





Abstract
Although covalent nucleotide modifications were first identified on the bases of transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs), a number of these epitranscriptome marks have also been found to occur on the bases of messenger RNAs (mRNAs). These covalent mRNA features have been demonstrated to have various and significant effects on the processing (e.g. splicing, polyadenylation, etc.) and functionality (e.g. translation, transport, etc.) of these protein-encoding molecules. Here, we focus our attention on the current understanding of the collection of covalent nucleotide modifications known to occur on mRNAs in plants, how they are detected and studied, and the most outstanding future questions of each of these important epitranscriptomic regulatory signals.
Introduction
Since the discovery of nucleotide modifications on numerous RNA molecules, an additional layer of post-transcriptional regulation is now being understood as the epitranscriptome. The growing breadth of annotated RNA modifications (Boccaletto et al. 2018) has revealed a complex and dynamic chemical landscape with a powerful influence over transcript processing and fate. Despite not being the most modified RNA species—those being transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs)—covalent chemical modification of nucleotides in messenger RNAs (mRNAs) are now understood to be essential features of post-transcriptional RNA regulation. Following the initial detection of modification within mRNA in mammals (Desrosiers et al. 1974) and later plants (Kennedy and Lane 1979; Nichols and Welder 1981), a new and rapidly growing field studying the epitranscriptome of mRNA has emerged; not without biases. In Arabidopsis (Arabidopsis thaliana), as an example, N6-methyladenosine (m6A) dominates as not just the most abundant internal mRNA modification but the most well-studied and annotated to date. Fundamental as it is, the gap in knowledge of the other RNA modifications now known to be present on mRNA molecules is stark, considering a given RNA is more than likely harboring a combination of multiple dynamic modifications within its life span.
Here, we focus on the multiple modifications now known to be present in plant mRNA molecules, highlighting understudied marks and recent findings for those that are more well-studied. A brief introduction, current understanding of plant systems, and available methodology used to survey each of the following modifications are reviewed for 5′-nicotinamide adenine diphosphate caps (NAD+ caps), 5-methylcytosine (m5C), N1-methyladenine (m1A), 3-methylcytosine (m3C), m6A, pseudouridine (Ψ), and 7-methylguanosine (m7G). A comprehensive list of current transcriptome-wide methodologies to catalog the highlighted modifications is presented in Supplemental Table S1.
5′-nicotinamide adenine diphosphate caps (NAD+ caps)
The most recently discovered modification discussed here, the 5′ addition of nicotinamide adenine dinucleotide (NAD+) (Fig. 1), was first identified in 2009 during profiling of unknown small molecule RNA conjugates in bacterial species (Chen et al. 2009; Kowtoniuk et al. 2009). Further experimentation revealed that NAD+ is attached to the 5′-end of RNA in a similar manner as the canonical eukaryotic m7G cap through the development of NAD+ capture sequencing, a chemoenzymatic-coupled next-generation sequencing-based method (Cahová et al. 2015). Following its initial characterization in prokaryotic RNA (Frindert et al. 2018), NAD+-capped RNAs (NAD-RNAs) were later described in yeast (Walters et al. 2017), human kidney tissue (Jiao et al. 2017), and plants (Zhang et al. 2019a). With its recent identification across biological transcriptomes, the influence of the NAD+ cap on transcript stability was subsequently interrogated. Initial studies reported that NAD-RNAs displayed increased stability in prokaryote transcriptomes (Bird et al. 2016). Conversely, studies in eukaryotic systems revealed connections to decapping-dependent mRNA decay in yeast (Jiao et al. 2017) and plant transcriptomes (Kwasnik et al. 2019; Wang et al. 2019; Zhang et al. 2019a; Yu et al. 2021a, 2021b). Current literature suggests that the connection between NAD+ cap presence and RNA half-life likely differs across kingdoms and conditions. Here, we will focus our discussion on the recent findings in plant systems and direct readers here (Julius and Yuzenkova 2019; Li et al. 2021; Wiener and Schwartz 2021; Doamekpor et al. 2022) for comprehensive reviews focused on NAD+ capping in other systems. Overall, the current literature in plant systems suggests a critical role for NAD+ caps in regulating mRNA stability, by acting as a destabilizing mark.
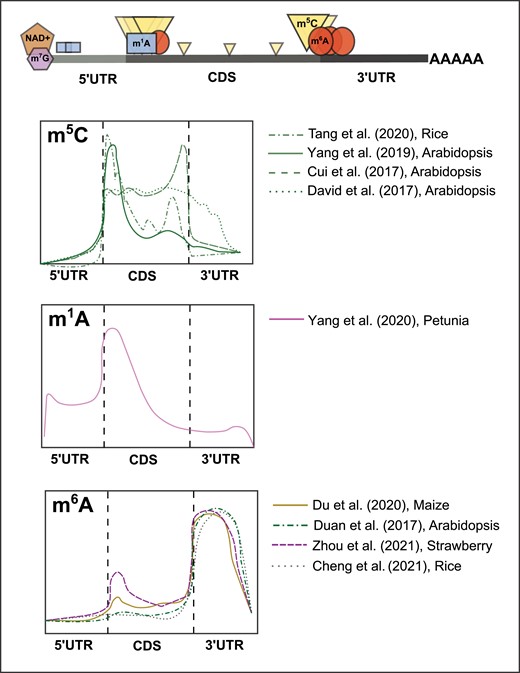
Previously detected RNA covalent modification profiles on plant mRNAs. Representation of gene structure showing typical locations of detected modifications (upper panel), and graphical representation of transcriptome coverage plots (lower panels) from previous experiments depicting the relative positional enrichments of the m5C, m1A, and m6A mRNA modifications in plant systems. Relative peak height/intensity are graphical interpretations from cited sources and do not represent actual data points.
