





Abstract
VESICLE-INDUCING PROTEIN IN PLASTID1 (VIPP1) is conserved among oxygenic photosynthetic organisms and appears to have diverged from the bacterial PspA protein. VIPP1 localizes to the chloroplast envelope and thylakoid membrane, where it forms homooligomers of high molecular mass. Although multiple roles of VIPP1 have been inferred, including thylakoid membrane formation, envelope maintenance, membrane fusion, and regulation of photosynthetic activity, its precise role in chloroplast membrane quality control remains unknown. VIPP1 forms an oligomer through its amino-terminal domain and triggers membrane fusion in an Mg2+-dependent manner. We previously demonstrated that Arabidopsis (Arabidopsis thaliana) VIPP1 also exhibits dynamic complex disassembly in response to osmotic and heat stresses in vivo. These results suggest that VIPP1 mediates membrane fusion/remodeling in chloroplasts. Considering that protein machines that regulate intracellular membrane fusion/remodeling events often require a capacity for GTP binding and/or hydrolysis, we questioned whether VIPP1 has similar properties. We conducted an in vitro assay using a purified VIPP1-His fusion protein expressed in Escherichia coli cells. VIPP1-His showed GTP hydrolysis activity that was inhibited competitively by an unhydrolyzable GTP analog, GTPγS, and that depends on GTP binding. It is particularly interesting that the ancestral PspA from E. coli also possesses GTP hydrolysis activity. Although VIPP1 does not contain a canonical G domain, the amino-terminal α-helix was found to be important for both GTP binding and GTP hydrolysis as well as for oligomer formation. Collectively, our results reveal that the properties of VIPP1/PspA are similar to those of GTPases.
VESICLE-INDUCING PROTEIN IN PLASTID1 (VIPP1), an essential protein in chloroplasts, is highly conserved among oxygenic photosynthetic organisms (Vothknecht et al., 2012; Zhang and Sakamoto, 2013, 2015). Multiple roles of VIPP1 related to the organization of chloroplast membranes have been proposed (Kroll et al., 2001; Lo and Theg, 2012; Zhang et al., 2012; Hennig et al., 2015; McDonald et al., 2015), along with the formation of photosynthetic protein supercomplexes (Zhang et al., 2014), through the provision of structural lipids (Nordhues et al., 2012) and thylakoid formation via vesicles (Aseeva et al., 2007). Despite pleiotropic roles in chloroplasts, these studies imply that the important function of VIPP1 converges into a lipid-binding property. Indeed, recent work suggests that VIPP1 induces membrane remodeling and destabilization (Hennig et al., 2015; Heidrich et al., 2017). This membrane-fusion function might explain the proposed roles of VIPP1 in the biogenesis and maintenance of chloroplast membranes. Actually, VIPP1 is homologous to bacterial PspA, which also is thought to play a role in the membrane integrity of plasma membranes in Escherichia coli. Specific to VIPP1 is an addition of the C-terminal tail, which seems to provide the flexibility of complex formation with its intrinsically disordered property (Zhang et al., 2016a).
Membrane fusion/remodeling, which is observed in various cellular events, is known to involve sophisticated protein machineries in the endoplasmic reticulum, Golgi apparatus, and mitochondria (Bonifacino and Glick, 2004; Montessuit et al., 2010; Youle and van der Bliek, 2012), which often require nucleotide binding and/or hydrolysis. For example, the membrane-anchored GTPase Atlastin (ATL), which is well conserved in eukaryotes, mediates the homotypic fusion of endoplasmic reticulum membranes (Park and Blackstone, 2010; McNew et al., 2013; Liu et al., 2015). The ATL of Drosophila melanogaster (D-ATL) as well as the distantly related yeast SEY1 and Arabidopsis (Arabidopsis thaliana) RHD3 orthologs can catalyze GTP-dependent fusion when purified and inserted into synthetic liposomes, which is consistent with the direct role of ATL in homotypic membrane fusion (Orso et al., 2009; Anwar et al., 2012; Zhang et al., 2013). The fusion is preceded by the transient tethering of ATL-containing vesicles, which requires not only GTP binding but also GTP hydrolysis (Liu et al., 2015). By contrast, dynamin is the most extensively studied component of the membrane fission machinery, which requires GTP binding and/or GTPase activities to implement the maturation of the clathrin-coated pit (Liu et al., 2013). Moreover, members of the Rab or ARF/Sar branches of the Ras GTPase superfamily have been shown to regulate intracellular membrane trafficking mediated by vesicular transport (Mizuno-Yamasaki et al., 2012). According to these examples, GTP is likely to be an indispensable molecule in most membrane-remodeling processes characterized to date.
In contrast to the well-studied membrane-remodeling machinery above, little is known about the mechanism in chloroplasts. For example, CPRabA5e was identified recently as a chloroplast-localized Rab GTPase protein in Arabidopsis (Karim et al., 2014). However, the detailed function of CPRabA5e in membrane remodeling remains elusive, although the knockout mutant showed irregularly shaped chloroplasts. FZL (FZO-like), a new protein class within the dynamin superfamily, is suggested to function in chloroplasts as a membrane fusion protein. It is particularly interesting that the GTPase activity-dependent formation of punctate structures of FZL-GFP near the chloroplast periphery (Gao et al., 2006) is similar to the case of VIPP1-GFP (Zhang et al., 2012). We have demonstrated that VIPP1-GFP, which fully complements the loss of VIPP1 in the vipp1 null mutant (vipp1-ko), forms large and various macrostructures along chloroplast envelopes that display dynamic movement that is especially responsive to hypotonic stress (Zhang et al., 2012). The connection between the dynamic VIPP1 movement and the membrane damage caused by various stresses (Zhang et al., 2016b) led us to postulate that VIPP1 plays a role in protecting membranes through fusion, which might require GTPase activity.
In this study, we prepared recombinant VIPP1-His protein and conducted analyses of the GTP hydrolysis activity of this protein. We demonstrated that VIPP1 is indeed capable of both GTP binding and GTP hydrolysis, dependent on the first α-helix-mediated precise complex formation. We also demonstrated that VIPP1 and its ancestral protein PspA from E. coli both have GTP hydrolysis activity. Our results suggest that VIPP1/PspA is a novel type of GTPase that lacks canonical G domains.
