





Abstract
Parasitism is a successful life strategy that has evolved independently in several families of vascular plants. The genera Cuscuta and Orobanche represent examples of the two profoundly different groups of parasites: one parasitizing host shoots and the other infecting host roots. In this study, we sequenced and described the overall repertoire of small RNAs from Cuscuta campestris and Orobanche aegyptiaca. We showed that C. campestris contains a number of novel microRNAs (miRNAs) in addition to a conspicuous retention of miRNAs that are typically lacking in other Solanales, while several typically conserved miRNAs seem to have become obsolete in the parasite. One new miRNA appears to be derived from a horizontal gene transfer event. The exploratory analysis of the miRNA population (exploratory due to the absence of a full genomic sequence for reference) from the root parasitic O. aegyptiaca also revealed a loss of a number of miRNAs compared to photosynthetic species from the same order. In summary, our study shows partly similar evolutionary signatures in the RNA silencing machinery in both parasites. Our data bear proof for the dynamism of this regulatory mechanism in parasitic plants.
Introduction
Haustorial parasites such as the dodders or broomrapes infect other photosynthetic hosts by way of a specialized infection organ that bears functional similarity to roots (Yoshida et al., 2016). Dodders are vine-like, rootless parasites that belong to the genus Cuscuta in the Convolvulaceae family, which is part of the order Solanales. Broomrapes (genus Orobanche, family Orobanchaceae) have arisen in the order Lamiales. Both genera have evolved their parasitic lifestyles independently (Nickrent, 2020). While Orobanche infects the roots of their hosts, Cuscuta is a shoot parasite that only grows on above-ground host plant parts. The organ, which both dodders and broomrapes have in common, is their “haustorium” that enables them to withdraw water, minerals, photosynthates, and other compounds from their hosts. Cuscuta vines and Orobanche shoots are both strong sinks for photosynthates and cause substantial losses in agricultural production by weakening their hosts. This is aggravated by the fact that many species of both genera, including Cuscuta campestris and Orobanche aegyptiaca can infect hosts of a wide range of families.
The recent publication of the genomic sequences from both C. campestris and Cuscuta australis represented a step forward in understanding how a parasitic lifestyle affects species at the genomic level (Sun et al., 2018; Vogel et al., 2018). These studies revealed that during the evolution to parasitism both genomes experienced gene losses associated with leaf and root development, nutrient uptake, photosynthesis, flowering, or defense (Sun et al., 2018; Vogel et al., 2018). Although this information is crucial to understanding plant–plant parasitism, it is equally paramount to comprehend the transcriptional and posttranscriptional regulation of the information contained within these genomes by mechanisms like RNA silencing (Almeida and Allshire, 2005).
RNA silencing (also known as RNA interference) is a conserved RNA regulatory mechanism that controls RNA expression both at transcriptional and posttranscriptional levels (Baulcombe, 2004; Eamens et al., 2008; Borges and Martienssen, 2015). Its effector molecules, small RNAs (sRNAs), which are typically between 20 and 25 nucleotides (nts) in length, guide the sequence specificity of the regulation through sequence complementarity with their target RNAs (Borges and Martienssen, 2015). There are several types of sRNAs depending on their biogenesis and role. In plants, the main sRNA classes are microRNAs (miRNAs) and small interfering RNAs (siRNAs) (Axtell, 2013; Borges and Martienssen, 2015). Both sRNAs have similar pathways that depend on double-stranded RNA (dsRNA) processing by DICER-LIKE (DCL) proteins and loading of the resulting sRNAs into ARGONAUTE (AGO) proteins, but differ with regard to the factors involved. For example, miRNAs are excised from single-stranded RNAs with high degree of self-complementarity that are processed by DCL1 and loaded into AGO1. siRNAs can be derived from heterochromatic regions (termed heterochromatic siRNAs or hc-siRNAs) or from amplification of RNA templates targeted by miRNA or siRNA (termed secondary siRNAs). hc-siRNAs are a product of the RNA-DEPENDENT RNA POLYMERASE 2 (RDR2) amplification of RNA POLYMERASE IV ssRNAs, and their processing by DCL3 and loading into AGO4, 6, or 9. On the other hand, secondary sRNAs are generated from RDR6 amplification of miRNA- or siRNA-targeted mRNAs that are processed by DCL4 or 2 and loaded into AGO1. AGO loading determines in great measure the sRNA activity and while AGO1, 2, 5, 7, and 10 induce the cleavage of their target mRNAs, AGO4, 6, and 9 direct DNA methylation (Axtell, 2013; Borges and Martienssen, 2015). sRNAs produced from these pathways control multiple processes like the stress response (Sunkar et al., 2012), developmental transitions (Li and Zhang, 2016; Martinez and Kohler, 2017), or the epigenetic regulation of the genome (Matzke and Mosher, 2014).
The availability of a complete genome sequence is necessary for the detailed and proper characterization of sRNA populations, but also sRNA data together with de novo transcriptomes can, in the absence of genome data, already reveal some important sRNA features. Much focus has been put on plants of high economic interest and especially on the characterization of their miRNA populations (Reinhart et al., 2002; Sunkar et al., 2005; Moxon et al., 2008; Zhang et al., 2009; Martinez et al., 2011). Cumulative data from related species have furthermore allowed the collection of hints on the evolution of very conserved miRNAs (Baldrich et al., 2018). For example, in grasses miRNAs are retained in connection with their target gene and their implication in stress response (Abrouk et al., 2012), and their domestication has been associated with the reduction of a miRNA-mediated RNA silencing cascade (Swetha et al., 2018). Data from the comparison between the miRNA populations from Arabidopsis (Arabidopsis thaliana) and Arabidopsis lyrata indicated that poorly conserved miRNAs are often lost or evolve rapidly compared to highly conserved miRNAs (Fahlgren et al., 2010; Ma et al., 2010). Together with the divergence of miRNA sequences per se, some miRNAs are conserved but diverge on the mRNAs that they target, a process termed neofunctionalization and exemplified by the diversity of targets of the miR482/miR2118 superfamily (Baldrich et al., 2018). Despite detailed insight into the overall characteristics of sRNAs of plants in general, little is known on parasitic plants with their substantially different lifestyles. Analysis of miRNA conservation in parasitic organisms is a fruitful aspect in the quest to understand miRNA evolution and function. The analysis of the miRNA repertoire in flatworms, parasitic cestodes, and parasitic nematodes indicated that these organisms suffered a loss of conserved miRNAs and a gain of new miRNAs (Wang et al., 2011; Winter et al., 2012; Fromm et al., 2013; Macchiaroli et al., 2015; Gu et al., 2017). Interestingly, RNAi plays an important role in the parasitic relation with the host and a common theme between parasites is the use of sRNAs to subvert their hosts to their own benefit (Hakimi and Cannella, 2011; Zheng et al., 2013; Martinez and Krause, 2018; Shahid et al., 2018). Whether the trend of loss and gain of miRNAs in parasitic organisms is also true for parasitic plants is currently unknown.
To shed light onto the characteristics of the sRNA repertoire of parasitic plants, we performed an in-depth investigation of the miRNA populations of the shoot-parasitic C. campestris, for which a reference genome is available. Since miRNAs from the host were identified in Cuscuta in other studies (Shahid et al., 2018; Johnson et al., 2019), we have avoided host contact to the best degree possible by using germinated seedlings, noninfecting stems, and artificially induced haustoria that have had no contact with a host. To investigate if some trends observed in Cuscuta may signify signatures of a parasitic lifestyle, we compared its miRNA population to that of the root-parasitic O. aegyptiaca. Our analyses revealed an apparent array of losses, retentions, and novel acquisition of miRNAs in the parasites compared to their photosynthetic relatives. The evolutionary pathways that yield new miRNAs and their targets in the C. campestris genome seem to include development from noncoding RNAs and acquisition by horizontal gene transfer (HGT). Overall, our study reveals that on top of the earlier described trans-acting Cuscuta miRNAs that exert control over the parasite’s hosts (Shahid et al., 2018; Johnson and Axtell, 2019), sRNAs can also be attributed important contributions to the parasite’s ontogenesis and phylogenesis.