The first reported detection of NAD-RNAs in a plant used copper-catalyzed azide–alkyne cycloaddition tagging followed by sequencing (CuAAC-NAD-seq), termed NADcapture-seq, for transcriptome-wide identification and relative quantification of this mark. More specifically, this methodology utilizes enzyme-catalyzed “click chemistry” to label NAD+ capped RNAs with a biotin molecule. These transcripts were then isolated using streptavidin-mediated pull-down and subsequently sequenced using Oxford Nanopore technology (Zhang et al. 2019a). This approach identified ∼2,000 polyadenylated NAD-RNAs that included 1,980 mRNAs, 12 noncoding RNAs (ncRNAs), and 8 long noncoding RNAs (lncRNAs) in 12-d-old Arabidopsis seedlings. Subsequent transcript-specific analyses of the 210 most highly abundant NAD-RNAs revealed that, on average, ∼1% of their total transcript population were found in a NAD+ capped form, with the highest levels of observed population NAD+ capping being ∼5% (Zhang et al. 2019a). A similar study compared NAD-RNAs identified using NADcapture-seq in seedling (5,687) and inflorescence tissue (6,582) and found that the majority of identified NAD-RNAs were encoded by the nuclear genome, some by the mitochondrial genome, and none by the chloroplast genome (Wang et al. 2019). Further analyses demonstrated that NAD-RNAs were mostly protein-coding mRNAs and were often associated with actively translating polysomes. Together, these results suggested a possible direct connection between NAD+ capping and translation (Wang et al. 2019). A more recent study used a variant of NADcapture-seq (Cahová et al. 2015) to characterize NAD-RNAs in Arabidopsis unopened flower buds by comparing NAD+ capped mRNAs in dxo1 mutant plants that lack the known decapping enzyme DXO1 to wild-type Col-0 (Yu et al. 2021b). This study identified 1,033 and 358 NAD+ capped transcripts, respectively, in dxo1 compared to Col-0 plants, confirming DXO1's role in active removal of NAD+ caps. Transcript analysis showed 92.1% of detected NAD-RNAs to be mRNA, as well as snoRNAs and other ncRNA species. The same group profiled the NAD+ capped transcriptome following abscisic acid (ABA) treatment and found significant remodeling of the NAD+ capped transcriptome, primarily independent of DXO1 function, suggesting condition- and tissue-specific addition of this mark (Yu et al. 2021b). Lastly, degradome profiling of NAD-RNAs in combination with smRNA-seq revealed that NAD+ capped transcripts tend to be more unstable and prone to RDR6-mediated post-transcriptional small RNA processing, which is likely to free the incorporated NAD+ caps in the absence of deNADing functionality (Yu et al. 2021b). More recently, strain-promoted azide–alkyne cycloaddition (SPAAC)-NAD-seq, a copper-free NAD-seq method, in combination with m7G depletion was developed for higher sensitivity and to eliminate false positive identification (discussed in more detail below). SPAAC-NAD-seq, a modification of CuAAC-NAD-seq, was adapted to Arabidopsis RNA to reveal a total of 5,642 NAD-RNAs following m7G depletion (Hu et al. 2021a). Initial experimentation has revealed tissue- and context-dependent signatures of this mark (Wang et al. 2019; Yu et al. 2021b), specifically during stress, with complementary work on decapping enzymes affirming its role in both stress and RNA regulation through RNA destabilization (Kwasnik et al. 2019; Pan et al. 2020; Yu et al. 2021b; Ma et al. 2022).
Methods and future directions: NAD+
Despite its recent discovery as an RNA modification, NAD+ capping has been detected and successfully mapped to the Arabidopsis transcriptome (Wang et al. 2019; Zhang et al. 2019a; Hu et al. 2021a; Yu et al. 2021b) using multiple variations of NAD+ capture-seq (Cahová et al. 2015; Winz et al. 2017) (Fig. 2). The resulting NAD+ transcriptome profiles vary significantly enough to note and discuss the experimental differences for future experimental consideration. In the original reaction, enzyme catalysis with adenosine diphosphate-ribosyl cyclase (ADPRC) of NAD+ to replace the nicotinamide moiety with an alkynyl alcohol enables the modified NAD+ cap to be biotinylated, bead-purified, and sequenced. Although successful, the use of copper ions for biotinylation causes RNA degradation, which can make it difficult to detect mRNAs and other low-abundance RNA species compared with abundant noncoding populations. A recent study addressed this issue and the other major issue of specificity through the implementation of SPAAC-NAD-seq (mentioned above) (Hu et al. 2021a). The SPAAC-based reaction does not require a Cu2+ catalyst and reduces potential RNA degradation. Going further, this group performed SPAAC-NAD-seq with and without ADPRC, the negative sample serving as a more specific background. Another notable improvement of this approach is the addition of an antibody-based depletion step for the m7G modification to remove the contamination of m7G-capped RNAs due to their potential enzymatic and chemical reactivity to the SPAAC reaction (Holstein et al. 2015). This combinatorial approach not only enables the detection of novel less abundant NAD-RNAs, but it also refines previous false-positive NAD+ calls, likely due to m7G cap contamination (Hu et al. 2021a). Moving forward, this focus on maintaining sample RNA stability and NAD+ cap specificity needs to be the future of standard practice and an example for other sequencing-based detection methods used to identify various RNA modifications. Additionally, designing and implementing experiments in mutant backgrounds of deNADing enzymes (hyper-NAD+ capped plants), such as dxo1 mutants (Yu et al. 2021b), provides a means to improve specificity and confidence in calling bona fide NAD-RNAs. A major research focus for the future of the NAD+ capping field is identifying the machinery and corresponding mechanism by which this modification is added to plant transcripts. Only by uncovering this information can a more holistic picture of the importance of NAD+ capping in the context of the plant cell be achieved. Furthermore, the identification of the factors will also allow mechanistic insights into tissue- and condition-specific NAD+ capping, and how this activity is balanced with available NAD+/NADH pools. In general, the NAD+ RNA cap stands as an example of the potential of plant systems to be the driving force to progress work on this and other RNA modifications. Specifically, in a relatively short time period, both the methodology and biochemistry of detection as well as determining the biological significance of the mark are advancing, with research in plant systems leading the way. Thus, work on this modification provides a model for plant researchers for targeting further investigation of the understudied epitranscriptome marks mentioned herein.
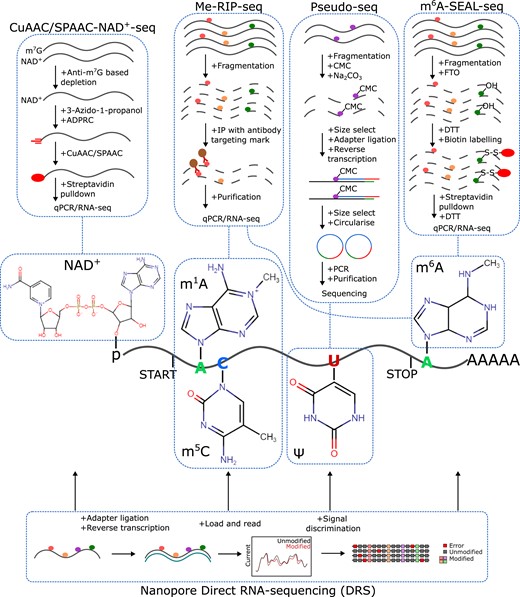
Detecting post-transcriptional mRNA modifications in plants. Key steps in a few of the approaches for investigating covalent nucleotide modifications in plant mRNAs include NAD+ capping (NAD+), m1A, m5C, pseudouridine (Ψ), and m6A. These approaches include the click chemistry-based NAD+ -seq, the antibody-based immunocapture approaches for numerous modifications (Me-RIP-seq), the chemical-based probing of pseudouridine (Pseudo-seq), and the demethylase-mediated pulldown of m6A sites (m6A-SEAL-seq). Continual developments in nanopore-seq offer the potential to simultaneously detect numerous mRNA modifications with a single experimental approach. It is notable that this sequencing technology is already being used in studies of various RNA modifications as noted throughout the text herein.