RESULTS
Evaluation of the GTP Hydrolysis Activity of VIPP1-His Protein
To demonstrate that VIPP1 possesses GTPase activity in vitro, we prepared C-terminally His-tagged VIPP1 proteins (VIPP1-His) without signal peptide. Figure 1 depicts the schematic structures of recombinant proteins that were used in this study (Fig. 1). The VIPP1-His protein was expressed in BL21(DE3)pLysS cells at 27°C. In the soluble proteins obtained from E. coli cells, an IPTG-inducible protein around the 25- to 37-kD marker can be found by SDS-PAGE (Fig. 2A, top), which immunoblot analyses confirmed as VIPP1 (Fig. 2A, bottom). The overexpressed proteins were purified by an Ni2+-affinity column (Supplemental Fig. S1). The purity of the recombinant protein was evaluated using SDS-PAGE. All the preparations of recombinant proteins contained the VIPP1-His protein as the major component (Fig. 2B; Supplemental Fig. S2). First, we tested whether wild-type VIPP1-His has GTP hydrolysis activity using a colorimetric detection system as described in “Materials and Methods.” We observed the increase of released inorganic phosphate (Pi) by increasing amounts of VIPP-His in the reaction mixture, which was readily detectable by its deep-green coloration (Fig. 3A). When quantified using a standard curve, the level of released Pi was found to be proportional to the amount of VIPP1-His for less than 1 µg in the reaction mixture (Fig. 3B). Consequently, to obtain quantitative results for our further analyses, we chose to use 0.5 µg of VIPP1-His protein in the reaction mixture as our standard condition (see “Materials and Methods”).
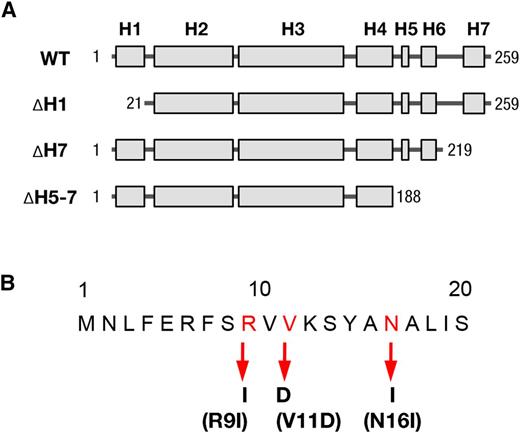
Schematic model for the structure of recombinant VIPP1 proteins used in this study. A, Schematic illustration of Arabidopsis VIPP1 secondary structure, indicating the total length and different truncated VIPP1 proteins. H denotes the predicted α-helix structure of VIPP1. The numbers indicate the positions of first (left) and last (right) amino acids of VIPP1 proteins of different lengths. WT, Wild type. B, Point mutations of amino acids in the first α-helix at the N terminus of VIPP1. The amino acids are positioned with the corresponding number, in which mutated residues are red. The substituted amino acids are listed below and are indicated with arrows.
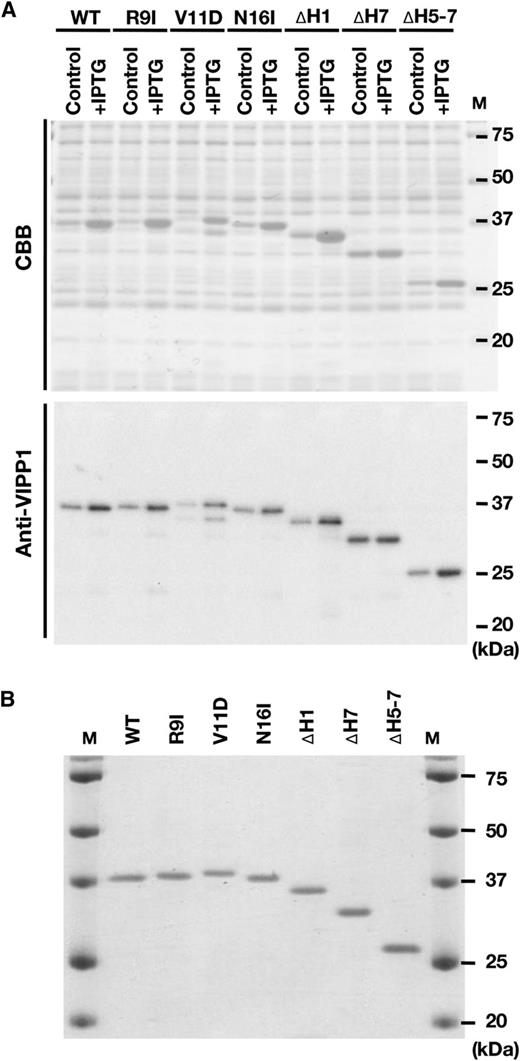
Preparation of recombinant VIPP1-His proteins. A, Expression of recombinant VIPP1-His proteins in E. coli cells, which was confirmed by SDS-PAGE and either Coomassie Brilliant Blue (CBB) staining or immunoblot analyses. B, Composition of protein solutions used for this study. The final protein preparation (0.2 µg protein lane−1) was subjected to SDS-PAGE and Coomassie Brilliant Blue staining to confirm most VIPP1-His proteins.
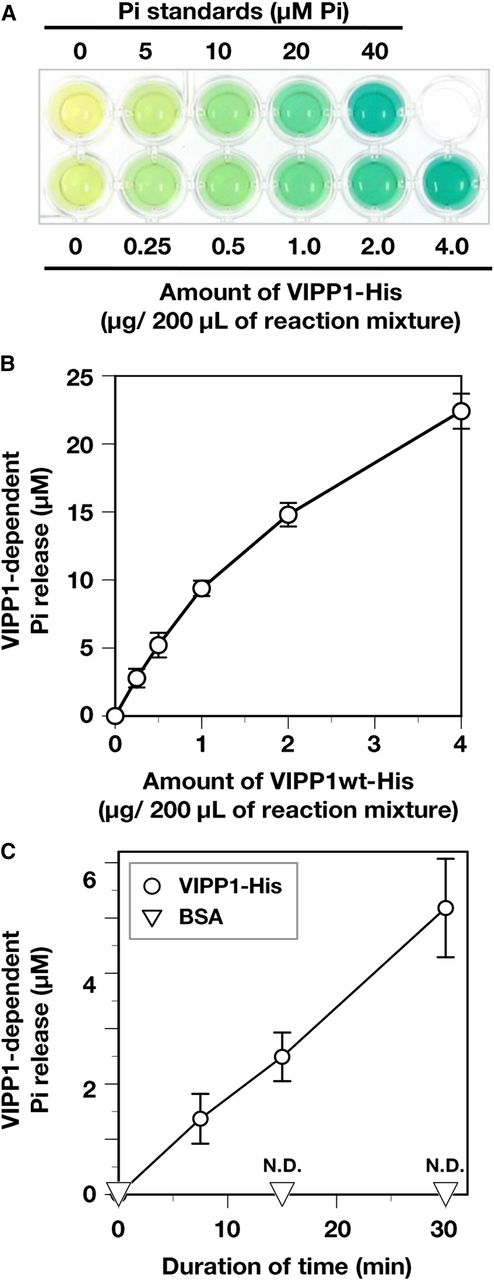
Evaluation of GTP hydrolysis activity of His-tagged recombinant proteins. A, Photograph of a typical assay for GTP hydrolysis using a GTPase assay kit (Innova Biosciences). Green denotes the presence of free Pi. Absorption at 635 nm of reaction mixture without VIPP1-His (the left end of bottom row wells) was subtracted as background in each measurement. B, VIPP1-His dependency of GTP hydrolysis. The level of released Pi was analyzed with various amounts of wild-type (wt) VIPP1 protein (0.25–4 µg in 200 µL of reaction mixture), which was quantified based on the absorption of Pi standard solutions. C, Time-dependent release of Pi. All the analyses were performed in the presence of 0.5 µg of protein (VIPP1-His and BSA) in each reaction mixture of 200 µL. The reaction mixtures were incubated for 0, 7.5, 15, and 30 min at 37°C. Then, free Pi was quantified. Data points and error bar represent means and sd, respectively, of results obtained from four to six independent experiments (Student’s t test, for which P < 0.01 was inferred as significant). N.D., Not detected.