Results
The 24-nt sRNAs are dominant in noninfective tissues but not in haustorial tissues of C. campestris
To gain insight into the sRNA characteristics and their activity in C. campestris, we generated sRNA libraries from tissue pre- and postexposure to a host plant but without direct contact to the host. These included (1) germinating seedlings (Figure 1A), (2) apical regions from shoots that were in search of a new host (20–80 cm distant to closest infection site to minimize the proportion of host-derived miRNAs) (Figure 1B), and (3) early and late stages of artificially induced host-independent haustoria (Figure 1C) (Olsen et al., 2016). For the haustorial tissues of C. campestris, degradome libraries were prepared in addition (Supplemental Table S1). For the purpose of comparison with a root parasite, we generated, sequenced, and analyzed sRNA libraries from the tubercle and the host/parasite interface of the root parasite O. aegyptiaca (Figure 1D). Overall, our C. campestris libraries produced more than 28 million filtered and mapped reads (>70 million raw reads) that represent a diverse set of sRNAs, while the O. aegyptiaca tissues yielded over 8 million filtered and mapped reads (>47 million raw reads) (Supplemental Table S1). Principal component analysis (PCA) of global sRNAs in our C. campestris libraries demonstrated that biological replicates and tissues of similar origin clustered together (Figure 1E), with surprisingly little differences between the different haustorial stages. To explore the characteristics of the sRNA population of C. campestris within the context of the Solanales order, we retrieved sRNA datasets for different members of the nightshade family (Solanaceae, including eggplant [Solanum melongena], tomato [Solanum lycopersicum], potato [Solanum tuberosum], currant tomato [Solanum pimpenillifolium], tobacco [Nicotiana tabacum], Nicotiana benthamiana, pepper [Capsicum annum], petunia [Petunia x hybrida], and sweet potato [Ipomea batatas]) from public databases (Supplemental Table S2). A general overview of the global sRNA profile from the autotrophic plant species compared to the heterotrophic parasite revealed that in C. campestris the contribution of 24-nt sRNAs to the global sRNA profile is most prominent, and the presence of 21-nt sRNAs is reduced (Figure 1F;Supplemental Figure S1), similar to what was found in many other Solanales with the exception of S. melongena, S. lycopersicum, and N. tabacum (Figure 1G;Supplemental Figure S1). In the root parasitic O. aegyptiaca, on the other hand, 24-nt sRNAs did not represent the majority of the sRNAs, while sRNA datasets that were retrieved for a photosynthetic member of the same order (Lamiales) as O. aegyptiaca, Erythranthe guttata (Supplemental Table S2) showed a strong dominance of this size (Figure 1G). We detected a considerable variation in the sRNA size ratios in our C. campestris sRNA libraries depending on the tissue or origin, with haustorial tissues producing a higher percentage of 21- and 22-nt sRNAs than seedling and stem tissues, which produced a higher amount of 24-nt sRNAs (Figure 1H), which agrees with the data previously published (Johnson et al., 2019). Both the tubercles of O. aegyptiaca and the interface have considerable amounts of 22-nt sRNA, which seem to play a more dominant role than in any other species in our set (Figure 1H). The 22-nt sRNAs do not typically play a role in root tissues (Herranz et al., 2015), and in some species are produced from clusters in response to viral infection (Baldrich et al., 2022). We could not detect the presence of viral-derived sRNAs in our Orobanche libraries compared to viral-infected samples (Annacondia and Martinez, 2021) (Supplemental Figure S2), indicating that these populations of 22-nt sRNAs were not produced in response to viral infection. It is plausible that this unusual size distribution could be connected to the root-parasitic lifestyle.
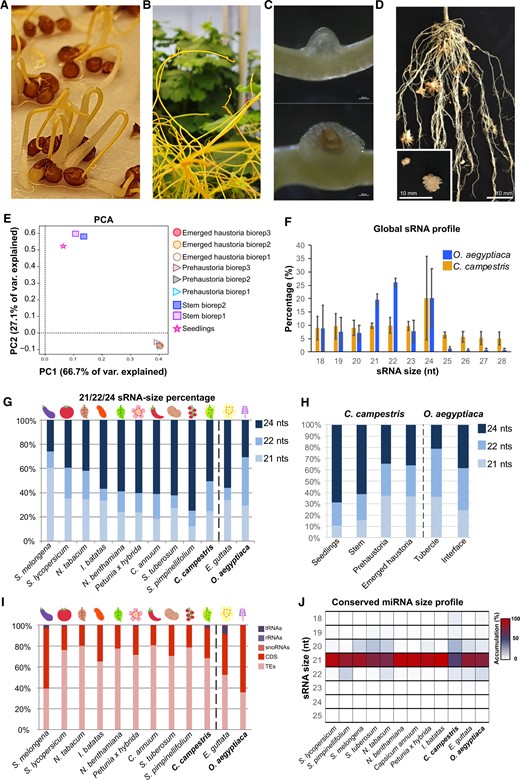
Overview of the C. campestris sRNAs. A–D, Representative pictures of the tissues analyzed in this work. A, Germinating seedlings. B, Apical regions from shoots that were in search of a new host (20–80 cm distant to closest infection site). C, (Top) Early (prehaustorium) and (bottom) late (emerged) stages of artificially induced host-independent haustoria. D, Tubercle and the host/parasite interface of the root parasite O. aegyptiaca. E, PCA of the sRNA libraries produced from different C. campestris tissues. F, sRNA profile for total sRNAs from an average of all the libraries analyzed. G, Comparison of abundance of 21-, 22-, and 24-nt total sRNAs in different species of the Solanales order. Values shown are the average between multiple sRNA libraries (two bioreplicates for Orobanche and nine for Cuscuta) from different tissue origins with error bars representing the standard deviation between bioreplicates. H, Abundance of 21-, 22-, and 24-nt total sRNAs in different C. campestris and O. aegyptiaca tissues. I, Comparison of the categorization of total sRNAs in different species of the Solanales order. Categories were defined as tRNAs, ribosomal RNA, small nucleolar RNAs, CDSs, and TEs. J, Heatmap of miRNA size and accumulation for conserved miRNAs in different species of the Solanales order. For comparison, E. guttata and O. aegyptiaca are included in the plots shown in (G), (I), and (J).
In plants, 24-nt sRNAs are mainly produced by RNA Polymerase IV and are involved in the control of transposable element (TE) expression through the RNA-directed DNA methylation (RdDM) pathway (Axtell, 2013). As expected, the high presence of 24-nt sRNAs in C. campestris matched a high percentage of sRNAs that mapped to TEs (77.1%; Figure 1I). In contrast, coding sequence (CDS)-derived sRNAs represented only 26.3% and sRNAs derived from functional noncoding RNAs represented a mere 2%, with transfer RNA (tRNA)-derived sRNAs being the most represented in this category (1.8%).
Subsequently, the miRNA population was compared with other species in the order Solanales using sequenced reference species where sRNA libraries were available from previous studies (Supplemental Table S3). The comparison of conserved miRNAs sequences between all these species indicated that, similar to the other Solanales members, a majority of miRNAs found in C. campestris were of 21 nt in size, followed by miRNAs of 20 nt (Figure 1J). In O. aegyptiaca also the 21-nt miRNAs were predominant, but unlike in C. campestris, the 22-nt miRNAs accounted for a larger proportion than the 20-nt miRNAs.
