 5B, both bait and prey constructs were transformed into Y2HGold, and interaction was assayed in this haploid strain. For Figure 5, B and C and Supplemental Figure S6, C–E, Y2HGold and the opposite mating type, Y187, were transformed with pDEST32 and pDEST22 constructs, using LiAc/PEG transformation, and plated on -Leu and -Trp media to select for the bait and prey plasmids, respectively. Mating experiments and selection for diploids were performed as described above. To test for autoactivation of bait plasmids Y2HGold/pDEST32 were plated in -Leu media and replica plated to media that selects for activity of the reporter genes HIS3 and AUR1-C respectively. Y2H assays for imaging were performed as described above. For analyzing bait protein expression, single colonies expressing pDEST32 constructs were grown at 30°C to saturation in media lacking leucine. Protein was extracted using the sodium hydroxide/trichloroacetic acid method and expression was monitored by Western blot using a 1:500 dilution of GAL4 [DBD] (RK5C1) mouse monoclonal antibody (sc-510 HRP, Santa Cruz Biotechnology, Dallas, TX, USA).
Y2H quantitative β-galactosidase assay
For Supplemental Figure S6, A and B, Gateway cloning was used to transfer genes for HD-Zip III and IV TFs from pENTR223.1 clones to pDEST22 and pDEST32 destination vectors (ProQuest Two-Hybrid System, Invitrogen). The bait and prey plasmids were transformed into yeast strain MaV203 (Invitrogen): MATα, leu2-3,112, trp1-901, his3Δ200, ade2-101, cyh2R, can1R, gal4Δ, gal80Δ, GAL1::lacZ, HIS3UAS GAL1::HIS3@LYS2, SPAL10::URA3 following the Clontech Yeast Protocol Handbook. Four independent colonies from each transformation were selected to perform quantitative β-galactosidase liquid assay as previously described (Schrick et al., 2014).
BiFC in N. benthamiana
To generate constructs for split GFP, Gateway compatible BiFC vectors 125-NXGW and 127-CXGW for N-terminal fusions of split GFP (a gift from Adrienne Roeder) were used for LR recombination reactions of the corresponding wild-type and mutant genes in pENTR/D-TOPO plasmids. Agroinoculation of N. benthamiana was performed as described in Sparkes et al. (2006). Seeds of N. benthamiana were germinated on wet filter paper for 7 days, and then the seedlings were planted on above described soil mix. For transient expression of fluorescent protein, leaves of 3- to 4-week-old plants grown at 22°C under 12-h light/12-h dark were used for agroinfiltration. Plasmids were electroporated into A. tumefaciens GV3101 and transformants were selected on agar plates containing the required antibiotics. Transformation of N. benthamiana leaves was performed with Agrobacterium taken directly from agar plates. Cultures were re-suspended in 10-mM MgCl2, 10-mM 2-(N-morpholino)ethanesulfonic acid pH 5.6, and 150-μM acetosyringone to an optical density of 0.2, and incubated for 2–4 h at 28°C in the dark. BiFC constructs were co-expressed with the p19 suppressor of gene silencing, derived from tomato bushy stunt virus (Voinnet et al., 2003). Leaves were infiltrated with a needle-less syringe into the abaxial side of leaves of 3- to 4-week-old N. benthamiana plants and examined after 2–3 days by confocal laser scanning microscopy using a Zeiss LSM-5 Pascal microscope with an 488-nm Argon laser for excitation (30% intensity), collecting emission with a 510–540 nm band pass filter (Gain = 800). Mean pixel intensities of three different circular regions of interest (ROIs) were quantified from 10 images for each construct combination using Image J software.
In vivo protein stability assay
Wild-type and mutant seedlings were germinated on 0.5× MS media (Murashige and Skoog, 1962) for 4 days. On Day 5, 30 seedlings of each genotype were transferred to 24-well plates containing 1 mL 0.5 × MS supplemented with 50-µM MG132 (474787, Sigma-Aldrich). Plates were sealed with surgical tape and placed on a shaker at 55 rpm for 16 h at 23°C under continuous light. After 16 h, the seedlings were washed 3× with 1-mL fresh media. Cycloheximide (C1988, Sigma-Aldrich) (400 µM in DMSO) or DMSO control was added at 0 h, and harvesting occurred at 0, 2, 4, 5, 8, 10, or 24 h, respectively. Samples of 30 seedlings were flash frozen in liquid nitrogen and stored at −80°C prior to protein extraction. Tissue was homogenized in liquid nitrogen, and hot sodium dodecyl sulfate (SDS) buffer (8-M urea, 2% SDS, 0.1-M DTT, 20% glycerol, 0.1-M Tris–Cl pH 6.8, 0.004% bromophenol blue) was added prior to SDS–polyacrylamide gel electrophoresis (PAGE) and Western blotting. Anti-GFP (1:2,000; 11814460001, Roche, Basel, Switzerland) served as the primary Ab, followed by secondary Goat Anti-Mouse IgG [HRP] (1:3,000; A00160, Genscript). Proteins were detected with SuperSignal West Femto Maximum Sensitivity Substrate (ThermoFisher Scientific) using an Azure 300 chemiluminescence imager (Azure Biosystems, Dublin, CA, USA), and blots were stained with Bio-Safe Coomassie Blue G-250 (Bio-Rad) for loading controls. Band intensities were quantified with ImageJ software.