Both VIPP1 and PspA Possess GTP Hydrolysis Activity in Vitro
VIPP1 has no consensus motif as a G protein. Therefore, we decided to evaluate the observed GTPase-like activity carefully. We first examined whether the release of Pi increased as incubation time proceeded. Figure 3C shows that, during 30 min of incubation, the amount of Pi increased in a time-dependent manner and reached about 5 µm in the presence of 0.5 µg of VIPP1-His, whereas BSA did not raise the level of Pi at all in the reaction mixtures. Next, we examined whether the same activity was observed using a protein unrelated to GTPase activity. DPD1 is an exonuclease targeted to plastids and mitochondria, which was purified as a His-tagged protein. In an earlier study, VIPP1-His without its putative targeting signal was shown to exhibit Mg2+-dependent exonuclease activity in vitro (Matsushima et al., 2011). Therefore, we performed our GTP hydrolysis assay using purified DPD1-His (Supplemental Fig. S3, A and B) and observed no detectable level of released Pi (Supplemental Fig. S3C). Our result obtained using DPD1-His as a negative control, therefore, suggested that GTP hydrolysis activity is unlikely to result from a residual contamination of GTPase from cell extracts.
Given the structural and functional similarities between VIPP1 and the bacterial PspA, we reasoned that PspA might well display GTP hydrolysis activity in our in vitro assay. Our earlier study showed that VIPP1 expression in the E. coli pspA mutant rescues the defect in maintaining membrane potential (Zhang et al., 2012). The expression of PspA, fused to an N-terminal chloroplast-targeting signal and the C-terminal tail of VIPP1, did not rescue the defective growth of vipp1-ko in Arabidopsis, whereas PspA appeared to form large macrostructures resembling VIPP1. They are affected by the C-terminal extension specific to VIPP1 (Zhang et al., 2016a). It is conceivable that VIPP1 and PspA overlap their functionality in membrane-remodeling activity. Our results showed that PspA-His was purified successfully (Supplemental Fig. S2 and S3B) and that it showed GTP hydrolysis activity comparable to that from VIPP1-His (Supplemental Fig. S3C). Based on these results, we inferred that both VIPP1 and PspA might represent a novel type of GTPase, although lacking a canonical nucleotide-binding domain. As described hereinafter, we specifically examined VIPP1-His and further characterized its biochemical properties.
GTPase-Like Properties of VIPP1-His
Because Mg2+ has been established as an essential cofactor for GTP-binding protein functions (John et al., 1993; Sprang and Coleman, 1998), we tested whether the GTP hydrolysis activity of VIPP1-His depends on Mg2+. According to Figure 3C, 0.5 µg of VIPP1-His was able to release 5.21 ± 0.9 µm Pi during 30 min of reaction. Consequently, the GTP hydrolysis activity was calculated as 0.35 ± 0.06 µm Pi release µg−1 protein min−1 (Fig. 4A). The GTP hydrolysis activity was 0.02 ± 0.01 µm Pi release µg−1 protein min−1 in the absence of Mg2+, which was less than 10% of that in the presence of Mg2. These results indicated that the GTP hydrolysis activity of VIPP1-His is dependent on the presence of Mg2+, which is similar to typical GTPases. It is also reported that Ca2+, another divalent metal cation, can function as a cofactor of some GTPase, such as tubulin (Soto et al., 1996). We prepared a reaction mixture containing VIPP1-His and CaCl2 without Mg2+. The absorption at 635 nm of the mixture was always less than the background, indicating that no detectable GTP hydrolysis activity exists without Mg2+, even in the presence of Ca2+.
![Characterization of GTP hydrolysis activity. A, Substrate specificity and cofactor requirement of GTP hydrolysis activity of VIPP1-His. GDP and GTPγS were added to 0.5 mm instead of GTP. Both species of divalent cation (Mg2+ and Ca2+) were supplemented in 2.5 mm. N.D., Not detected. B, Inhibitory effects of GTPγS. GTP hydrolysis activity with various concentrations of GTP was analyzed in the presence (right graph) or absence (left graph) of 0.5 mm GTPγS. Bar graphs and error bars show means and sd, respectively, of results obtained from three to six independent experiments. C, Analyses for V max and K m for GTP hydrolysis activity in the presence or absence of 0.5 mm GTPγS based on the results in B. The concentration of GTP and average of VIPP1-dependent Pi release are shown on graphs according to the Cornish-Bowden plot method. The x and y values of intersection points represent K m and V max, respectively. D, Direct binding test of GTP to VIPP1-His protein. VIPP1-His, ∆H1, and BSA were dotted on a nitrocellulose membrane and then incubated with radiolabeled GTP ([α-32P]GTP) at room temperature for 1 h. The signals were detected using the BAS1000 system.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/plphys/177/1/10.1104_pp.18.00145/2/m_plphys_v177_1_328_f4.jpeg?Expires=1750284766&Signature=QGtw6IkR2IQo6VvU8jUju0zbDDL70u9I1SS6RjKTR9OJE8qo91D-kmbl1mxJJfVB1pg~ARchkENgeAsg29dxHhhqG~svuSSfZAQ9cYorJ3Jx~clglsDYmOY2tYu5oBw9jj8IP2fPBo3oaqJYJRTWKyZ1LUh08UOklYk-xIdNqn5NzhX--zAQZMhLUilPiwjjATgr-WAVClLahJ0jEkSzk8ypZT~uicRrrjKREEjFOfYUqRo-cfvsR5iz77a4JLfB7TpHDYOuthl6qbgCz3lJvA1jFemNrF6lzEYoiGWYeYzBGsDRs-GtYdVefOeB0YO0TfpKwVy7ql~qgpxXcYQXlg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Characterization of GTP hydrolysis activity. A, Substrate specificity and cofactor requirement of GTP hydrolysis activity of VIPP1-His. GDP and GTPγS were added to 0.5 mm instead of GTP. Both species of divalent cation (Mg2+ and Ca2+) were supplemented in 2.5 mm. N.D., Not detected. B, Inhibitory effects of GTPγS. GTP hydrolysis activity with various concentrations of GTP was analyzed in the presence (right graph) or absence (left graph) of 0.5 mm GTPγS. Bar graphs and error bars show means and sd, respectively, of results obtained from three to six independent experiments. C, Analyses for V max and K m for GTP hydrolysis activity in the presence or absence of 0.5 mm GTPγS based on the results in B. The concentration of GTP and average of VIPP1-dependent Pi release are shown on graphs according to the Cornish-Bowden plot method. The x and y values of intersection points represent K m and V max, respectively. D, Direct binding test of GTP to VIPP1-His protein. VIPP1-His, ∆H1, and BSA were dotted on a nitrocellulose membrane and then incubated with radiolabeled GTP ([α-32P]GTP) at room temperature for 1 h. The signals were detected using the BAS1000 system.