Overall, the sRNA analyses of both root and shoot parasites point toward an importance of 24-nt sRNAs that, at least in C. campestris, are TE derived. The sRNA populations, however, appear to display dynamic changes during haustoriogenesis.
The miRNA repertoire of C. campestris lacks some miRNA families while others are conserved
MiRNAs are important regulators of biological processes in eukaryotes. In plants, miRNAs regulate important developmental processes including leaf and flower development (Nag and Jack, 2010; Yang et al., 2018), juvenile to adult transition (Guo et al., 2017), or meristem identity (D'Ario et al., 2017). To better understand the miRNA population in the context of C. campestris parasitization, we performed a categorization of the conserved miRNA families found in the different Solanales members (see Figure 1J). The results indicated that a core group of 24 miRNAs was shared between all of them (Figure 2A). Thirty-five miRNA families that were found in C. campestris were common to seven or more photosynthetic Solanales species. Among these are highly conserved plant miRNAs like miR156, miR172, or miR390 (Figure 2B). The overall number of miRNA families in C. campestris was 130, which is lower than the number of conserved miRNA families found in the cultivated representatives of most Solanales families with the exception of I. batatas (71) from the Convolvulaceae (Figure 2B). Interestingly, C. campestris appeared to lack several miRNAs that are involved in the development and resistance at different levels. Notably, miR482, a miRNA family that targets NBS–LRR genes to produce phased siRNAs (de Vries et al., 2015) and that is highly conserved in Solanales, was not found in C. campestris. The same applied to the following miRNA families: miR394, a contributor to the regulation of leaf morphology through the targeting of a F-box gene (Song et al., 2012), miR395 that regulates sulfate concentration by targeting the ATP sulfurylase family in Arabidopsis (Matthewman et al., 2012; Ai et al., 2016), miR162 and miR403 which target the RNA silencing members DCL1 and AGO2/3, respectively (Xie et al., 2003; Allen et al., 2005), and miR477 and miR827 that are involved in the control of immunity mediating the targeting of a phenylalanine ammonia-lyase and a ubiquitin E3 ligase gene, respectively (Hewezi et al., 2016; Wang et al., 2020) (Figure 2C;Supplemental Table S4). Of these, miR394, miR477, and miR827 are also missing in O. aegyptiaca (Figure 2C;Supplemental Table S5). Nevertheless, only miR394 might have been lost in Orobanche, since it was found in E. guttata, a photosynthetic member of the Lamiales, which was the only species within Lamiales where sRNA datasets could be retrieved. In contrast, miR482, miR395, miR162, and miR403 were found to be present in the root parasite.
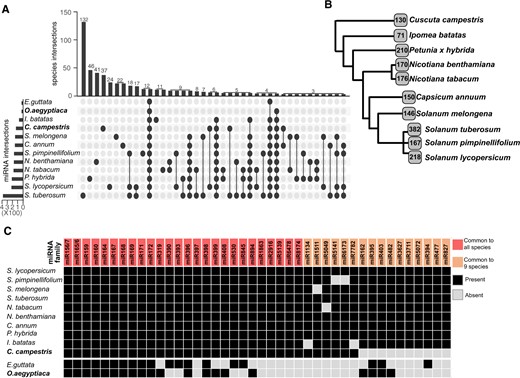
Overview of the C. campestris conserved miRNAs. A, Upset plot showing the conservation of miRNA families between different species of the Solanales order. B, Simplified phylogenetic tree showing the number of conserved miRNA families identified in this study. C, Comparison of highly conserved miRNAs in members of the Solanales order, O. aegyptiaca and E. guttata.
MiRNAs miR156/7 and miR165/6 that are highly conserved in the Solanales had the largest number of coding loci in the C. campestris genome as well as in phylogenetically more distant species like Arabidopsis, rice (Oryza sativa), or maize (Zea mays) (Supplemental Figure S3, A and B; Supplemental Table S4). This was in line with earlier reports that linked the conservation and size of the miRNA families (Fahlgren et al., 2010; Ma et al., 2010). Interestingly, other typical large and conserved miRNA families like miR169 or miR399, which are involved in regulation of stress-induced flowering (Kim et al., 2011; Xu et al., 2013), had a reduced number of members in C. campestris (Supplemental Figure S3A and S3B and Supplemental Table S4).
Briefly, our results indicated that parasitic plants seem to have retained miRNA families important for the regulation of developmental processes and lost miRNA families that control the defense responses and organ development, which could be an indication that their lifestyle has made these obsolete.
Expression of conserved miRNAs in C. campestris does not reveal pronounced organ-specific signatures
Next, we made use of our sRNA libraries generated from different tissues to explore the organ-specific expression of conserved and newly identified miRNAs. The origin of our libraries allowed us to explore the differences in miRNA accumulation in haustorial tissue in comparison to noninfective tissues but also a tentative investigation of the effect of host contact on miRNA profiles by including tissue from germinating seedlings that lacked any exposure to a host (Figure 1A). The accumulation pattern of conserved miRNAs showed some differences between C. campestris tissues, but also revealed that more than half of the miRNAs were not differentially regulated (Figure 3A). A PCA of conserved miRNAs in our libraries demonstrated that only the seedling sample differed from the rest of the biological replicates and tissues that clustered fairly closely together (Supplemental Figure S4). Nevertheless, miRNA size profiles in the different samples (Figure 3B) indicated that 20-nt miRNA sequences were mainly found in stem tissues, while both germinated and haustorial tissues produced miRNAs of mainly 21 nt in size (Figure 3B;Supplemental Figures S5 and S6). This result partially explained the relatively high presence of 20-nt miRNA sequences in C. campestris’ conserved miRNA profile (Figure 1J) which corresponded with the preferential accumulation of miR156 (which is 20 nt in length) in stem tissues (Figure 3A;Supplemental Figure S5; Supplemental Table S6). The O. aegyptiaca samples also revealed a dominance of 21-nt miRNA sequences in both haustorial tissues analyzed and showed an identical accumulation pattern for all identified miRNAs (Figure 3, A and B).
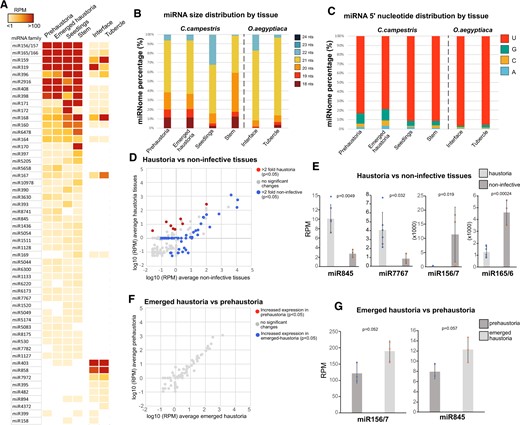
Tissue-specific expression of C. campestris conserved miRNAs. A, Accumulation of conserved miRNAs in different tissues. Values are shown as a heatmap of RPM values for each miRNA family. B, miRNA size distribution in the different tissues per species analyzed here. C, 5′-nt presence in miRNAs with different tissues of origin. D, Comparison of the accumulation values of conserved C. campestris miRNAs between noninfective tissues (germinated and stem) and haustorial tissues (emerged haustoria and prehaustoria). Only miRNAs with significant differences (P < 0.05 calculated through an unpaired t test) and 2-fold up/downregulation are highlighted. E, Accumulation values for selected miRNAs with noninfective or haustoria-specific accumulation patterns. Error bars show the standard deviation of the RPM values for each sample. Dots indicate the individual values from each sample included in the tissue analysis. P-values were calculated using an unpaired t test. F, Comparison of the accumulation values of conserved miRNAs between emerged C. campestris haustoria and prehaustoria. G, Accumulation values for selected miRNAs with emerged haustoria-specific accumulation patterns. Error bars show the standard deviation of the RPM values for each sample. Dots indicate the individual values from each sample included in the tissue analysis. P-values were calculated using an unpaired t test.