Statistical analysis
Trichome differentiation on the first leaves
Total number of trichomes and branching patterns were counted on the first leaves of 20 individual plants per genotype. Values are represented as mean ± sd. Ordinary one-way analysis of variance (ANOVA) using Tukey’s multiple comparisons test was applied to establish significant differences between genotypes (P < 0.05).
Root hairs of young seedlings
Numbers of root hairs on primary roots of 4- to 5-day-old seedlings were counted for 6–10 individuals per genotype. Values are represented as mean ± sd. Ordinary one-way ANOVA using Tukey’s multiple comparisons test was applied to establish significant differences (P < 0.05).
BiFC assays
For each combination of paired constructs, 10 images were analyzed. Mean pixel intensity of three different circular ROIs was quantified by the ImageJ software. An unpaired t test was used to indicate statistically significant differences between wild-type and mutant using data from two independent experiments in two trials (P < 0.0001).
Protein stability assays
The graphed data represent two or three independent cycloheximide experiments with whole seedlings. Numerical values for protein levels were obtained from band intensity quantification and were normalized to the mock 10 (M10) and mock 24 (M24) samples depending on experiment, which were designated a value of 1.0. Protein half-life was determined by the protein level at a Y-axis value of 0.5. Values are represented as mean ± sd. *P < 0.05 indicates statistical significance using a two-tailed t test with unequal variance.
Accession numbers
The ChIP-seq data are deposited in the NCBI Gene Expression Omnibus (Edgar et al., 2002) under the GEO Series accession number GSE186860 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSExxx). The A. thaliana genes described in this study are as follows: GL2 (At1g79840); PDF2 (At4g04890); ATML1 (At4g21750); ANL2 (At4g00730); FWA/HDG6 (At4g25530); HDG1 (At3g61150); HDG2 (At1g05230); HDG3 (At2g32370); HDG4 (At4g17710); HDG5 (At5g46880); HDG7 (At5g52170); HDG8 (At3g03260); HDG9 (At5g17320); HDG10 (At1g34650); HDG11 (At1g73360); HDG12 (At1g17920).
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Structural alignments of HD and START domain from HD-Zip IV TFs (related to Figures 1, A and 3, A).
Supplemental Figure S2. Root hair phenotypes and nuclear localization in various START domain missense mutants (related to Figure 3).
Supplemental Figure S3. Characterization of START domain missense mutants in the N-terminus of the START domain (related to Figure 3).
Supplemental Figure S4. START domain is required for in vitro DNA binding as a dimer (related to Figure 4C).
Supplemental Figure S5. GL2 binding motif and ChIP-seq peak distance from TSS (related to Figure 4, D–G).
Supplemental Figure S6. The START domain is required for GL2 homodimerization (related to Figure 5C).
Supplemental Figure S7. Protein expression of Bait plasmids in Y2H experiments (related to Figure 5).
Supplemental Figure S8. BiFC negative assays (related to Figure 5, D–F).
Supplemental Figure S9. Cycloheximide chase experiments reveal that the START domain is critical for protein stability of GL2 (related to Figure 6).
Supplemental Figure S10. Cycloheximide chase experiments with double mutants affecting both HD and START domain of GL2 (related to Figure 6; Supplemental Figure S9).
Supplemental Table S1. EYFP expression is independent of phenotypic rescue (related to Figures 1 and 2).
Supplemental Table S2. Distribution and number of significant peaks from ChIP-seq data (related to Figure 4F).
Supplemental Table S3. Oligonucleotides used in this study.
Supplemental Data Set 1. Mapping statistics of sequencing reads from ChIP-seq data.
Supplemental Data Set 2. Functional descriptions of GL2 targets from ChIP-seq data.
Acknowledgments
We thank Joel Sannemann for help with confocal microscopy and Timothy Durrett for valuable scientific input.
Funding
This research was funded by the National Science Foundation (MCB1616818), National Institute of General Medical Sciences of the National Institutes of Health under Award no. P20GM103418 (K-INBRE) and P2-RR-17686 (Confocal Microscopy Core, Kansas State University), USDA National Institute of Food and Agriculture Hatch/Multi-State project 1013013, and Johnson Cancer Research Center at Kansas State University. This is contribution no. 21-279-J from the Kansas Agricultural Experiment Station.
Conflict of interest statement. None declared.
T.M., B.S., A.K., and K.S. designed the research, analyzed data, and wrote the paper. T.M., B.S., A.K., E.M.P., and P.M.S. performed experiments and analyzed data. T.M., R.L.R., A.L.W., E.M.P., A.K., and K.A.T. generated transgenic lines. S.G. performed ChIP-seq informatic analyses. K.S. oversaw the study.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://dbpia.nl.go.kr/plphys/pages/general-instructions) is: Kathrin Schrick ([email protected]).
References
Author notes
These authors contributed equally (T.M., B.S., and A.K.).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}