We next tested other guanosine residues, GDP and guanosine 5-O-[γ-thio]-triphosphate (GTPγS), in our reaction. In neither case did we observe any detectable VIPP1-dependent release of Pi (Fig. 4A). This result indicates that VIPP1 can release only the Pi located at the γ-position, which is similar to typical GTPases. GTPγS causes competitive inhibition if VIPP1-His has a similar property to those of typical GTPases (RayChaudhuri and Park, 1992). Therefore, we further characterized the inhibition mode of GTPγS. The level of released Pi increased along with increasing GTP (0.2–4 mm; Fig. 4B, left). We also measured GTP hydrolysis activity in the presence of 0.5 mm GTPγS in the same range of GTP concentrations. The GTP hydrolysis activity was lower in all conditions than those in the absence of GTPγS (Fig. 4B, right), indicating that GTPγS acts as an inhibitor of this hydrolysis reaction.
We analyzed V max and K m of these reactions based on the average values of the GTP dose-dependent assay (Fig. 4B) conducted with a Cornish-Bowden plot (Eisenthal and Cornish-Bowden, 1974). According to the graphs presented in Figure 4C, K m and V max were taken as approximately 2.2 mm and approximately 1.9 to 2 µm Pi release µg−1 protein min−1, respectively, in the control condition (Fig. 4C, left). By contrast, K m and V max in the presence of GTPγS (Fig. 4C, right) were 5 mm and 2 to 2.1 µm Pi release µg−1 protein min−1, respectively. Given that only K m tended to change markedly by the presence of GTPγS, the inhibition by GTPγS probably occurs in a competitive mode against GTP. By contrast, k cat (V max mol−1 protein) of this GTP hydrolysis activity was calculated as 12 min−1 based on the theoretical V max described above. This value is in the range of GTPase activity of the dynamin family, which is 1 to 20 min−1 (Hinshaw, 2000).
To confirm GTP hydrolysis activity implemented by VIPP1-His, we examined the interaction between VIPP1-His and GTP. A dot-blot assay with radiolabeled GTP ([α-32P]GTP) revealed that VIPP1-His was capable of interacting with GTP, although the negative control BSA had almost no signal (Fig. 4D). This result suggests strongly that the observed GTP hydrolysis occurred on VIPP1-His protein. It is noteworthy that ∆H1, an N-terminally truncated version of VIPP1-His (Fig. 1; see below), showed almost no signals to the same degree as BSA, revealing the requirement of the N-terminal region of VIPP1 for GTP binding (see below).
Critical Regions of VIPP1 Necessary for GTP Hydrolysis Activity
Because VIPP1 has no canonical nucleotide-binding domain, we investigated protein regions that are crucially important for GTPase activity. The VIPP1 protein of Arabidopsis putatively consists of seven α-helices that show amphipathic features (Zhang and Sakamoto, 2015). In an earlier study, we demonstrated that the C terminus of VIPP1 negatively regulates the association of VIPP1 protein particles and that it is necessary for establishing the tolerance of Arabidopsis against heat shock stress (Zhang et al., 2016b). It was also reported that the C-terminal region of cyanobacterial VIPP1 (IM30 in Synechocystis sp.PCC6803) is necessary for membrane binding (Hennig et al., 2017). However, the N terminus is a prerequisite of the function of VIPP1 through oligomerization (Aseeva et al., 2007). It is also involved in lipid binding (Otters et al., 2013). To detect the presence of any critical domain for the GTP hydrolysis activity, we prepared either C-terminally or N-terminally truncated VIPP1-His proteins, ∆H1, ∆H5-7, and ∆H7 (Fig. 1, A and C), as described in an earlier study (Otters et al., 2013). The results of our in vitro assay demonstrated that the C-terminally truncated versions of VIPP1 proteins (∆H5-7 and ∆H7) were all capable of releasing nearly wild-type levels of Pi, despite the lack of 40 and 71 amino acid residues (Fig. 1A). By contrast, the Pi release by ∆H1 was reduced significantly to approximately 13% of that of wild-type VIPP1-His (Fig. 5). These results demonstrate clearly that the N-terminal α-helix, rather than the C-terminal region, is crucially important for GTP hydrolysis.
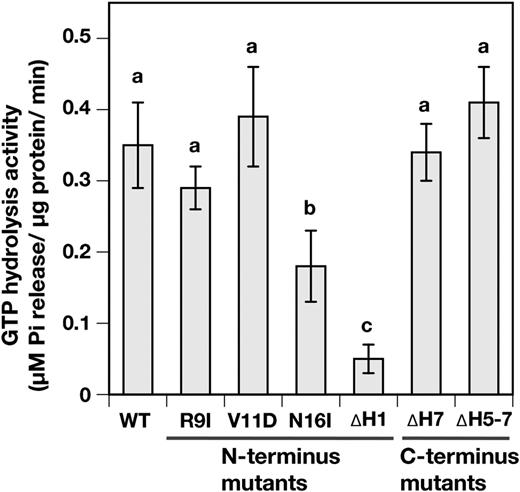
Analyses for the important domains and amino acids on VIPP1 for GTP hydrolysis activity. The GTP hydrolysis activities of wild type (WT) and N-terminally and C-terminally mutated VIPP1-His proteins were analyzed. All analyses were conducted under standard conditions (see “Materials and Methods”). Bar graphs and error bars show means and sd, respectively, of results obtained from four to six independent experiments. Different letters denote significant differences between GTP hydrolysis activity of His-tagged proteins (Student’s t test, for which P < 0.05 was inferred as significant).