The 5′-nt of miRNAs determines which AGO protein is loaded with the miRNA (Mi et al., 2008), so we explored the prevalent 5′-nt in miRNAs of the different tissues to infer potential differential AGO tissue-specific activity. Although U was found to be the prevalent 5′-nt in all samples of both, C. campestris and O. aegyptiaca miRNAs (Figure 3C), both haustorial tissues of C. campestris showed a noticeable increase in the amount of 5′-terminal Cs, which points to a loading in potential AGO5 homologs (Figure 3C). Interestingly, analysis of RNA sequencing data (Bawin et al., 2022) indicated that prehaustorial and emerged haustorial tissues showed increased expression of two AGO5 homologs present in the C. campestris genome (Supplemental Figure S7).
The comparison of libraries of different origins indicated that the main differences were found between haustorial and noninfective tissues, which contained 66 miRNA families with significant accumulation changes (more or equal to two-fold expression change and P < 0.05, Figure 3D). From these 17 miRNA families, 8 were exclusively present in haustoria or upregulated more than two-fold in haustorial tissues, while 58 were not detected or downregulated more than two-fold in haustorial sRNA libraries (Figure 3D). miRNA families that were upregulated in haustorial tissues compared to noninfective stems and seedlings included miR845 and miR7767 (Figure 3E) but also miR5083, miR8741, miR5044, or miR5658 (Supplemental Table S6). On the other hand, miRNAs repressed in haustoria included families like miR156/7 and miR165/6 (Figure 3E) but also miR159, miR171, miR390, mi396, or miR397 (Supplemental Table S6). In contrast, our analysis of differential miRNA accumulation between artificially induced prehaustoria and emerged haustoria revealed only minor differences in the accumulation of conserved miRNA families (Figure 3, F and G). Only two miRNA families, miR845 and miR156/7, showed nearly significant upregulation in the later haustorial stage compared to the earlier one (Figure 3G).
In summary, our analysis revealed that the conserved miRNAs in C. campestris showed characteristics that indicate potential tissue-specific biogenesis effects. Not surprisingly, differences in miRNA accumulation were higher between haustorial versus noninfective tissues than among the two haustorial stages.
New miRNA families in both parasites show tissue-specific expression patterns
Beside widely conserved miRNAs, species-specific miRNAs are often found that can serve new functions (Axtell, 2013). In C. campestris, new miRNAs are an important part of the host colonization process, and have been shown to induce the production of secondary sRNAs in the host (Shahid et al., 2018). An exploration of our sRNA libraries for the presence of new miRNAs in haustorial and noninfective tissues revealed the presence of 30 predicted novel miRNAs (Figure 4A;Supplemental Figure S8; Supplemental Table S7), four of which were previously identified as haustorially induced sRNAs (Johnson et al., 2019). In contrast, the O. aegyptiaca dataset revealed a total of only 12 new miRNAs present in both interface and tubercle samples (Figure 4D;Supplemental Table S8). Similar to the conserved miRNAs, the novel miRNAs in C. campestris showed differential accumulation patterns, with some miRNAs appearing to accumulate predominantly in noninfective stems (miR17), seedlings (miR9 and miR10), or both (e.g. miR20 and miR23) and other miRNAs showing a preferential accumulation in haustorial tissues (miR2, miR6, and miR4). Interestingly, one of these novel miRNAs, miR15, has nine members potentially revealing the start of the formation of a new miRNA family within the C. campestris genome (Figure 4A). New miRNAs in O. aegyptiaca also showed tissue-specific accumulation in interface (miR1, miR6, miR7, miR8, miR9, and miR10) or tubercle (miR2, miR3, miR4, miR5, miR11, and miR12) tissues. The analysis of total sequences from new miRNAs showed that >40% of novel C. campestris miRNAs exhibit a 5′-terminal A, indicating a potential loading in AGO2, followed by C (AGO5) (Figure 4B). The presence of a terminal U (loading into AGO1) was found to be higher in unique (nonredundant) sequences compared to the total sequence set, independent of the nature of the sampled tissues (Figure 4B). Additionally, new miRNAs in C. campestris were of mostly 21 nt in length (Figure 4C). The same reduced amount of 5′-terminal Us (relative to conserved miRNAs) was also observed in O. aegyptiaca, where 5′-terminal Cs even exceeded Us at the interface, but not in the tubercles (Figure 4E). Interestingly, this difference coincided with differences in the sizes of the novel miRNAs, with 22 nt being predominant at the interface and 21 nt in the tubercle (Figure 4F).

Overview of Cuscuta and Orobanche new miRNAs. A, Accumulation of new C. campestris miRNAs in different tissues. Values are shown as a heatmap of RPM values for each miRNA family. B, 5′-nt presence in new miRNAs within different tissues of C. campestris. C, Size distribution of new miRNAs in the analyzed C. campestris tissues. D, Accumulation of new O. aegyptiaca miRNAs in different tissues. Values are shown as a heatmap of RPM values for each miRNA family. E, 5′-nt presence in new miRNAs within different tissues of origin in O. aegyptiaca. F, Size distribution of new miRNAs in the analyzed O. aegyptiaca tissues.
All these characteristics indicated that new miRNAs are most likely bona fide novel miRNAs that may have arisen in the C. campestris and O. aegyptiaca genomes to accomplish novel regulatory functions.
Degradome sequencing identifies potential targets of miRNAs in C. campestris haustoria
Following the analysis of miRNA sequence conservation and identification of novel miRNAs we explored the regulatory potential of those populations. We initially made a bioinformatic prediction of the genes potentially targeted by conserved and new miRNAs (Supplemental Table S9). To confirm these predictions and corroborate the regulatory potential of C. campestris miRNAs, a degradome sequencing approach was employed in which we produced PARE/degradome libraries from haustorial tissues (Supplemental Table S1). In plants, miRNAs can induce the cleavage or translational repression of their target sequences (Axtell, 2013). miRNA cleavage activity leaves a “fingerprint” cleavage event between the nts 10 and 11 of their region of homology with their target RNA that is used to identify miRNA-targeted RNAs (Addo-Quaye et al., 2008; German et al., 2008; Gregory et al., 2008). Our degradome libraries showed differential clustering according to their tissue of origin (Supplemental Figure S9A). The analysis of high-stringent miRNA-cleavage events allowed us to identify 642 miRNA targets, 187 of which correspond to genes (Supplemental Figure S9B; Supplemental Table S10). Importantly, the alignment of PARE reads to the confirmed miRNA/mRNA degradation events indicated that most of the miRNAs in C. campestris might exert their function through cleavage of their target mRNAs (Figure 5A). It also revealed that highly conserved miRNAs like miR156/7, miR414, miR172, miR165/6, miR167, miR164, miR168, miR390, and miR393 accounted for 43.85% of all identified target sites in genes (Supplemental Figure S9B).