5-methylcytosine (m5C)
Discovered in the 1970s, m5C was identified in smaller quantities as compared to m7G and m6A on eukaryotic RNAs (Dubin and Taylor 1975). Modern liquid chromatography-mass spectrometry (LC-MS) has confirmed the additional methyl group on carbon-5 in cytosine pyrimidine rings, both in DNA and RNA species across kingdoms (Motorin et al. 2010). The distribution of m5C appears to display species- and cell-type-specific enrichment across mRNAs. For instance, human cells show striking enrichment at the translation start codon (Yang et al. 2017), which was also observed, to a lesser extent, in mice (Amort et al. 2017). Interestingly, mouse brain-derived RNAs also showed enrichment in the 3′ UTR (Amort et al. 2017). In plants, multiple studies portray conflicting m5C enrichment patterns, however, the most consistent enrichment is observed around the translation start codon and across the coding sequence (CDS) (Cui et al. 2017; David et al. 2017; Yang et al. 2019; Tang et al. 2020) (Fig. 1). Most of the current research conducted on m5C has focused on its deposition, removal, and function in rRNA and tRNA species, specifically in mammalian systems, covered in detail here (Sharma and Lafontaine 2015; Sloan et al. 2017; Bohnsack and Sloan 2018; Chen et al. 2021). For an in-depth review of m5C on mRNA, we direct readers here (Trixl and Lusser 2019). Here, we will discuss the major findings from work in plant systems and future directions for this understudied mark.
The first report of m5C in Arabidopsis utilized RNA bisulfite sequencing to map over 1,000 m5C sites predominantly in mRNA, but also in long noncoding RNAs, small noncoding RNAs, and other noncoding species (David et al. 2017). Sites containing m5C were reported to be evenly distributed along the CDS and beginning of the 3′ UTR in mRNA (Fig. 1). Comparison of methylated sites between silique, seedling shoots, and roots, revealed tissue-specific m5C profiles, suggesting tissue-specific regulation and function of this mark. Loss of RNA m5C methyltransferase, tRNA-specific methyltransferase 4B (TRM4B), reduced the number of m5C sites on both mRNA and noncoding RNAs, and trm4b mutants displayed shorter primary roots and reduced cell division in the root apical meristem. Another study profiled m5C across plants and tissue types through liquid chromatography-tandem mass spectrometry (LC-MS/MS) and antibody-based dot blot analysis (Cui et al. 2017). Detection and quantification of m5C/C ratios in Arabidopsis, Medicago, Rice, Maize, and Foxtail Millet showed conservation of this mark across plants, while the tissue-specific resolution provided by this study in Arabidopsis rosette and cauline leaves, stems, flower buds, open flowers, roots, and siliques expanded the above-reported tissue-specific detection of this mark. m5C RNA immunoprecipitation sequencing (m5C-RIP-seq) from the same group revealed around 6,000 m5C peaks concentrated immediately after start codons and before stop codons (Fig. 1) (Cui et al. 2017). They also confirmed the results from David et al. (2017) showing mutation of writer TRM4B (trm4b mutant) affects m5C peak enrichment and negatively impacts root development, while also finding a potential effect of m5C on RNA stability (Cui et al. 2017). The first mechanistic report of m5C in plants recently linked this mark with mRNA transport through phloem cells in Arabidopsis. m5C RIP-seq in seedlings and rosette leaves, complemented with nanopore sequencing in seedlings, revealed a total of 562 m5C peaks, 16 overlapping between all three (Yang et al. 2019). This study reported that methylation deposition was concentrated immediately after the translation start codon, decreasing in intensity across the CDS (Fig. 1). Overlapping of the significant m5C peaks with previously reported mobile transcripts revealed enrichment of m5C in mobile mRNA. The authors confirmed this correlation, focusing on the mobility of 2 mRNAs through phloem-mediated transport across graft junctions. TRANSLATIONALLY CONTROLLED TUMOR PROTEIN 1 (TCTP1) and HEAT SHOCK COGNATE PROTEIN 70.1 (HSC70.1) mRNA transport was diminished upon removal of their respective m5C methylated regions, specifically +133–183 after the start codon. Furthermore, grafted transgenic shoots expressing YFP-tagged TCTP1 mRNA with Col-0 rootstocks in the presence and absence of m5C inhibitor 5-azacytidine (5-azaC) confirmed m5C-mediated transport, which was supported by the observation that both YFP-tagged mRNA and protein were transported to WT rootstocks. The most recent profiling of m5C in plants was conducted in rice in the context of heat stress using a combination of genetic analysis and bisulfite RNA sequencing (Tang et al. 2020). A total of 6,889 m5C sites within 1,450 mRNAs were detected in tissue from the seedling shoot. Analysis of the site locations revealed ∼47% of m5C harboring transcripts contained a single modified site and ∼80% of identified m5C sites only had a level of 24% or less methylation of the transcript population. Enrichment for m5C was found along the CDS, with higher enrichment around 88 nucleotides downstream of the translation start site. They found these transcripts to be enriched in pathways involved in stimulus-response and development. The authors investigated the role of m5C further in the context of heat stress utilizing a mutant in OsNSUN2, an RNA m5C methyltransferase in rice. Disruption of the methyltransferase resultes in severe temperature- and light-dependent phenotypes, specifically heat stress hypersensitivity. Continuing, they showed m5C-dependent protein synthesis phenotypes in selected transcripts involved in photosynthesis and detoxification (Tang et al. 2020). Overall, these findings in plant systems have revealed important functionality for m5C sites located just downstream of mRNA start codons.