Next, we specifically examined the N-terminal α-helix and attempted to compromise GTP hydrolysis activity by introducing amino acid substitutions. Our search for any characterized domain feature failed. As a consequence, based on multiple alignment analysis and helical wheel projection (Supplemental Fig. S4), we randomly selected several conserved amino acids positioned on hydrophobic and hydrophilic surfaces of this region. Of these mutated VIPP1 proteins, we were able to prepare single-amino acid-mutated VIPP1 proteins of three kinds: R9I, V11D, and N16I (Fig. 1, B and C). We observed that N16I-His exhibited a marked decrease of GTP hydrolysis activity (Fig. 5) to approximately 50% of the wild-type level. By contrast, no significant alteration in activity was detected in the other two.
Oligomerization State of Mutated VIPP1/N16I-His
The N terminus-mediated oligomerization (Otters et al., 2013) is a prerequisite for the proper function of VIPP1 (Aseeva et al., 2007). Therefore, we questioned whether GTP hydrolysis activity was related to the oligomerization state of our recombinant proteins. The decreased GTP hydrolysis activity in N16I might correlate with the altered oligomerization state differently from VIPP1-His. We first used Suc density gradient centrifugation to separate VIPP1 complexes. Subsequently, the fractions obtained were subjected to immunoblot analysis. VIPP1-His was detected in the fractions with high densities of Suc. By contrast, ∆H1 remained at the top of the gradient (Fig. 6A), confirming the crucial role of the first helix to form the VIPP1 complex (Otters et al., 2013). N16I was detected in the fractions similar to those of VIPP1-His. However, it migrated slightly to an upper phase (Fig. 6B). The theoretical concentrations of Suc in the fractions where VIPP1-His (fraction 22) and N16I (fraction 21) were detected most abundantly were 1.45 and 1.4 m, respectively. Moreover, VIPP1-His was distributed broadly from fraction 20 to fraction 25, whereas N16I appeared to be condensed in fraction 21, suggesting a more rigid structure of N16I.
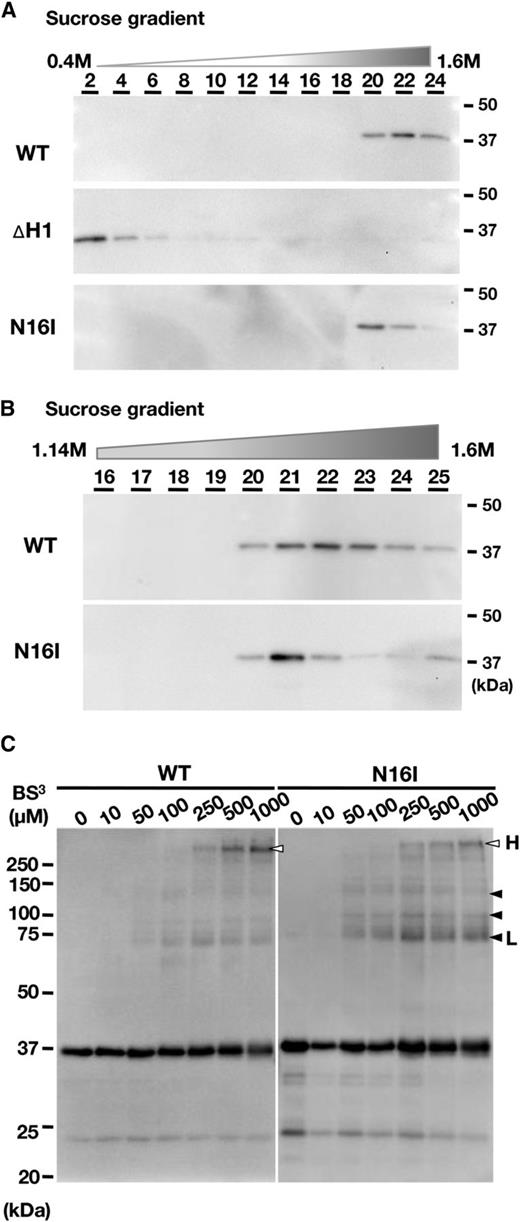
Evaluation of the oligomerization state of wild-type (WT) and mutated VIPP1-His proteins. A and B, Analyses for the size and density of the VIPP1 complex by linear Suc density gradient centrifugation (0.4–1.6 m). Among the 25 obtained fractions, fractions of an even number (A) or bottom half (B) were subjected to immunoblot analyses with the specific antibody against VIPP1. C, Chemical cross-linking of recombinant proteins of wild-type VIPP1 and N16I. Recombinant proteins were treated with BS3 and then subjected to SDS-PAGE followed by immunoblot analyses with a specific antibody against VIPP1. Arrowheads and letters are as described in “Results.”
We next attempted to analyze the oligomerization state using a chemical cross-linker, bis[sulfosuccinimidyl]suberate (BS3), according to an earlier study that cross-linked VIPP1 on chloroplast envelopes successfully (Aseeva et al., 2004). As expected, several chemical cross-linking products appeared when VIPP1-His and N16I preparations were used (Fig. 6C). The cross-linked products appeared to increase proportionally to the concentration of BS3. A low concentration of BS3 produced mainly low-molecular-mass products (shown with black arrowheads in Fig. 6C), whereas high-molecular-mass products (white arrowheads) were detectable when more than 250 mm BS3 was added. It is particularly interesting that N16I exhibited a cross-linked product of about 75 kD (black arrowhead L in Fig. 6C) even at the lower concentration, which was barely detectable in VIPP1-His. However, the largest product of ∼250 kD (white arrowhead H) more efficiently appeared in VIPP1-His than in N16I at greater than 250 µm. Overall, large products were readily detected in VIPP1-His, whereas small products were abundant in the N16I reaction mixture. Quantification of the signals corresponding to H and L on the lanes of 1,000 µm BS3 for both wild-type VIPP1-His and N16I indicated that the H:L ratios were calculated as 5.13 ± 0.94 and 1.88 ± 0.6, respectively (means ± sd from three independent experiments), confirming the efficient appearance of high-molecular-mass products in the wild-type preparations. The increase in small products of N16I showed that small or destabilized complexes are more abundant than those of the wild type. However, N16I protein was not detected in fractions of low density on the Suc gradients (Fig. 6A). Therefore, small complexes or destabilization may be a minor part. Chemical cross-linking by BS3 is dependent on the distance between amino groups (-NH3) exposed to the surface of the complex; distinct cross-linking patterns show differences not only in the number of cross-linkable protein molecules in oligomers but also in the interaction manner between proteins. Together, these results demonstrated that N16I forms supercomplexes in some irregular modes compared with wild-type VIPP1, which correlated with GTP hydrolysis activity (Fig. 5).