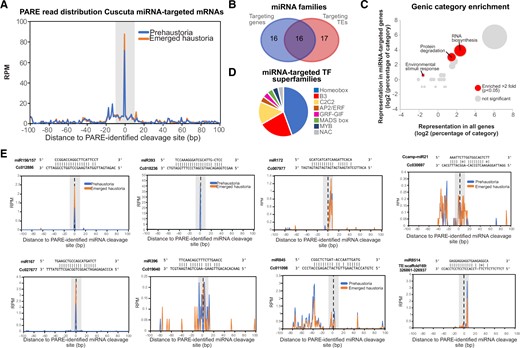
Analysis of conserved and new miRNA targets in Cuscuta. A, Overall profile of PARE read 5′-nt position relative to the identified cleavage site (position 0 on the x-axis) for early haustoria and emerged haustoria libraries. Number or reads are expressed as RPM. B, Venn diagram showing the number of miRNAs targeting genes and/or TEs. C, Bubble graph showing the comparison of categories in miRNA-targeted genes versus all genes in the C. campestris genome. At least two-fold upregulated significant categories are highlighted. Significance was calculated through a Fisher’s exact test with P < 0.05. Diameter of the bubble indicates the number of genes within each category. D, Pie chart showing the number of different categories of TF superfamilies targeted by miRNAs. TF superfamilies are defined as homeobox, B3-domain containing (B3), Cys2 Cys2 zinc-finger motif (C2C2), APETHALA2/ETHYLENE RESPONSIVE FACTOR, GROWTH REGULATING FACTOR(GRF)-GRF-INTERACTING FACTOR (GRF-GIF), MCM1-AGAMOUS-DEFICIENS-SRF box (MADS box), myeloblastosis viral oncogene homolog (MYB), and No Apical Meristem (NAM)-A. thaliana Activation Factor 1 (ATAF1)-Cup-shaped Cotyledon (CUC2) (NAC). E, Examples of PARE reads 5′-nt position relative to the cleavage site (position 0 on the x-axis) for selected targets of the miRNAs indicated. Number of reads are expressed as RPM.
Of the total targeted events, 70.87% corresponded to TEs, showing that the role of miRNAs in controlling TE expression is tentatively crucial, at least in Cuscuta’s haustorial tissues (Supplemental Table S10). Seventeen miRNA families targeted exclusively TEs, while 16 targeted exclusively genes, and 16 appeared to target both TEs and genes (Figure 5B).
A categorization of miRNA-targeted protein-coding genes indicated that there was a significant enrichment of genes involved in RNA biosynthesis, protein degradation and environmental stimuli response (Figure 5C). The category “RNA biosynthesis” included a majority of transcription factor (TF) superfamilies, as expected from conserved miRNA functions (Figure 5D). Other conserved targeting events in C. campestris included two homologs of AGO1 (targeted by miR168) and homologs of ARFs 6 and 8 (targeted by miR167) (Supplemental Figure S9B).
Intriguingly, some miRNAs seem to have followed neofunctionalization events (Baldrich et al., 2018). This can be concluded from the identification of cleavage sites that were uniquely found in C. campestris. These included a remorin protein homolog (Cc005763) that was identified as a target of the conserved miR164 (which targets CUP-SHAPED COTYLEDON TFs in Arabidopsis), and a plasma membrane intrinsic protein (Cc011098) targeted by miR845 (a miRNA known to target TEs in Arabidopsis) (Figure 5E). We also detected target sites for four of our identified novel miRNAs, pointing toward an active role of these: miR15, miR17, miR21, and miR29. These novel miRNAs targeted several TEs, and from them, miR21 had an additional genic target, Cc030697 a RAB-GTPase GDP-dissociation inhibitor (Figure 5E;Supplemental Figure S9B).
Overall, the analysis of the targets of miRNAs in C. campestris indicated that, while a number of conserved miRNAs still exert their expected functions, some miRNAs may have experienced a neofunctionalization. In addition, new miRNAs did arise and are potentially associated with new regulatory functions possibly associated to the regulation of development and not only the regulation of the parasitic relationship with the host.
Relationship between new miRNAs and horizontally acquired genes in C. campestris
Finally, we explored how new miRNAs might have arisen in C. campestris. The parasitic lifestyle has enabled C. campestris to obtain DNA fragments with intact and expressed genes by HGT (Vogel et al., 2018). We therefore explored the genomic environment of new miRNAs identified in our study and searched for potential sequence homologies of those regions within the genomes of phototrophic host plant species. The majority of new miRNAs had a similar pattern, with sequences being conserved in a majority of other species from the order Solanales. For example, Ccamp-miR15 (Figure 6A) is derived from a sequence that is highly conserved in both closely related species like S. lycopersicum and more distant species like O. sativa (Figure 6B). However, one of the newly identified miRNAs, Ccamp-miR17 (Figure 6C) did not have high levels of similarity with other related plant species (Figure 6D). This prompted us to look within the reported HGT events to inquire whether these could have led to the acquisition of a new miRNA. For this, we retrieved HGT sequences identified in Vogel et al. (2018) and compared them with respect to their homology to the Ccamp-miR17 precursor transcript. Our analysis, indeed, revealed that this miRNA is highly similar to an HGT event in C. campestris (Figure 6E). Interestingly, the horizontally transferred gene suffered several mutations and insertions that brought stability to the produced hairpin, which lost many of its bulges (Figure 6E). The original HGT sequence may have experienced one and two sequence insertions during this process that might have resulted in the generation of the new miRNA (Figure 6F). Based on these data, we propose that HGT events not only are a source for new CDSs to be used in evolutionary tinkering, but can at least occasionally also lead to the acquisition of new noncoding RNAs, in this case a miRNA.
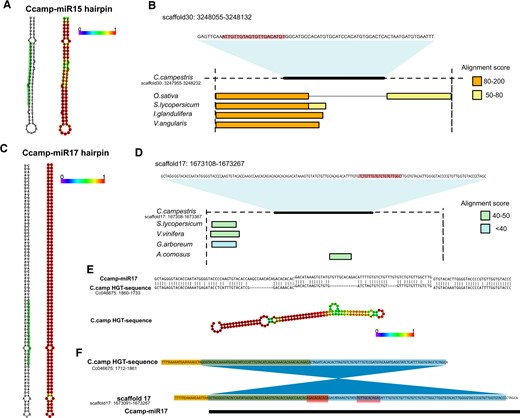
Exploration of the origin of new miRNAs in Cuscuta. A, Secondary structure of the precursor miRNA identified for the new miRNA Ccamp-miR15 (left) from C. campestris and secondary structure with base pair probability for each nucleotide (right). The position of the mature miRNA sequence is highlighted in the hairpin (left). B, Conservation of the genomic environment (pre-miRNA sequence including 100-bp upstream and downstream) of pre-miRNA Ccamp-miR15 in different plant species. Alignment scores from National Center for Biotechnology Information (NCBI) nucleotide blast are shown. Position of the mature miRNA sequence within the pre-miRNA is highlighted. C, Secondary structure of the precursor miRNA identified for the new miRNA Ccamp-miR17 (left) and secondary structure with base pair probability for each nucleotide (right). The position of the miRNA is highlighted in the hairpin. D, Conservation of the genomic environment (pre-miRNA sequence including 100-bp upstream and downstream) of pre-miRNA Ccamp-miR17 in different plant species. Alignment scores from NCBI nucleotide blast are shown. Position of the mature miRNA sequence within the pre-miRNA is highlighted. E, Comparison of the homologous sequences for pre-miRNA Ccamp-miR17 and a HGT event described in Vogel et al. (2018) (upper) and RNA secondary structure with base pair probability for each nucleotide of the HGT sequence (lower). Note than in the upper panel the HGT sequence is in 3′–5′-orientation. F, Potential sequence changes during the acquisition of Ccamp-miR17. The original HGT genomic sequence might have suffered one microinversion (marked in blue). Sequences spanning the pre-miRNA (marked in yellow) might also be conserved from the HGT event. Red blocks indicate novel insertions that are only present in the pre-miRNA sequence and not in the HGT event. Green areas indicate overlap between the microinversion and the conserved sequences.
