





Abstract
Reproductive tissues are a rich source of small RNAs, including several classes of short interfering (si)RNAs that are restricted to this stage of development. In addition to RNA polymerase IV-dependent 24-nt siRNAs that trigger canonical RNA-directed DNA methylation, abundant reproductive-specific siRNAs are produced from companion cells adjacent to the developing germ line or zygote and may move intercellularly before inducing methylation. In some cases, these siRNAs are produced via non-canonical biosynthesis mechanisms or from sequences with little similarity to transposons. While the precise role of these siRNAs and the methylation they trigger is unclear, they have been implicated in specifying a single megaspore mother cell, silencing transposons in the male germ line, mediating parental dosage conflict to ensure proper endosperm development, hypermethylation of mature embryos, and trans-chromosomal methylation in hybrids. In this review, we summarize the current knowledge of reproductive siRNAs, including their biosynthesis, transport, and function.
Introduction
The most abundant small RNAs in most plant genomes are 24-nt short interfering (si)RNAs that are key components in RNA-directed DNA methylation (RdDM) (Matzke and Mosher, 2014). RdDM begins when RNA polymerase IV (Pol IV) is recruited to DNA through the action of a CLASSY (CLSY) protein (Zhou et al., 2018). Pol IV transcribes a short (25–40 nt) non-coding RNA before backtracking along the DNA, which causes the 3′ end of the transcript to be taken up by RNA-DEPENDENT RNA POLYMERASE 2 (RDR2) (Blevins et al., 2015; Zhai et al., 2015a; Fukudome et al., 2021). Using the Pol IV transcript as a template, RDR2 transcribes a complementary strand, producing double-stranded RNA (Singh et al., 2019; Huang et al., 2021). These short double-stranded RNAs are trimmed to 24-nt siRNA duplexes by DICER-LIKE 3 (DCL3) (Wang et al., 2021; Loffer et al., 2022). One strand of the siRNA duplex is bound by an ARGONAUTE (AGO) and directs these effector proteins to complementary transcripts produced by RNA polymerase V (Pol V), or possibly to single-stranded DNA resulting from Pol V transcription (Wierzbicki et al., 2008, 2009; Lahmy et al., 2016; Liu et al., 2018). The siRNA/AGO complex then recruits DOMAINS REARRANGED METHYLTRANSFERASE (DRM) to catalyze cytosine methylation in all sequence contexts (Zemach et al., 2013; Stroud et al., 2014; Zhong et al., 2014). This DNA methylation helps recruit Pol IV for additional siRNA production (Law et al., 2013; Choi et al., 2021), thereby creating a stable feedback loop. Because non-symmetric DNA methylation is lost from one daughter strand during semi-conservative DNA methylation, RdDM's feedback loop is important for maintaining CHH methylation (where H = A, T, or C) in euchromatin (Law and Jacobsen, 2010; Law et al., 2013). In addition to this canonical mechanism, variations of RdDM initiate methylation at unmethylated sequences (Nuthikattu et al., 2013; McCue et al., 2015; Cuerda-Gil and Slotkin, 2016; Sigman et al., 2021).
RdDM primarily functions at transposons, including both DNA- and RNA-based elements (Kasschau et al., 2007; Zhang et al., 2007; Mosher et al., 2008), and is responsible for transcriptional silencing of transposons at the boundary between heterochromatin and euchromatin (Li et al., 2015; Böhmdorfer et al., 2016). RdDM of transposons that are in proximity to protein-coding genes can influence expression of such genes (Hollister and Gaut, 2009) and remnants of transposons can create gene-specific regulatory structures that silence gene expression (Chan et al., 2006; Kinoshita et al., 2007; Henderson and Jacobsen, 2008). Differing activity of RdDM in alternate developmental contexts thus provides epigenetic control of gene expression (Vu et al., 2013).
In angiosperms, 24-nt siRNAs are most abundant in reproductive tissues (Mosher et al., 2009; Grover et al., 2020; Zhou et al., 2022a), which are a complex mix of somatic, germ line, and zygotic cells. The female and male germ lines are established when megaspore or microspore mother cells, respectively, are specified from surrounding somatic cells (Berger and Twell, 2011). The megaspore and microspore mother cells undergo meiosis to generate the haploid megaspore and microspore, which then undergo mitosis to generate female and male gametophytes, respectively. In most angiosperms, the megaspore goes through three rounds of nuclear division before cytokinesis to generate the 7-celled female gametophyte, while the microspore has an asymmetric division, followed by a second division of the smaller cell to create the male gametophyte (pollen grain). The two haploid sperm cells in the pollen grain fertilize the haploid egg cell and diploid central cell to create the zygotes that develop into the embryo and endosperm, respectively. Mega/microspore specification, meiosis, gametogenesis, fertilization, and zygotic development all happen in intimate connection with diploid somatic cells, providing the potential for intercellular, and indeed intergenerational, movement of siRNAs during reproduction (Feng et al., 2013).