Methods and future directions: m5C
Current reporting in plant systems demonstrates successful m5C detection using LC-MS/MS, antibody-based dot blot analysis, antibody-based immunoprecipitation-seq, bisulfite RNA-seq, and nanopore-seq (Fig. 2). The reported difference in total m5C site discovery, the lack of overlap between methods, and differences in distribution across mRNAs in plants suggest issues with consistency and potential accuracy between detection methods. For example, overlap between m5C-RIP-seq and nanopore-seq (Yang et al. 2019) revealed extremely low m5C site overlap within the same experiment. In future experiments, using a combination of methods may provide a pool of high-confidence m5C sites, although smaller, that will facilitate more meaningful biological questioning regarding the function of this mark. Additionally, the adaptation of methods from mammalian systems will increase our detection of bonafide m5C harboring transcripts. For instance, Aza-IP-seq, whereby the 5-azaC analog is used to covalently trap methyltransferases interacting with target RNAs, which are then purified and sequenced should be applied to plant systems (Khoddami and Cairns 2013). Similarly, adapting miCLIP with tagged mutant methyltransferases will allow for direct target RNA identification (Hussain et al. 2013). For a comprehensive list of m5C detection methods along with their advantages and disadvantages, we direct readers here (Owens et al. 2021). From the original discovery of the m5C mark to the most recent findings related to mRNA transport, the potential role of m5C in regulating key biological processes is clear. More focus directed at profiling global m5C marks in the plant epitranscriptome coupled with genetic and biochemical experimentation to unravel their impact on RNA metabolism, including stability and translation, is needed.
N1-methyladenine (m1A)
Since its discovery in the 1960s (Dunn 1961), m1A has been annotated within mRNA, tRNA, rRNA, and mitochondrial mRNAs (RajBhandary et al. 1966; Sharma et al. 2013; Dominissini et al. 2016; Li et al. 2017). Most of the previous research on this mark has focused on its deposition and function in tRNA where it is critical for maintaining structure and is found at positions 9, 14, and 58 across kingdoms (Agris 1996; Grosjean 2005). Compared with its abundance in tRNAs, m1A’s abundance in mRNA is extremely low (Li et al. 2016). Its unique arrangement blocks Watson–Crick base pairing, harbors a positive charge, and impacts protein–RNA interaction and internal secondary structure (Roundtree et al. 2017). The distribution of this mark within mRNA has been reported to occur within the 5′ UTR, with higher concentrations detected right before or at the start of the CDS in mammalian systems (Dominissini et al. 2016; Li et al. 2016, 2017) and at, or right after, the transcription start site in plants (Yang et al. 2020) (Fig. 1). Transcriptomic bulk analyses of m1A in mammalian systems (Dominissini et al. 2016; Legrand et al. 2017; Li et al. 2017) and one report in plants (Yang et al. 2020) have used combinations of antibody-based sequencing methods as well as misincorporation-based signatures to detect global m1A deposition. Despite multiple efforts, there is a serious lack of consistency in reporting of m1A sites within mRNA. More specifically, there are reports showing thousands of sites detected (Dominissini et al. 2016; Li et al. 2016; Yang et al. 2020) and contradictory reporting of hundreds or fewer sites (Li et al. 2017; Safra et al. 2017). With little work conducted in plant systems, we direct readers here (Zhang and Jia 2018; Wiener and Schwartz 2021) for comprehensive reviews discussing the controversial detection and reporting of this mark. Here, we will focus on the only current study conducted in plant systems and the potential for future exploration in plants.
The only current report in plants surveyed the global m1A methylome of Petunia mRNA and reported the detection of the mark across various other species. Utilizing antibody-based dot blot analysis and LC-MS/MS, m1A was detected at different levels in total RNA and mRNA from roots, stems, leaves, and corollas at different development stages in Petunia (Yang et al. 2020). LC-MS/MS analysis confirmed the detection of m1A in Brunfelsia latifolia, Solanum lycopersicum, Capsicum annuum, Arabidopsis, Phalaenopsis aphrodite, and Oncidium hybridum, with the highest signal detected in total RNA from B. Latifolia. Transcriptome-wide profiling was conducted using m1A-RIP-seq with and without ethylene treatment. From this analysis, the authors identified around ∼4,900 m1A peaks in ∼3,200 transcripts in the Petunia corolla transcriptome. Ethylene- and control-specific m1A peak discovery in combination with mRNA-seq suggested condition-specific m1A profiles that may influence transcript abundance. Peaks identified were preferentially located in the CDS, concentrated at or right after the start codon (Fig. 1). Silencing of the Petunia tRNA-SPECIFIC METHYLTRANSFERASE 61A (PhTRMT61A) mRNA reduced total m1A levels in mRNA and resulted in abnormal leaf development, suggesting the modification's influence on regulating development.
Methods and future directions: m1A
Successful m1A detection, quantification, and transcriptome-wide mapping were reported across various plant species (Yang et al. 2020) utilizing a combination of dot blot, LC-MS/MS, and m1A-RIP-seq (Fig. 2). The dot blot and transcriptome-wide mapping relied on an AMA-2 monoclonal antibody that was assumed to be specific for m1A. However, recent reporting of the cross-reactivity of this antibody with the m7G cap demonstrated that a significant amount of previously called m1A peaks were false positives in mammalian studies (Grozhik et al. 2019). The concentrated detection of m1A peaks toward the 5′ end of transcripts is thus likely biased by this finding. Moving forward, Li et al. (2017) demonstrated the successful use of a base-resolution m1A profiling method, where m1A-induced misincorporation during reverse transcription can be used to detect bona fide sites. This method can be easily translated to plant systems and can be used to compare and validate antibody-based method detection. However, as noted previously, both antibody-based and misincorporation detection-based methods produce variable maps of m1A in mammalian systems, with recent publications refining the search (Zhou et al. 2019a). Taken together, more studies in plant systems need to be conducted, specifically mapping using multiple methods in a model such as Arabidopsis. The use of antibody-based detection and mapping methods should be done with an understanding of the potential promiscuity of each antibody used. Future studies should also control for false-positive m1A site discovery by performing m1A-IP-seq and misincorporation reverse transcription sequencing in genetic models of m1A transferase mutants, which would reveal the bona fide methyltransferase-dependent targets. Although detected at low stoichiometric levels, mutational analysis and condition-specific profiling suggest that m1A may influence core biological processes, such as normal plant development, and warrant continued study.