Discussion
The use of sRNA high-throughput sequencing in combination with degradome sequencing has led to the identification of sRNA activity and their targeted RNAs in a multitude of plant species (Djami-Tchatchou et al., 2017). Here we have used the same approach to analyze miRNA populations of the parasitic plant C. campestris. This strategy has allowed us to (1) compare sRNA populations between this parasite and other members of the order Solanales, (2) examine the miRNAs of the shoot parasite C. campestris in comparison to that of a root parasite, O. aegyptiaca, (3) predict the putative biological roles of these noncoding RNA molecules, and (4) explore their acquisition of new miRNA sequences. Overall, our analysis offers an extensive overview of the miRNA populations and activity in plants with a parasitic lifestyle with their ensuing differences in body plan, metabolism, and interaction with their environment.
Cuscuta campestris has a large fraction of 24-nt sRNAs, which seem to be part of the RdDM pathway and are derived from TEs. The relative proportions between 21-nt and 24-nt sRNA appear to differ strongly between infective and noninfective tissues of C. campestris, which is supported by the observation that 24-nt sRNAs are also less abundant in the root parasite O. aegyptiaca. However, the proportion of 24-nt sRNAs seems to vary greatly among related species, for example from within the Solanales (see Figure 1G) leaving it open whether this is of importance for the lifestyle of the parasitic species, but it is tempting to speculate on this. Epigenetic mechanisms have been proposed as a key component for the plasticity of genomes especially due to its influence on gene expression under environmental conditions (Duncan et al., 2014). Dodders are constantly challenged with the stresses connected to their biotic interaction with a host and with the ensuing struggle for nutrients. Epigenetic regulation could be involved in adapting the parasite to different hosts, that in turn provide different nutritional contributions or respond with different defense strategies. Whether parasites have an epigenetic “memory” to preadapt them to recently encountered hosts or whether the epigenetic modifications help them in other ways to adapt is not investigated yet. Furthermore, parasitic plants are known to promote HGTs (Mower et al., 2004), a process that could be mediated by TEs (Panaud, 2016). We found that 24-nt sRNAs, which mediate DNA methylation at TE regions, were lowest in haustorial tissues, which could be connected to a need for a higher TE activity to mediate HGT during parasitism. This aspect deserves deeper investigation in the future. It must be noted, nevertheless, that since our analysis relied on multiple sequencing datasets from different origins and technical characteristics it is difficult to draw final conclusions from our data and future analysis of sRNA conservation are advisable.
The parasitic lifestyle of C. campestris had considerable consequences on the configuration of its genome. Several genes that are very conserved in other Solanales were lost in Cuscuta genomes (Sun et al., 2018; Vogel et al., 2018). Among them are genes involved in nutrient uptake, development, or defense. Likewise, our analysis showed that a number of miRNAs that are important for the overall development and nutrient sensing in normal green plants, including miR162, miR394, miR395, miR403, and miR482 (Xie et al., 2003; Vidal et al., 2010; Si-Ammour et al., 2011; Matthewman et al., 2012; Song et al., 2012; de Vries et al., 2015), were not found among the miRNAs that are expressed in any of the tissues under study. Two scenarios can potentially explain this finding: For some miRNAs it seems plausible that they were lost because their targets are either missing or do not require regulation due to a different set of conditions in the parasite. miR394 is a putative example for this group, being involved in leaf development (Vidal et al., 2010; Si-Ammour et al., 2011; Song et al., 2012), which is nonexistent in Cuscuta. Other RNAs may be repressed in the parasite due to its different nutrient supply situation. For miR395, being involved in the control of sulfate assimilation (Matthewman et al., 2012), this possibility may apply. Another example for a plausible loss of regulatory roles is represented by miR482 (also known as miR2118), which in Solanaceae targets several NBS–LRR defense genes (de Vries et al., 2015). The loss of this miRNA might be tentatively connected to the reduction of NBS–LRR genes in Cuscuta (Sun et al., 2018). Furthermore, we detected some differential expression of conserved and new miRNAs (Figure 7). Several miRNAs regulating different TF families and other genes showed a more enhanced expression in haustorial tissues. These included miR845 and miR7767, which assumedly regulate a plasma membrane intrinsic protein (Aquaporin) and two heat shock protein 70 (HSP70) family members, respectively. Aquaporins are involved in the transport of water, small solutes, and gases (Maurel et al., 2008), while HSP70 proteins play important roles in the protection against abiotic stresses (Wang et al., 2004). Genes associated with transporters and heat shock proteins are differentially expressed in infective stages of another Cuscuta species compared to stems and seedlings (Ranjan et al., 2014), so it is plausible to speculate that expression of these miRNAs in this tissue will add another layer of tissue-specific transcriptional regulation. On the other hand, noninfective tissues had higher accumulation of miRNAs like miR156/7, miR159, miR165/6, miR396, miR171, and miR319 which regulate MADS box, MYB, HD-ZIP III, GRF, SCL, and TCP TFs. While it is theoretically possible that not all miRNAs were detected in our approach, it is still plausible and likely that C. campestris has experienced some losses of miRNAs that are important for obsolete processes while it has obtained (or retained) other miRNAs that are involved in other levels of development and metabolism that are relevant to the parasitic lifestyle.
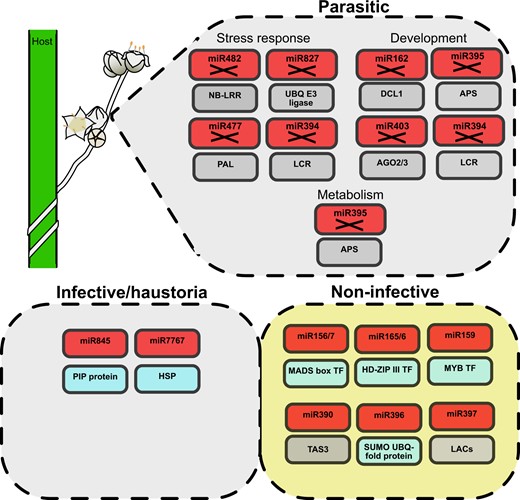
Graphic conclusion. Parasitic plants (drawing in the upper left corner) lack several miRNA families (boxes marked with X) regulating different gene families (boxes underneath) associated with the response to stress and developmental and metabolic processes. Furthermore, infective/haustoria and noninfective tissues show differential miRNA regulation both for confirmed genic targets (blue boxes) and predicted genic targets (gray boxes).
Novel miRNAs are generally believed to originate by de novo emergence or expansion and neofunctionalization (Baldrich et al., 2018). The miRNA pool identified by us bears indications for another origin of sRNAs in parasitic plants: namely by HGT. One of the new miRNAs (Ccamp-miR17) appears to have originated from an HGT event. The Ccamp-miR17 is more strongly expressed in noninfective tissues and targets a TE. HGT has been shown to be a source of mobile sRNAs and miRNAs (Johnson and Axtell, 2019; Yang et al., 2019). Our analysis confirms that this mechanism could be an acquisition strategy for new miRNA sequences involved in the regulation of processes other than the control of infection.
Conclusions
Overall, our data indicate that the sRNA populations of C. campestris and O. aegyptiaca offer valuable information of how the parasitic lifestyle is regulated at the posttranscriptional level. Mechanisms like miRNA targeting could be an important part of the regulation of developmental plasticity. Such information will help to understand the regulation of parasitic behavior in plants and its dynamism during development, but also highlights that genomic information is crucial to interpret these data.