Consistent with the abundance of 24-nt siRNAs in reproductive tissues, RdDM is required for reproductive development in a variety of species, including tomato (Gouil and Baulcombe, 2016), other Brassicaceae (Grover et al., 2018; Wang et al., 2020), rice (Xu et al., 2020; Zheng et al., 2021; Chakraborty et al., 2022; Wang et al., 2022), and maize (Erhard et al., 2009). Here, we will discuss the role of RdDM in establishing and maintaining epigenetic marks during plant reproduction, with particular emphasis on the role of small RNA movement from somatic cells.
Male germ line
Although there is no defect in Arabidopsis (Arabidopsis. thaliana) when RdDM is eliminated, research in multiple other species suggests that 24-nt or transposon-associated siRNAs are required for male germ line development (Walker et al., 2018; Bélanger et al., 2020; Teng et al., 2020; Wang et al., 2020; Nan et al., 2022). During development, microspore mother cells, microspores, and immature pollen are surrounded by a layer of sporophytic cells called the tapetum or tapetal nurse cells (Figure 1A). Likewise, after pollen development, sperm cells are encased within the vegetative cell. While they do not contribute genetically to the next generation, these companion cells frequently produce male-reproductive specific siRNAs that move intercellularly to trigger methylation or gene silencing in the germ line, causing an epigenetic impact on the subsequent generation.
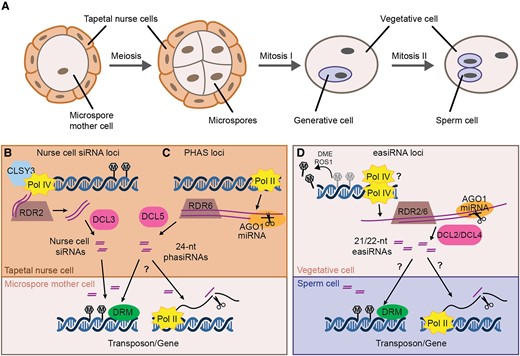
Male reproductive development, siRNA biogenesis, and proposed intercellular movement. A, Male germ line development. A microspore mother cell undergoes meiosis to generate four haploid microspores (shown in tetrad stage). Each microspore has an asymmetric mitotic division to create the vegetative cell and the generative cell. The generative cell later divides to create the two sperm cells. Together, the vegetative cell and sperm cells form the male gametophyte or mature pollen grain. Microspore mother cells, microspores, and immature pollen are surrounded by a layer of sporophytic cells called the tapetum or tapetal nurse cells. B, The Arabidopsis tapetum produces 24-nt nurse cell siRNAs through the canonical RdDM pathway. Pol IV and CLSY3 produce RNA precursors which are converted into dsRNA by RDR2 before processing by DCL3 to generate 24-nt nurse cell siRNAs. Nurse-cell siRNAs move into microspore mother cells, inducing DNA methylation with the aid of DRM1 and/or DRM2 (DRM). C, In the tapetum of many other angiosperms, reproductive phasiRNAs are produced from PHAS loci. Pol II transcribes PHAS loci to generate precursor transcripts, which are targeted by miRNAs for cleavage. The cleaved transcripts are converted into dsRNA by RDR6, and those targeted by miR2275 are further processed by DCL5 to generate 24-nt phasiRNAs. These phasiRNAs move intercellularly into germ cells, and might induce DNA methylation (left arrow) or post-transcriptional gene silencing through transcript cleavage (right arrow). D, Epigenetically-activated (ea)siRNAs are produced from reactivated transposons in the vegetative nucleus, where DME and ROS1 actively demethylate transposons and some protein coding genes. The demethylated loci are transcribed by either Pol II or Pol IV and the resulting RNAs are cleaved by miRNA/AGO1 complexes, triggering their conversion into dsRNA by RDR2 or/and RDR6. DCL2 and DCL4 then produce 22- and 21-nt easiRNAs from the dsRNA, respectively. These vegetative cell-derived easiRNAs can induce gene silencing in the sperm cell, either through transcriptional gene silencing (left arrow) or post-transcriptional gene silencing (right arrow).