N6-methyladenosine (m6A)
Since its discovery on mRNA in the 1970s (Desrosiers et al. 1974), methylation of the adenosine base at the nitrogen-6 position, m6A, has dominated not only as the most abundant internal mRNA modification but also as the most studied. Following the first annotation in plants, specifically in Arabidopsis (Zhong et al. 2008), m6A has been mapped and annotated across a wide range of plant species with a focus on crop species. In fact, this mark is significantly more well annotated than all other RNA modifications combined (Supplemental Table S1). In plants, the distribution of the mark across mRNA is enriched within the final exon of the CDS and the beginning of the 3′ UTR, similar to mammalian systems (Ke et al. 2015; Shen et al. 2016; Du et al. 2020; Parker et al. 2020; Zhang et al. 2021; Hu et al. 2022a) (Fig. 1). Recent analysis in Arabidopsis refined m6A localization nearly exclusively to the 3′UTR (Parker et al. 2020) using a combinatorial approach to verify bona fide m6A harboring nucleotides. Interestingly, tissue- and condition-specific profiling in particular species has shown deposition in the CDS as well (Liu et al. 2020; Zhou et al. 2021; Guo et al. 2022) (Fig. 1), although it is important to note the use of traditional antibody-based methods in these experiments, which have limitations discussed below. Within plant m6A transcriptomes, a number of m6A motifs were originally identified using the traditional m6A-seq methods, including the highly conserved RRACH (R = A or G and H = U, A, or C) (Luo et al. 2014; Shen et al. 2016; Zheng et al. 2021; Zhou et al. 2021), a plant-specific motif URUAH (Wei et al. 2018; Yang et al. 2021; Hu et al. 2022a), a UGUAHH variation (Hou et al. 2022), and UGWAMH (W = U or A and M = C or A). Importantly, the use of mapping m6A with individual nucleotide resolution using crosslinking and immunoprecipitation (miCLIP) (Grozhik et al. 2017; Parker et al. 2020), discussed in more detail below, individual nucleotide resolution UV crosslinking and immunoprecipitation (iCLIP) with known m6A-binding proteins, and nanopore direct sequencing (Huppertz et al. 2014; Parker et al. 2020; Arribas-Hernández et al. 2021) in Arabidopsis have been used to differentiate between bona fide m6A containing motifs and m6A-associated (neighboring) motifs. These collective studies confirm the major m6A methylation site DRACH (D = A, G, or U and R = A or G), GGAU as a minor m6A site, and DRACH/GGAU islands in between U-rich regions in Arabidopsis (Parker et al. 2020; Arribas-Hernández et al. 2021). Despite the abundance of research focused on mapping m6A, its function and impact on RNA fate and functionality in plants are complex. In plants, there are clearly demonstrated roles of m6A in regulating mRNA stability (Shen et al. 2016; Duan et al. 2017; Anderson et al. 2018; Wei et al. 2018; Kramer et al. 2020; Arribas-Hernández et al. 2021; Yang et al. 2021; Zhou et al. 2021), RNA secondary structure (Kramer et al. 2020), alternative polyadenylation (Parker et al. 2020; Song et al. 2021), miRNA maturation (Bhat et al. 2020), translation (Luo et al. 2020; Miao et al. 2020; Zheng et al. 2021; Zhou et al. 2021, 2022), and transcriptome integrity (Pontier et al. 2019). Complementary to transcriptome profiling of m6A is the growing amount of research focused on m6A writers, readers, and erasers as the modulators of m6A signatures in each species (Zhong et al. 2008; Duan et al. 2017; Martínez-Pérez et al. 2017; Arribas-Hernández et al. 2018, 2021; Frindert et al. 2018; Scutenaire et al. 2018). With the bounty of m6A research and review, we direct readers here (Fray and Simpson 2015; Bhat et al. 2018; Shen et al. 2019; Yue et al. 2019; Arribas-Hernández and Brodersen 2020; Kim et al. 2020; Shao et al. 2021; Zhou et al. 2022) for comprehensive reviews on m6A in plants. We will briefly highlight a few recent findings and discuss the advent of new profiling technology.
As the collection of m6A methylomes grows, one recent study provided a comprehensive analysis of m6A across a range of plant species. Using an antibody-based m6A-RIP-sequencing approach they performed transcriptome-wide mapping and evolutionary analysis of m6A in 13 species: A. thaliana, Gossypium arboreum, Gossypium hirsutum, Glycine max, Phaseolus vulgaris, Sorghum bicolor, Zea mays, Aegilops tauschii, Triticum dicoccoides, Triticum aestivum, Oryza sativa, and Physcomitrella patens (Supplemental Table S1) spanning half a billion years of evolutionary time (Miao et al. 2022). This offered the first comprehensive analysis of m6A evolutionary conservation and divergence in plant transcriptomes as well as profiles in under-studied plant systems. The group uncovered retention of m6A on ancient orthologous genes while showing less conservation between relatively newer orthologous gene pairs, suggesting its presence on specific transcripts is highly conserved. They also annotated methylation ratios and variations in transcript expression and translation efficiency across species, offering a comprehensive data set for future m6A studies in an evolutionary context. Recent work in nonclimacteric strawberry fruit showed a direct link between m6A deposition and ripening (Zhou et al. 2021). In fact, they demonstrated a potential dual role of m6A, both positively and negatively affecting mRNA stability, hypothesizing context and location (within the mRNA) as 2 leading factors contributing to the overall effect m6A has on a given mRNA (Supplemental Table S1). Continuing, they show specific transcripts encoding proteins fundamental in the ABA biosynthesis and signaling pathways as bona fide m6A harboring mRNAs whose fate is directly altered in the presence or absence of the mark (Zhou et al. 2021). Similarly, work in tomato profiling m6A revealed m6A-mediated regulation of tomato fruit ripening (Zhou et al. 2019b) and fruit expansion (Hu et al. 2022a). Interestingly, Hu et al. (2022a) utilized direct injection of 3-deazaneplanocin A (an m6A writer inhibitor) and meclofenamic acid (an m6A eraser inhibitor) into tomato fruit as alternative approaches to observe the tissue-specific effects of m6A. Their work uncovered a global increase in m6A methylation during fruit expansion and a positive association between methylation and mRNA abundance. In Arabidopsis, the recent advent of nanopore technology to detect and quantify m6A, discussed more below, demonstrated the successful use of the new sequencing platform to map this mark in plants. In 2020, a group successfully used nanopore direct RNA sequencing (DRS) to annotate Arabidopsis m6A transcriptome, utilizing an orthogonal dataset as validation (Parker et al. 2020). Using nanopore DRS they were able to obtain full-length mRNA reads, map 5′ cap positions, estimate the length and position of the poly(A) tail, alternative splicing patterns, sites of internal cleavage, and m6A modification estimations in a wild-type and m6A deficient background. Importantly, they validated their findings by overlapping miCLIP-m6A sequencing data. This offers a comprehensive transcriptome view whereby estimated m6A presence and absence can be directly compared to other transcript signatures within the same experiment. Using these data, they showed that loss of m6A from the 3′ UTR in a population of transcripts was associated with an overall decrease in relative transcript abundance and altered 3′ end formation (Parker et al. 2020). A complementary study used nanopore DRS to map m6A in Western Balsam Poplar. By comparing nanopore DRS with m6A-RIP-seq and endonuclease MazF-based m6A-REF-seq, this group was able to map the m6A transcriptome in Poplar with high accuracy, noting differential alternative polyadenylation usage compared with m6A ratios in stem-differentiating xylem (Gao et al. 2021). Another group reported utilizing nanopore sequencing to annotate m6A methylation on Moso bamboo, Phyllostachys edulis, developing a protocol to sequence the nonpolyadenylated RNAs of this plant species, and providing additional plant datasets for this important RNA modification (Wang et al. 2020a). Overall, the studies in plants have demonstrated the massive biological significance of m6A to transcriptome regulation, and research on this modification is unlikely to slow down anytime soon.