DISCUSSION
VIPP1/PspA Possesses GTPase Activity
Although multiple functions of VIPP1 have been proposed in earlier reports (Zhang and Sakamoto, 2015), it is conceivable that VIPP1 acts primarily in membrane fusion and remodeling through its feature of lipid binding (Otters et al., 2013; Heidrich et al., 2016; Hennig et al., 2017; McDonald et al., 2017; Saur et al., 2017). Supporting the role of VIPP1 in chloroplast membrane protection, overexpression of VIPP1-GFP in Arabidopsis was shown to produce increased tolerance against high temperature and improved cotyledon development under photooxidative damage (Zhang et al., 2016a, 2016b). For this study, we postulated that VIPP1 possesses GTPase-like traits similar to those of other membrane fusion proteins. GTPases constitute a large family and play important biological functions in membrane remodeling and traffic (Mizuno-Yamasaki et al., 2012). We demonstrated that (1) VIPP1 possesses Mg2+-dependent GTP hydrolysis activity (Figs. 3 and 4A), (2) GTP hydrolysis occurred on neither GDP nor GTPγS (Fig. 4A), (3) GTP hydrolysis activity is competitively inhibited by GTPγS (Fig. 4, B and C), and (4) VIPP1 can bind GTP through the N-terminal region (Fig. 4D). The calculated k cat of the GTP hydrolysis activity was in the range of that of dynamin, one of the protein families exhibiting GTPase activity. Collectively, VIPP1 can bind GTP and hydrolyze it at the γ-position at a certain rate. We conclude that VIPP1 possesses GTPase activity.
In general, GTP binding and hydrolysis on GTPases take place in the highly conserved G domain, which consists of four conserved motifs designated as G1 (P-loop), G2 (switch 1), G3 (switch 2), and G4. Although tubulin and FtsZ do not contain typical G domains, they have conserved amino acid residues for GTP binding and hydrolysis (Scheffers et al., 2002). We attempted alignment analyses between known GTPase proteins and VIPP1, which revealed no common sequence on the VIPP1 protein, even on the N terminus that is necessary for GTP hydrolysis activity, as described above. Although further analyses are required, we demonstrate that PspA has GTP hydrolysis activity similar to that of VIPP1. These results together imply that VIPP1/PspA belongs to a new class of untypical GTPases.
Properties of GTP Hydrolysis Activity in VIPP1
GTPases require divalent cation (usually Mg2+) to coordinate the phosphates of GTP molecules to precede efficient catalysis. The metal ion usually bridges between the oxygen atoms of the terminal two phosphates of nucleotides and oxygens from bulk solvent or protein side chains (Bourne et al., 1991). In some cases, GTPases show specificity against the divalent cation that performs this coordination. For example, the GTPase activity of FtsZ was abolished completely in the presence of Ca2+ instead of Mg2+ (Marrington et al., 2004). The GTP hydrolysis activity of VIPP1 appears to have this characteristic: the absence of Mg2+ induced a significant loss of GTP hydrolysis activity, although the addition of Ca2+ had no effect on VIPP1 in hydrolyzing GTP (Fig. 4A). In the case of FtsZ, GTP and Mg2+ can form a macrochelate conformation of GTP-Mg2+, which is the productive pathway leading to GTP hydrolysis (Marrington et al., 2004). The GTP hydrolysis activity of VIPP1 might be a similar case.
In our experiment, GTP hydrolysis activity was assayed in the condition of 2.5 mm Mg2+. Schröppel-Meier and Kaiser (1988) reported that total Mg2+ concentration in spinach (Spinacia oleracea) chloroplasts is around 13 to 18 mm. The stroma Mg2+ concentration also was estimated as being on the order of 5 mm (Portis and Heldt, 1976). According to these reports, the Mg2+ concentration in this study can occur in vivo. As an essential cofactor of GTPase, Mg2+ has been shown to be necessary for both guanine nucleotide binding and GTP hydrolysis. For example, in the presence of Mg2+, Ras, a small GTPase superfamily protein, exhibits an extremely high binding affinity to the guanine nucleotides, with a dissociation constant of approximately subnanomolar concentration (Feuerstein et al., 1987; John et al., 1990, 1993). The data in this study still cannot discriminate the precise role of Mg2+ in the GTP hydrolysis activity of VIPP1. For that reason, more experiments are necessary to characterize the role of Mg2+ in the GTP binding and hydrolysis by VIPP1.
Interrelation between Oligomerization and GTP Hydrolysis Activity
Our in vitro assay of truncated VIPP1 proteins revealed the essential role of the first 20 amino acids at the N terminus rather than the C terminus for GTP hydrolysis activity (Fig. 5). The dispensability of the C terminus has been implicated previously (Zhang et al., 2016a). It correlates well with the fact that the ancestral PspA in bacteria does not carry the C-terminal region (corresponding to Lys-222 to Phe-259 of Arabidopsis VIPP1) yet presents an ability to hydrolyze GTP (Fig. 3C). These results led us to infer that the oligomer formation of VIPP1 through its N-terminal α-helix might be crucially important. Among the point mutations of three kinds, N16I significantly reduced the GTP hydrolysis activity of VIPP1 (Fig. 5) and modified the complex formation state (Fig. 6). Consequently, oligomerization appeared to be necessary for the GTP hydrolysis activity. The reaction center for GTP binding and hydrolysis on VIPP1 might be structured by oligomerization similarly to that in multimeric FtsZ, of which the reaction site together with the nucleotide-binding site were formed on adjacent monomers (Scheffers et al., 2002). Separation of the wild-type VIPP1 supercomplex on a Suc density gradient showed a broad distribution (Fig. 6B), probably reflecting various oligomers with 10- to 18-fold internal symmetric structure (Fuhrmann et al., 2009; Hennig et al., 2017; Saur et al., 2017). By contrast, N16I was condensed in only a few fractions, suggesting a more rigid structure and/or a smaller variety of oligomers (Fig. 6B). As discussed in an earlier report (Saur et al., 2017), structural flexibility is probably necessary for the proper function of VIPP1, which also might involve GTP hydrolysis activity and which might result in a dynamic assembly/disassembly in chloroplast (Zhang et al., 2012).