Materials and methods
Plant material
Seeds from Cuscuta (C. campestris) were harvested from a culture of the parasite that was grown in a glasshouse at the phytotron of the University of Tromsø, Norway, in 24 h of light at 21°C on geranium (Pelargonium zonale) as host. To increase germination percentage, the seeds were scarified with concentrated sulfuric acid (96% v/v) for 20 min before being rinsed with water and placed on moist sterile filter paper. Young seedlings were harvested when they were 3–6 cm long (Figure 1A). For the non-infective parts of mature C. campestris vines, the top 3 cm measured from growing tips that were 20–80 cm distant to the closest infection site (Figure 1B) were harvested directly from the C. campestris culture on P. zonale. Similarly, shoots remote from the host were used to induce the early stage and emerged haustoria by far-red light and a tactile stimulus as described by Olsen et al. (2016) (Figure 1C). Haustorial sites harvested by cutting the stems immediately to the left and right of each site were divided according to their stage (early and emerged, respectively) and pooled into batches of 15–20 per replicate.
Orobanche (O. aegyptiaca) seeds collected in bulk from several emerged shoots in a highly infested tomato (S. lycopersicum) field were grown in the controlled research greenhouse (15-h natural light, temperature of 25°C, and humidity of 25%) of the Razi University-Kermanshah, Iran, on S. lycopersicum as host. Tissue samples (Figure 1D) were harvested when O. aegyptiaca was at the “stage 4.2” of its life cycle (“late post-vascular connection stage” or “spider stage”) described by Westwood et al. (2012).
Total RNA and sRNA/PARE library construction
Total RNA of the different C. campestris tissue types was isolated using TRIzol reagent (Life Technologies, Carlsbad, CA, USA). sRNAs were gel purified in a 15% (v/v) acrylamide/8-M urea gel using as input 10 µg of total RNA. Purified sRNAs were used as input to generate sRNA libraries using the NEBNext sRNA Library Prep Set for Illumina (New England Biolabs, Ipswich, MA, USA) following the manufacturer’s instructions. This procedure is identical to the one described in Martinez et al. (2016). PARE libraries were constructed following the protocol described in Zhai et al. (2014) using 20–30 µg of total RNA as input. For O. aegyptiaca, 12 biological replicates each containing materials from three attachment sites were pooled for total RNA isolation using TRIzol reagent (Life Technologies). Extracted RNAs were sent to the Novogene Co., Ltd. (Beijing Nuohe Zhiyuan Biological Information Technology, China) for downstream steps toward sRNA sequencing on an Illumina Hiseq 2500 platform.
Transposon annotation
Transposons were annotated by a homology search against the REdat_9.8_Eudicot section of the PGSB transposon library (Spannagl et al., 2016). Next to transposon sequences from different dicot species, the library also contains 691 nonredundant C. campestris-specific full-length LTR retrotransposons which had previously been identified (Vogel et al., 2018). The program vmatch (http://www.vmatch.de), a fast and efficient matching tool, was used with the following parameters: identity ≥ 70%, minimal hit length 75 bp, seedlength 12 bp (exact commandline: d p -l 75 identity 70 seedlength 12 exdrop 5). The vmatch output was filtered for redundant hits via a priority-based approach, which assigns higher scoring matches first and either shortens (<90% coverage and ≥50-bp rest length) or removes lower-scoring overlapping hits to obtain an overlap free annotation.
Analysis of sRNA libraries
sRNA libraries were trimmed using Trim Galore (https://github.com/FelixKrueger/TrimGalore). Reads were aligned using bowtie (Langmead et al., 2009) with the command “–v2” that allows two mismatches unless otherwise indicated. The r0.32 version of the C. campestris genome (Vogel et al., 2018) and the miRbase version 21 (https://www.mirbase.org/) were used in this analysis. Reads were normalized to reads per million (RPM) to the total reads mapped to the genome. PCA was performed using the plotPCA tool from deepTools2 (Ramirez et al., 2016) through the Galaxy platform (Jalili et al., 2020). Input BAM files were generated from SAM files produced using Bowtie2 with the option “-v2.” Coverage in the BAM files used to perform the PCA was calculated using sRNAs ranging from 18 to 28 nts in 10,000-bp bins for whole-genome coverage (Figure 1E) or limited to the coordinates of conserved miRNAs (Supplemental Figure S4). For the analysis of the presence of sRNAs of viral origins, sRNA libraries were mapped against the nonredundant (clustered at 90%) plant viral sequences contained in the Virosaurus database (https://viralzone.expasy.org/8676).
Analysis of PARE libraries
For PARE library analysis, miRNA cleavage events were identified using PARESnip (Stocks et al., 2018) using as input the miRNA sequences identified in this analysis and the transcript sequences of the r0.32 genome version. PARESnip parameters were as follow:
min_sRNA_abundance = 1
subsequences_are_secondary_hits = false
output_secondary_hits_to_file=false
use_weighted_fragments_abundance = true
category_0 = true
category_1 = true
category_2 = true
category_3 = true
category_4 = true
discard_tr_rna=true
discard_low_complexity_srnas=true
discard_low_complexity_candidates=true
min_fragment_length = 20
max_fragment_length = 21
min_sRNA_length = 20
max_sRNA_length = 22
allow_single_nt_gap=true
allow_mismatch_position_11 = false
allow_adjacent_mismatches=false
max_mismatches = 4
calculate_pvalues=true
number_of_shuffles = 100
pvalue_cutoff = 0.05
do_not_include_if_greater_than_cutoff=true
number_of_threads = 2000
auto_output_tplot_pdf=false
Targets for miRNAs that excluded the maximum number of targets of miR156/7 (145) were discarded. These only included targets of miRNAs miR5658 and miR1134 which had a marginal contribution to the overall profiles. For genome-wide plots of PARE reads, Bowtie (Langmead et al., 2009) was used to align PARE libraries to the whole transcriptome using the command “-v2.” For the generation of the plots, reads were aligned according to the predicted cleavage site coordinates identified in PARESnip. PARE library coverage in the BAM files used to perform the PCA was calculated in 10,000-bp bins. Gene category enrichment of target genes was performed using the annotation of C. campestris genes in the genome version r0.32. Gene category enrichment analysis was performed using the categories included in the CDS annotation of the C. campestris genome 0.32. Gene categories for the genes detected as miRNA targets was compared to global gene categories for all genes in the C. campestris genome.
Identification of conserved and new miRNAs
Conserved miRNAs in C. campestris were identified following three steps. First, genome-filtered sRNA sequences (identified with Bowtie1 using “-v0”) were mapped allowing 2 mismatches (“-v2”) to all plant miRNA mature sequences from miRbase release 22.1 (Kozomara and Griffiths-Jones, 2011). Only miRNA matches with at least 1 read of 20–22 nts in length were considered, so miRNA matches with only matches shorter or larger than 20–22 nts were discarded. Additionally, we mapped all plant pre-miRNA sequences from the same miRbase release (22.1) to the C. campestris genome using the blast tool of NCBI’s Genome Workbench with default parameters (Kuznetsov and Bollin, 2021). Only matches with an e-value lower than 0.005, an identity over 70% and a coverage over 50% were considered. These positive matches were then manually check using the blast suite included in miRbase (https://www.mirbase.org/search.shtml) to confirm the presence of a mature miRNA within the potential pre-miRNA sequence. Sequences that did not contain matches to any mature miRNA were discarded. Additionally, miRNA sequences with matches to the miRbase identified with miRCat (see below) were included in our analysis of conserved miRNAs. Finally, negative matches for conserved miRNAs were confirmed by mapping mature miRNA (or 5p) and their complementary sequences (miRNA* or 3p) present in miRbase release 22.1 (matching at least 20 bp in the genome) using Bowtie, checking the distance between miRNA/5p and miRNA*/3p matches (lower than 200 bp) and calculating the MFE values (lower than −0.02/nt).