Methods and future directions: m6A
The overwhelming majority of m6A sequencing and annotation in plant systems has been done using antibody-based immunocapture followed by sequencing, m6A-RIP-seq (Dominissini et al. 2013) (Supplemental Table S1). We will highlight 4 additional methods proven in plant systems for successful transcriptome mapping of m6A: m6A-SEAL-seq, m6A-REF-seq, miCLIP, and nanopore DRS for m6A detection (Fig. 2), suggest additional methods, which can be adapted to plant systems, and direct readers here for comprehensive reviews on m6A detection methodology (Shen et al. 2019; Owens et al. 2021). m6A-SEAL-seq is an antibody-free, FTO (fat mass and obesity-associated m6A erasing protein from mammals)-assisted chemical labeling method used to detect and map high-resolution m6A sites across organisms (Wang et al. 2020b). SEAL utilizes the ability of the FTO protein to bind to and recognize m6A in RNA combined with a dithiothreitol (DTT)-mediated thiol-addition chemical reaction converting unstable N6-hydroxymethyladenosine (hm6A) to the more stable N6-dithiolsitolmethyladenosine (dm6A), thereby generating stable m6A labeled RNA. The labeled m6A-RNA can then be isolated and sequenced using a streptavidin pull-down and DTT-mediated cleavage protocol. m6A-SEAL-seq was used to successfully map the m6A-methylome in adult rice (Wang et al. 2020b), offering advantages over the other antibody-free methods of REF-seq, discussed here, and MAZTER- and DART-seq. Unlike the other antibody-free methods, SEAL is not dependent on m6A sequence or cellular transfection; the key protein utilized, FTO, is both commercially available and readily synthesized in the lab (Wang et al. 2020b). Alternatively, m6A-sensitive RNA endoribonuclease-facilitated sequencing (m6A-REF-seq) utilizes the RNA endoribonuclease ChpBK (or ChpAK/MazF) which specifically recognizes ACA motifs in RNA molecules. ChpBK-mediated cleavage is sensitive to adenosine methylation, leaving m6A-modified sites intact and cleaves all nonmodified sites, allowing for computational comparison and detection of m6A harboring nucleotides transcriptome-wide (Zhang et al. 2019b). Side-by-side application of MazF-based m6A-REF-seq in Populus trichocarpa, with m6A-RIP-seq and nanopore DRS detection, affirmed its validity in detecting bona fide m6A sites in plants (Gao et al. 2021). As with other enzyme cleavage-based detection methods, detection is limited to m6A sites within the targeted motif of the given enzyme, e.g. ACA in the case of m6A-REF-seq. Furthermore, another current field standard for antibody-based high-throughput detection of m6A is miCLIP, which allows mapping of m6A at individual nucleotide resolution using crosslinking and immunoprecipitation (Grozhik et al. 2017). This is a modification of the traditionally used meRIP-seq, where m6A antibodies are UV crosslinked, creating covalent RNA-antibody bonds. Subsequent reverse transcription of the crosslinked RNA causes highly specific mutations and truncations in the cDNA at the site of antibody-m6A-RNA crosslinks. These “errors” are computationally identified and inferred as precise m6A positions with single nucleotide resolution. miCLIP enables transcriptomic profiling of m6A with only a slight variation from the commonly used meRIP-seq approach. In practice, miCLIP has been used to identify m6A sites in Arabidopsis and validate both nanopore- and meRIP-seq-called sites (Parker et al. 2020). Alternatively, with little to no manipulation necessary, nanopore DRS m6A detection and quantification offers a glimpse at the future of RNA modification profiling. The ability to detect modifications using nanopore is based on the unique electrical signal fingerprints of the single base being assessed as well as the peripheral effects of the 4 other nucleotides occupying a nanopore at any one time. Since RNA modification can affect the signal detected, error-rate-based approaches can approximate m6A within a limited range (Parker et al. 2020). Using this methodology, nanopore DRS was used to profile m6A in Arabidopsis (Parker et al. 2020), Moso bamboo (Wang et al. 2020a), and western poplar (Gao et al. 2021). Recently, a refined methodology to call m6A using nanopore more accurately was developed using Arabidopsis RNA. For instance, deep learning explore nanopore m6A (DENA) implements training on direct RNA-seq data of in vivo transcribed mRNAs from Col-0 and multiple m6A-deficient Arabidopsis lines (Qin et al. 2022). This method, which trains with data containing naturally occurring m6A profiles, allows for m6A coverage across a combination of m6A motifs (with multiple being detected in plants as discussed above), does not rely on m6A prediction models (which may distort detection), and allows for detection on multiple isoforms of the same gene. With the continued discovery and characterization of proteins that add and remove RNA modifications, the manipulation of which can serve as reference m6A-altered transcriptomes, this approach offers a framework for identifying other types of RNA modifications in sequencing datasets. Moving forward, nanopore DRS has obvious advantages over all the other existing m6A detection methods. Furthermore, developing additional analysis methods to not only improve site-calling but also determine m6A stoichiometry in plant transcriptomes continues to advance. For instance, programs including xPore (Pratanwanich et al. 2021), nanocompare (Leger et al. 2021), CHEUI (Mateos et al. 2022), and Yanocomp (Parker et al. 2021) have all been recently created to interpret modification miscalls from DRS data. Worth noting are recent benchmarking analyses comparing current software used for detection, revealing relatively high false discovery rates for programs like xPore as an example (Mateos et al. 2022). Understanding the limitations of each program is important to interpreting transcriptome-wide data. For example, profiling of m6A using alternative m6A writer FIONA1 with nanopore data processed with xPore revealed unusually high levels of m6A in the CDS (Wang et al. 2022), which could be merely a result of this package's high false discovery rate. As mentioned, future work should focus on building pipelines to detect m6A more accurately from nanopore data. Furthermore, 2 new techniques, not yet utilized in plant systems, highlight recent advances that can be easily adapted for use in plants: m6A-selective allyl chemical labeling and sequencing (m6A-SAC-seq) (Hu et al. 2022b) and glyoxal and nitrite-mediated deamination of unmethylated adenosines (GLORI) (Liu et al. 2022). M6A-SAC-seq allows for quantitative whole-transcriptome mapping of m6A at single-nucleotide resolution with low input, ∼30 ng of poly(A) or rRNA-depleted RNA (Hu et al. 2022b). Using this method, m6A sites are converted to allyl-labeled and cyclized adducts that are detected by having a ∼10-fold higher mutation rate with HIV-1 reverse transcriptase (RT) at the resulting a6m6A (labeled m6A site) compared with a6A (unmodified A) bases. GLORI is similar in principle to bisulfite sequencing and was developed as a transcriptome-wide sequencing technology where all unmodified adenosines in mRNA are converted to inosines based on the deamination by glyoxal and nitrite (Liu et al. 2022).Thus, all converted unmodified adenosines are read as G and m6A is “protected” and read as A in the resulting sequencing reads, which allows for nucleotide-specific resolution and quantification of m6A methylation.