Based on the partial crystal structure of PspA (Osadnik et al., 2015), at least one N-terminal helix of a monomer, out of a tetramer basic ring block in an oligomer, is located at the periphery of a large ring structure and, therefore, is free from interacting with the membrane (Saur et al., 2017). Therefore, although the N-terminal helix region of VIPP1/PspA is involved in the binding of lipids and membranes (Otters et al., 2013; McDonald et al., 2017), GTP molecules can access this region on an oligomer of the ring structure. The residue Asn-16 can be part of the reaction site of GTP hydrolysis. Because VIPP1 includes neither the canonical GTPase domain nor the tubulin/FtsZ-type G-box, it is difficult to find a key domain from its amino acid sequence. Although the electron microscopic study unveiled a fine whole structure of the IM30 oligomer, the exact assembly of tetramer basic building blocks remains to be determined (Saur et al., 2017). Further structural analyses must be conducted to identify GTP-binding site(s) on oligomer. In any case, our results revealed an interrelation between VIPP1 oligomerization and its GTP hydrolysis activity.
Possible Physiological Role of the GTP Hydrolysis Activity
We calculated the V max of the GTP hydrolysis of VIPP1 as 12 min−1 (Fig. 4C). This value is not only in the range of those from the dynamin family, it is also higher than that of already reported chloroplast-targeted GTPase proteins: approximately 0.5 min−1 for RbgA (Jeon et al., 2017) and 0.6 to 1.2 min−1 for chloroplastic FtsZ2 (Shaik et al., 2018). Consequently, it is reasonable to consider that the GTP hydrolysis activity of VIPP1 is sufficient in planta. Further study must be conducted to elucidate how the GTP hydrolysis activity contributes to the function of VIPP1. Among typical GTPases, the ones involved in the action of biological membranes (e.g. Arf/Sar and Rab) often show conformational changes associated with the nucleotide binding and nucleotide hydrolysis cycle, which is coupled with membrane association and dissociation (Mizuno-Yamasaki et al., 2012). However, VIPP1 might not be in this case. According to an earlier study, highly ordered oligomers and tetramers are able to bind to membranes (Heidrich et al., 2016). Because ∆H1, which forms a trimer or tetramer (Otters et al., 2013), shows neither binding nor hydrolysis of GTP, it is unlikely that the GTP hydrolysis activity is necessary for membrane binding. However, the polymerization of tubulin and FtsZ is driven by GTP (Battaje and Panda, 2017). It is particularly interesting that the dynamics of tubulin and FtsZ in vivo resembles the dynamic assembly/disassembly visualized by VIPP1-GFP in response to environmental stress (Zhang et al., 2012, 2016a). Conceivably, the GTP hydrolysis activity can drive the dynamic movement of VIPP1 on the chloroplast envelope and can regulate the membrane integrity maintenance function.
CONCLUSION
We demonstrated that the recombinant VIPP1-His protein has GTP hydrolysis activity that is comparable to those of typical GTPases. We concluded that VIPP1 is a novel type of GTPase acting in chloroplast membrane fusion/remodeling. Although the mechanism by which the activity is controlled and the manner in which it contributes to the physiological function of VIPP1 remain unclear, precise oligomerization through the N terminus is involved in the GTP hydrolysis activity.
MATERIALS AND METHODS
Preparation of His-Tagged Recombinant Proteins
The expression vectors for wild-type and truncated Arabidopsis (Arabidopsis thaliana) VIPP1 (∆H1, ∆H7, and ∆H5-7) were kindly provided by Dr. Ute C. Vothknecht (Otters et al., 2013). Those for mutated VIPP1 (R9I, V11D, and N16I) were prepared using a mutagenesis kit (QuickChange II XL Site-Directed Mutagenesis Kit; Agilent Technologies) based on the vector of wild-type VIPP1. To prepare His-tagged PspA, a PCR fragment of the PspA gene containing material from the initiation codon to immediately upstream of the termination codon (+1 to +677, counted from A of the initiation codon) was obtained by using pEC1 plasmid (Engl et al., 2009) and then introduced to pTrc-His-2c (Invitrogen) in the frame after digestion by NcoI and SnaBI. Primers for the mutagenesis of VIPP1 and the PCR of PspA are summarized, respectively, in Supplemental Tables S1 and S2. The expression vector of DPD1-His was described previously (Matsushima et al., 2011). All His-tagged recombinant proteins were obtained as described earlier (Matsushima et al., 2011) with a slight modification. Overexpression of proteins was conducted at 27°C in Escherichia coli strain BL21(DE3)pLysS. The cells carrying overexpressed protein were pelleted, frozen in liquid nitrogen, and then stored at −30°C until starting Ni2+-affinity purification. The following procedures (Ni2+-affinity purification, exchange of buffer, concentration of protein, and preparation of glycerol-containing stock solution) were done within 1 d (typically 10 h) to avoid the reduction of GTP hydrolysis activity. The soluble fraction extracted from E. coli cells was mixed with 0.5 mL of Ni-NTA agarose (GE Healthcare) and then incubated at 4°C for 2 to 2.5 h. After the collection of flow through as an unbound fraction, the Ni-NTA column was washed with 10 mL of 25 mm imidazole-containing buffer (25 mm imidazole, 20 mm Tris-HCl [pH 8], and 500 mm NaCl). Subsequently, proteins bound to the Ni-NTA column were stepwise eluted by 0.5 mL of each imidazole-containing buffer (50–1,000 mm, 20 mm Tris-HCl [pH 8], and 500 mm NaCl). Based on elution profiles (Supplemental Fig. S1), to minimize the amount of impurities, 500 to 1,000 mm fractions were used for further preparation of VIPP1-His, PspA-His, and mutated VIPP1-His proteins, except for ∆H1-His, while 200 to 250 mm imidazole fractions were collected for ∆H1-His and DPD1-His. After purification, the imidazole-containing buffer was exchanged with 2× VIPP1 storage buffer (100 mm HEPES-NaOH [pH 7.5] and 100 mm KCl) using a gel filtration column (midiTrap G-25; GE Healthcare). The obtained fraction containing the desired protein was confirmed by SDS-PAGE and following Coomassie Brilliant Blue staining (see below), then subjected to centrifugation (7,500g at 4°C) for more than 2 h with AmiconUltra-4 (10K; Merck Millipore) to concentrate the recombinant protein. The concentration of protein was estimated using the Bradford Protein Assay Kit (Bio-Rad Laboratories). When the concentration was more than 1 µg µL−1, the protein solution was diluted to 1 µg µL−1 with 2× VIPP1 storage buffer and then mixed with an equal amount of glycerol to make 0.5 µg µL−1 stock solution. The protein solution of low yield (less than 1 µg µL−1) was mixed with an equal amount of glycerol without further dilution. In all cases, the concentration of glycerol stock was kept at more than 0.2 µg µL−1 to avoid the reduction of GTP hydrolysis activity. The stock solutions were stored at −30°C and used within 2 weeks for further analyses.