New miRNAs were identified using miRCat (Stocks et al., 2018), only considering sequences with the following characteristics: 21–22 nt in length, detection of miRNA* complement, at least 10 reads in total, identification in at least two bioreplicates and a either a processing precision (defined as the number of miRNAs plus miRNA* sequences divided by the total number of sRNAs produced from the pre-miRNA sequence) of at least 75% in any of the individual sRNA libraries analyzed (criteria recommended in Axtell and Meyers (2018)) or a combination of at least a 30% processing precision with high levels of miRNA accumulation (at least 30 RPM in any library).
For O. aegyptiaca, sRNA libraries were trimmed using Trim Galore and first aligned to S. lycopersicum reference genome (SL 3.0) using Bowtie with “–v0” (https://plants.ensembl.org/) to remove possible host-originated reads. The filtered reads were then subjected to mapping against the available transcriptome reference of O. aegyptiaca (http://ppgp.huck.psu.edu/) using Bowtie with the command “–v2.” The same applied to identify conserved and new miRNAs based on the miRbase version 22.1 (https://www.mirbase.org/) using miRProf and miRCat, respectively, with ignoring criteria regarding bioreplicates. miRProf and miRCat are part of the UEA sRNA Workbench (Stocks et al., 2018). Conserved miRNA matches to the genome were confirmed using Bowtie allowing up to three mismatches (“-v3”) using the miRProf-identified miRNA sequences.
Other bioinformatic analysis
miRNA target prediction was performed using the psRNATarget server (https://www.zhaolab.org/psRNATarget) (Dai et al., 2018), with the default parameters of the 2011 release against the transcript sequences of the C. campestris genome version 0.32. Folding of RNA secondary structures was performed using the RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) (Gruber et al., 2008) using default parameters. Upset plots were generated using UpSetR (Conway et al., 2017) with default parameters and the option order.by= “freq.” For comparisons with other species, sRNA datasets were retrieved from the Gene Expression Omnibus (GEO) database repository (https://www.ncbi.nlm.nih.gov/geo/) from the accession numbers indicated in Supplemental Table S2. The following genome versions were used in our analysis: Nicotiana benthamiana version 0.4.4., N. tabacum version 4.5, S. lycopersicum build 4.00, S. melongena HQ-1315, S. tuberosum version 3.4, C. annum version 1.55, Solanum pimpinellifolium LA0480 and E. guttata version 2.0. Petunia x hybrida mapping was performed against the concatenated genomes of Petunia axilaris (version 1.6.2) and Petunia inflata (version 1.0.1). Ipomoea batatas mapping was performed against the concatenated genomes of Ipomoea trifica (version 3) and Ipomoea triloba (version 3). For TE mapping analysis, sRNA libraries from Solanales members (other than C. campestris) were matched against their respective TE annotations or a custom annotation combining the tomato, potato, and pepper TE annotations. For E. guttata and Orobanche aegyptica, reads were mapped against a custom annotation including the sequences contained within the plant TE databases Inpactor (Orozco-Arias et al., 2021), TREP (Wicker et al., 2002), PlantSat (Macas et al., 2002), and PGSB (Spannagl et al., 2016).
Accession numbers
All raw and processed sequencing data generated in this study have been submitted to the NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/) under accession numbers: GSE181576 (Cuscuta) and GSE181578 (Orobanche). The TE annotation data can be accessed temporarily under: https://hmgubox2.helmholtz-muenchen.de/index.php/s/qQZ6McDsAfKePkB.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Global sRNA profiles for the species shown in Figure 1.
Supplemental Figure S2. Presence of viral-derived sRNAs in Orobanche sRNA libraries compared to Arabidopsis mock and cucumber mosaic virus-infected sRNA libraries.
Supplemental Figure S3. Comparison of miRNA conservation between Cuscuta, Arabidopsis, maize and rice.
Supplemental Figure S4. PCA of the sRNA libraries produced from different Cuscuta tissues restricted to miRNAs.
Supplemental Figure S5. Size distribution of individual miRNA families for each Cuscuta sRNA library produced in this study.
Supplemental Figure S6. Size and 5′-nt distribution for each replicate of the C. campestris sRNA libraries shown in Figure 3, B and C.
Supplemental Figure S7. Heat map showing differential expression of AGO proteins in young and emerged haustoria.
Supplemental Figure S8. Secondary structure of new miRNAs identified in this study.
Supplemental Figure S9. Identification of miRNA-targeted mRNAs in Cuscuta through degradome sequencing.
Supplemental Table S1. Libraries generated in this study.
Supplemental Table S2. Public sRNA libraries from Solanales members used in this study.
Supplemental Table S3. List of conserved miRNA families in Solanales members.
Supplemental Table S4. Coordinates and sequences of pre-miRNA homologs identified in the C. campestris genome.
Supplemental Table S5. Coordinates of mature miRNA homologs identified in the O. aegyptiaca transcriptome.
Supplemental Table S6. Expression of conserved miRNAs (RPM) in different C. campestris tissues.
Supplemental Table S7. New miRNAs identified in C. campestris with their expression and processing precision values in different tissues.
Supplemental Table S8. New miRNAs identified in O. aegyptiaca and their expression values in different tissues.
Supplemental Table S9. Bioinformatic prediction of C. campestris miRNA targets.
Supplemental Table S10. PARE-identified miRNA targets in C. campestris.
Z.Z., M.L.A., A.D., and G.M. performed the experiments. Z.Z., H.G., J.B., and G.M. performed the bioinformatic analysis. H.S., K.K., and G.M. designed the experiments and analyzed the data. K.K and G.M. wrote the article with contributions of all the authors. K.K. and G.M. agree to serve as the authors responsible for contact and ensure communication.
The authors responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://dbpia.nl.go.kr/plphys/pages/general-instructions) are Kirsten Krause ([email protected]) and German Martinez ([email protected]).
Acknowledgments
We thank Dr. Rainer Schwacke and Prof. Björn Usadel (both from the Research Center Jülich, Germany) for valuable comments on the manuscript. Sequencing of sRNA and PARE libraries was performed by the SNP&SEQ Technology Platform in Uppsala. The facility is part of the National Genomics Infrastructure (NGI) Sweden and Science for Life Laboratory.
Funding
The SNP&SEQ Platform is supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation. Research in the Martinez group is supported by SLU, the Carl Tryggers Foundation (CTS 18:251), the Swedish Research Council (VR 2021-05023), Formas (2021-01161), and the Knut and Alice Wallenberg Foundation (KAW 2019.0062). The Tromsø Research Foundation (16-TF-KK) and the Norwegian Research Foundation (NFR 301175) have supported this work in the Krause group. H. Gundlach acknowledges funding from the German Ministry of Education and Research (031A536B, de.NBI). Z.Z. was supported by a sabbatical grant from Ministry of Science, Research and Technology of Iran.
Conflict of interest statement. None declared.
References
Author notes
Current address for Maria Luz Annacondia: Copenhagen Plant Science Centre, Department of Plant and Environmental Sciences, University of Copenhagen, Frederiksberg, 1870, Denmark.
Current address for Julien Bruckmüller: Solana Research GmbH, Windeby, 24340, Germany.
These authors contributed equally (Z.Z. and M.L.A.).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}