Here, as with other RNA modifications mentioned, it is becoming increasingly critical to generate better genetic models for temporal and stable m6A depletion and overexpression to be used as reference transcriptomes. Additionally, as nanopore and other detection methodology advances, combinatorial approaches using multiple methods and validation by site-specific mutation are critical in removing false-positive calls and building accurate RNA methylomes.
Pseudouridine (Ψ)
First discovered in the 1950s (Cohn and Volkin 1951) and deemed “the fifth nucleotide,” pseudouridine, the C5-glycoside isomer of uridine, is the most abundant modified RNA nucleotide (Davis and Allen 1957; Scannell et al. 1959). The 2 features distinguishing pseudouridine from uridine—the shift of the C–N glycosidic bond to a C–C bond and the addition of an extra hydrogen-bond donor on the non-Watson–Crick edge of the modification—create structurally and stably distinct nucleotide–nucleotide interactions and likely have specific effects on RNA–protein interactions as well (Cohn 1959; Arnez and Steitz 1994). Pseudouridine has mostly been studied in the context of tRNA (Hopper and Phizicky 2003), rRNA (Branlant et al. 1981), and snRNAs (Wu et al. 2011; Yu et al. 2011), where its effect on splicing and translation have been established (Ni et al. 1997; Darzacq et al. 2002; Karijolich et al. 2015). More recently, the focus for this modification has shifted towards its presence in mRNA, predominantly in mammal and yeast transcriptomes (Anderson et al. 2010; Carlile et al. 2014, 2019; Schwartz et al. 2014; Martinez et al. 2022), but also in Toxoplasma gondii (Nakamoto et al. 2017) and only recently in plants (Sun et al. 2019). The distribution of pseudouridine within mRNA has been reported across the entire mRNA, occurring predominantly in the CDS and, to a lesser extent, the 5′ and 3′ UTRs in yeast, humans, and plants (Carlile et al. 2014; Sun et al. 2019). Here, we will discuss the only work annotating pseudouridine in plants and direct readers here (Ge and Yu 2013; Spenkuch et al. 2014; Karijolich et al. 2015; Borchardt et al. 2020) for comprehensive reviews covering pseudouridine modification in mRNA of other organisms.
The only current report of transcriptome-wide analysis of pseudouridine in plants surveyed Arabidopsis total RNA and mRNA. Utilizing pseudouridine-seq (Carlile et al. 2014), discussed in more detail below, a total of 451 pseudouridine sites were discovered within 332 transcripts (Sun et al. 2019). Enrichment for this modification was found in the CDS specifically, along with potential positional bias in UUC, CUU, UUU, and UCU triplets, suggesting that the addition and function of this mark are likely position specific as seen with other marks. Genetic analysis of mutant SUPPRESSOR OF VARIEGATION1 (SVR1), a chloroplast pseudouridine synthase, resulted in a significant reduction of plastid ribosomal proteins and photosynthesis-related proteins relative to Col-0, suggesting a direct or indirect role of SVR1-mediated pseudouridinylation in regulating chloroplast protein abundance (Sun et al. 2019). Additionally, the homolog of mammalian pseudouridine synthase Dyskerin was annotated in Arabidopsis as AtNAP57 (Lermontova et al. 2007); a null mutation leading to loss of this protein results in lethality. Thus, highlighting the importance of this mark in plants (Kannan et al. 2008).
Methods and future directions: Ψ
The successful detection and mapping of pseudouridine in plant RNA was conducted utilizing pseudouridine-seq. In brief, pseudouridine-seq uses the ability of pseudouridine to be modified further with N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate (CMC), which blocks RT, one nucleotide 3′ to the original pseudouridylated adenosine (Bakin and Ofengand 1993; Carlile et al. 2014). Processing CMC(−) RNA samples in parallel with CMC(+) treated samples can thus be used to identify and remove false positive pseudouridine-independent RT stops occurring in the CMC(−) RNA samples. Widely used as the current standard, focus on detection using nanopore direct sequencing methods is being explored as with other modifications (Smith et al. 2019; Xu and Seki 2020). With only one current study conducted in plants compared to an abundance of work on both noncoding and coding RNA species in yeast and mammals, it is imperative that both pseudouridine profiling and functional characterization advance in plants in parallel with work in other eukaryotes. The biological significance of this mark is well established in other systems, as reviewed in Borchardt et al. (2020), leaving a significant knowledge gap in plants. Recent genome-wide identification of pseudouridine synthase family proteins in Arabidopsis and maize offers some insight into the potential modulators of this mark (Xie et al. 2022) as an example. Thus, robust profiling of this mark in combination with genetic analysis of its recently identified modulators should be initiated.
3-methylcytosine (m3C)
First discovered in the 1960s in yeast (Hall 1963), human cells (Iwanami and Brown 1968), and later in rat tRNA (Ginsberg et al. 1971), most of the research on m3C has been focused on annotating and understanding its presence in tRNAs in mammalian and yeast systems (Noma et al. 2011; El Yacoubi et al. 2012; Clark et al. 2016; Xu et al. 2017). However, m3C has been reported to be detected on mRNA species in mice and humans (Xu et al. 2017), with recent reporting using transcriptome-wide sequencing revealing very few m3C harboring mRNA transcripts. However, this study noted that more validation is needed to determine if the reported sites are bona fide modified nucleotides (Cui et al. 2021). In plants, there are currently no studies directly profiling m3C on mRNA despite sequencing methods being available (Cui et al. 2021; Marchand et al. 2021). However, prediction of m3C sites in plant RNA has been performed using high-throughput annotation of modified ribonucleotides, which uses base calling errors to predict RNA modification sites by taking advantage of sequencing data generated using a RT enzyme lacking proof-reading ability (Ryvkin et al. 2013). The predicted sites were later validated on a few candidate protein-coding transcripts using m3C-specific antibody-mediated immunocapture followed by qPCR (Vandivier et al. 2015). The lack of research profiling m3C in plants offers an opportunity and novelty in detecting and annotating this mark. As with other understudied modifications, it is imperative that plant transcriptomics continues to study this modification in parallel with mammalian and other eukaryotic systems.