GTP Hydrolysis Activity
Measurements of GTP hydrolysis activity were taken using a GTPase assay kit (Innova Biosciences), a dye-based detection system with PiColorLock, which is described as “superior malachite green reagent highly suppressing non-enzymatic GTP hydrolysis” in the manual provided. We conducted all the reactions with a 200-µL scale according to the manual. The reaction mixture contained 50 mm Tris-HCl (pH 7.5), 2.5 mm MgCl2, 0.5 mm GTP, and 0.5 µg of His-tagged recombinant protein. Reaction mixtures were incubated at 37°C for 30 min and then immediately cooled in ice water for 1 min. Subsequently, each reaction was terminated by adding the appropriate amount of PiColorLock at room temperature. The released Pi was quantified by measuring absorption at 635 nm according to standard solutions. Because the reaction mixture without only recombinant proteins often showed a pale green color, its absorption was subtracted as background in each experiment. Unless described specifically, the composition of a reaction mixture and the incubation time were used as standard conditions.
Dot-Blot Assay
Interactions between VIPP1-His and GTP were assessed using a dot-blot assay as described previously (Diekmann and Hall, 1995) with some modifications. In brief, 5 μg of wild-type VIPP1, ƊH1, or BSA was spotted in a volume of 5 μL onto nitrocellulose membranes. It was then air dried for 20 min at room temperature. The membranes were incubated in blotting buffer (100 mm Tris-HCl [pH 7.5], 1 mm MgCl2, 25 mm KCl, and 100 mm NaCl) for at least 1 h at room temperature. After supplementation with 20,000 μCi of [α-32P]GTP (specific activity, 3,000 Ci mmol−1), the incubation was continued at room temperature for 1 h. The membranes were washed with washing buffer (the blotting buffer supplemented with 0.2% (w/v) Tween 20) three times and then air dried. The signals were detected using a bioimaging analyzer (BAS1000; Fuji Photo Film).
SDS-PAGE and Immunoblot Analysis
Either aliquots of soluble proteins obtained from E. coli cells or purified recombinant proteins were solubilized by incubation at 75°C for 5 min in the presence of 2% (w/v) SDS and 0.1 m DTT. The protein samples were centrifuged for 1 min at greater than 20,000g and then subjected to SDS-PAGE with 12.5% (w/v) polyacrylamide gels. The proteins in gel were visualized subsequently by staining with CBB Stain ONE (Nacalai Tesque). In the case of immunoblot analyses, the separated proteins were blotted electrophoretically onto PVDF membranes and probed with polyclonal antibodies against VIPP1. Signals were visualized using a chemiluminescence reagent (Luminata Crescendo Western HRP Substrate; Merck kGaA) and detected using ChemiDoc analyzer (Bio-Rad Laboratories).
Chemical Cross-Linking
The chemical cross-linking reactions were conducted at 200-µL scale. BS3 was purchased from Thermo Fisher Scientific. Each mixture contained 0.5 µg of VIPP1-His proteins (wild type or N16I), 50 mm HEPES-NaOH (pH 7.5), and BS3 (0–1,000 µm). The reaction mixtures were incubated at 25°C for 30 min and then immediately cooled in ice water. Subsequently, 10 µL of 1 m Tris-HCl (pH 7.5) was added to the reaction mixtures. It was then incubated further at 25°C for 15 min to terminate cross-linking reactions. Aliquots of the reaction mixtures were subjected to SDS-PAGE and immunoblot analyses, as described above.
Suc Density Gradient Centrifugation
After 1 µg of recombinant proteins was diluted to 100 µL with 50 mm HEPES-NaOH (pH 7.5), it was overlaid on a continuous Suc density gradient (0.4–1.6 m) containing 50 mm HEPES-NaOH (pH 7.5) and 50 mm KCl. Each gradient was centrifuged at 30,000 rpm for 15 h at 4°C (SW50.1 rotor; Beckman Coulter). After centrifugation, the gradients were fractionated into 25 fractions (200 µL each) from top to bottom. An appropriate aliquot of the fractions was subjected to SDS-PAGE and subsequent immunoblot analyses with specific antibody against VIPP1 as described above.
Accession Numbers
Sequence data for VIPP1 can be found in the Arabidopsis Genome Initiative database under accession number At1g65260.1. Information for the nucleotide sequence of PspA can be found in the National Center for Biotechnology Information database under accession number NC_000913.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Imidazole elution profiles in Ni2+-affinity purification of His-tagged recombinant proteins.
Supplemental Figure S2. Confirmation of the majority of GTP hydrolysis-active proteins in solutions used for this study.
Supplemental Figure S3. Preparation and GTP hydrolysis activity of His-tagged PspA and DPD1 proteins purified from E. coli.
Supplemental Figure S4. Schematic illustration of the VIPP1 N-terminal region.
Supplemental Table S1. PCR primers used for mutagenesis of VIPP1 proteins.
Supplemental Table S2. PCR primers used for preparation of expression vectors of PspA-His protein.
ACKNOWLEDGMENTS
We thank Ute C. Vothknecht for providing the antibody against VIPP1 and some constructs used for this study. The plasmid DNA carrying pspA was kindly provided by Martin Buck of Imperial College. We also thank Dr. Kenji Nishimura for valuable assistance with the preparation of His-tagged recombinant proteins.
LITERATURE CITED
Author notes
This work was supported by the Core Research for Evolutional Science and Technology, Japan Science and Technology Agency (to W.S.), KAKENHI grants from the Japan Society for the Promotion of Science (16H06554 and 17H03699 to W.S.), the Oohara Foundation (to W.S.), and the National Natural Science Foundation of China (31660062 to L.Z.).
These authors contributed equally to the article.
Current address: School of Life Science and Technology, Inner Mongolia University of Science and Technology, Baotou 014010, China.
Address correspondence to [email protected].
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Wataru Sakamoto ([email protected]).
L.Z. and W.S. conceived the original research plans; N.O and L.Z. conducted experiments; W.S. supervised experiments; N.O., L.Z., and W.S. analyzed data and wrote the article.
Articles can be viewed without a subscription.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}