7-methylguanosine (m7G)
The m7G cap is a highly conserved feature of eukaryotic RNAs. First discovered in the 1970s (Both et al. 1975; Dasgupta et al. 1976; Furuichi 2015), the m7G cap has been annotated on the ends of most protein-coding and many noncoding RNAs and is critical for nearly all levels of RNA processing and therefore the ultimate fate of the RNA molecule (Filipowicz 1978; Cowling 2010; Galloway and Cowling 2019). As with other modifications, m7G capping is highly regulated (Jiao et al. 2010; Culjkovic-Kraljacic et al. 2020), can be influenced by development and differentiation (Cowling 2010), and is reversible (Schoenberg and Maquat 2009; Mukherjee et al. 2012). For comprehensive reviews covering this modification, we direct readers here (Topisirovic et al. 2011; Borden et al. 2021) and will instead focus on the controversial detection of internal m7G on mRNA in plants.
Interestingly, several groups have been investigating the presence of internal m7G modifications. One effort utilized differential enzymatic digestion with LC-MS/MS to differentiate and quantify internal as compared to terminal (e.g. RNA end) m7G modifications (Chu et al. 2018). With RNA from rice, A. thaliana, tobacco, and cotton, they detected internal m7G modification at comparatively higher levels than mammalian mRNA. Further exposure of rice to cadmium resulted in a decrease in global m7G capping, correlated with a cadmium-induced signature of inhibiting known m7G decapping enzymes. These findings were challenged, however, by the development and implementation of m7G mutational profile sequencing (m7G-MaP-seq), a sequencing method whereby m7G modified positions can be converted to abasic sites through a reduction reaction with sodium borohydride. Modified m7G can then be directly detected as mutations during reverse transcription and sequencing when compared to untreated RNA (Enroth et al. 2019). Using m7G-MaP-seq, internal m7G modifications were detected in Arabidopsis rRNA and tRNA but not in other small RNAs or mRNAs. Compared with high-sequencing depth m7G-MaP-seq from yeast and Escherichia coli mRNA, they also found no enrichment for internal m7G modifications (Enroth et al. 2019). Taken together, internal m7G was not detected using transcriptome-wide sequencing applications but was reported to be detected using a mass spectrometry-based methodology to separate external and internal modified residues on mRNA. These conflicting results, both in plants and other organisms, are intriguing and encouraging for future experimentation. Previously thought to be the sole cap on mRNA, now understood as one of several highly regulated end modifications, its presence elsewhere in mRNA is yet to be understood and requires further interrogation.
Concluding remarks
The epitranscriptome is rapidly being annotated and studied across many organisms. The field of plant biology is leading this charge in specific areas, for example in alternative NAD+ capping and the consistent contributions to m6A biology. Regarding other modifications, however, work in plant systems is either equally, or grossly disproportionately understudied compared to other eukaryotes. With advances in sequencing and computational mapping of modifications consistently improving, and the biological significance of RNA modifications more obvious than ever before, there is an urgency to invest in and expand research in the field of plant mRNA modifications. In fact, these marks appear to influence plant acclimatory responses to stressors (Anderson et al. 2018; Cheng et al. 2021; Hu et al. 2021b; Tian et al. 2021; Yang et al. 2021; Zhang et al. 2021; Zhou et al. 2022), thus elucidation of how this occurs offers potential new mechanisms to add to the crop engineering toolkit. As we expand our knowledge, it is important to begin investigating these modifications combinatorically, rather than as separately functioning regulatory moieties. This future direction is likely to be made easier by continual developments in nanopore-based detection of RNA modifications through DRS, which will undoubtedly play a key role in building this holistic RNA modification understanding (Mateos et al. 2022).
With advances in site detection and the steadily decreasing cost of sequencing, it is critical to focus on validating RNA modifications on individual transcripts of interest. Specifically, validation by mutational analysis of known or predicted m6A sites and interpreting the biological significance is pertinent to truly understand the direct impacts m6A (and other modifications) has on cellular biology. As an abundance of sequencing and annotation data sets are being generated across a range of plant species, there is little to no work correlating the biological impact of mRNA modification combinations in a given context, particularly in plants. As modifications appear to be critical across nearly all biological and physiological contexts in plants, we must begin to analyze the potential compensatory, redundant, or additive effects each mark may have on each other, and how these interactions culminate to produce a given RNA fate (e.g. translation, decay, etc.). Given the potential interactions with other metabolic pathways in the cell (e.g. balancing NAD+ pools as a coenzyme or RNA cap), there is more to unpack concerning how a plant cell balances conflicting biochemical demands. While some key factors involved in writing, reading, and erasing epitranscriptome marks have been discovered for better-studied modifications (e.g. m6A), new discoveries await for the machinery governing the distributions of lesser studied epitranscriptome marks (e.g. NAD+ caps). Although a challenge, the advantages of plant systems and the diversity of biology in these different models offer incredible opportunities to understand epitranscriptomic modifications across an exciting range of organisms. Exciting possibilities also await expanding these technologies into native ecosystems, which would offer opportunities to understand the extent to which these marks may contribute not only to plant environmental acclimation but also adaptation.
Acknowledgments
The authors would like to thank members of the B.D.G. lab both past and present for helpful discussions.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Table S1. Tabular list of published transcriptome-wide sequencing and annotation of the following mRNA modifications in plant systems: m5C, m1A, m3C, m6A, Pseudouridine (Ψ), m7G, and 5′ nicotinamide adenine diphosphate (NAD+ caps).
Funding
This work was funded by United States National Science Foundation grants Division of Integrative Organismal Systems (IOS)—2023310 and IOS—1849708 to B.D.G. The funders had no role in study design, literature collection and analysis, decision to publish, or preparation of the manuscript.
References
Author notes
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://dbpia.nl.go.kr/plcell) is: Brian D. Gregory ([email protected]).
Conflict of interest statement. The authors declare no conflicts of interest related to this work.

{kind=link}
{kind=link}