Nurse cell siRNAs
During Arabidopsis male germ line development, abundant 24-nt siRNAs accumulate from several hundred loci (Figure 1B) (Long et al., 2021). These siRNAs require RDR2 and CLSY3, the latter being expressed in the tapetal nurse cells surrounding the microspore mother cells (Long et al., 2021). Tapetal-specific expression of RDR2 in the rdr2 background is sufficient for siRNA accumulation in the microspore mother cells, indicating that these “nurse cell siRNAs” are produced specifically in the tapetum before moving intercellularly into microspore mother cells (Long et al., 2021). As expected, the nurse cell siRNAs induce DNA methylation in cis, but they also trigger methylation in trans at sites with up to three mismatches (Long et al., 2021). This results in CHH hypermethylation of hundreds of loci in microspore mother cells relative to somatic tissues (Walker et al., 2018; Long et al., 2021). These sites remain hypermethylated in microspores, sperm cells, and vegetative cells, likely through an absence of demethylation as well as continued transport of nurse cell siRNAs throughout germ line development (Walker et al., 2018; Long et al., 2021).
Unlike somatic RdDM, which mostly occurs at transposons and transposon fragments, most male germ line-specific hypermethylated loci overlap genes, suggesting that nurse cell siRNAs regulate gene expression during germ line development (Walker et al., 2018). In support of this hypothesis, some of the hypermethylated genes are transcriptionally upregulated in drm microspore mother cells but not in drm leaves (Walker et al., 2018). In addition to transcriptional silencing of targets, drm mutation also causes mis-splicing of MPS1, a gene targeted by RdDM in the male germ line. This mis-splicing results in defective meiosis (Walker et al., 2018), highlighting the functional significance of nurse cell siRNA-mediated RdDM in the male germ line. However, it is not clear whether nurse cell trans-methylation is developmentally significant for most target genes.
Reproductive phasiRNAs
While nurse cell siRNA loci have been described only in Arabidopsis to date, a different class of highly expressed siRNA loci are present during male germ line development in many other angiosperms—phased, secondary, small interfering RNAs (phasiRNAs) (Figure 1C) (Johnson et al., 2009; Song et al., 2012). Biosynthesis of phasiRNAs is distinct from Pol IV-dependent siRNAs. First, Pol II transcribes PHAS loci and generates long precursor transcripts, which are targeted by microRNAs for cleavage. The cleaved transcripts are converted into dsRNA by RDR6, and further processed by DCLs to generate phasiRNAs. There are two major classes of reproductive phasiRNAs: 21-nt phasiRNAs are usually produced by DCL4 following miR2118 cleavage, while miR2275 and DCL5 are responsible for generating most 24-nt phasiRNAs (Song et al., 2012; Zhai et al., 2015b; Pokhrel et al., 2021). Originally identified in rice and other monocots, reproductive phasiRNAs are also present in many eudicot lineages, although they have been lost from some families, including the well-studied Brassicaceae and Fabaceae (Xia et al., 2019). Their presence in Amborella trichopoda indicates conservation of this pathway since the emergence of angiosperms, however the number and sequence of PHAS loci is variable across species and some lineages use different microRNA triggers, or other strategies to induce dsRNA production (Kakrana et al., 2018; Xia et al., 2019; Bélanger et al., 2020; Pokhrel and Meyers, 2022). This conservation of phasiRNA production but variation in phasiRNA sequences is reminiscent of Pol IV-dependent siRNAs.
Both 21- and 24-nt reproductive phasiRNAs display distinct spatiotemporal distribution throughout male germ line development. While 21-nt phasiRNAs are abundant before meiosis and localize in all anther cell layers, most 24-nt phasiRNAs express during meiosis and persist throughout post-meiotic development (Zhai et al., 2015b; Araki et al., 2020). In maize, 21-nt phasiRNAs are absent from mutants that lack an epidermis, suggesting that they are synthesized in these cells, while the tapetum, but not microspore mother cells, is required for producing 24-nt phasiRNAs (Zhai et al., 2015b). While 24-nt phasiRNAs are detected in tapetum and other somatic cells by fluorescent in situ hybridization, their levels are highest in microspore mother cells, indicating that these tapetal-derived siRNAs move intercellularly into germ cells (Zhou et al., 2022b). Whether 24-nt reproductive phasiRNAs have the same function in most species as nurse cell siRNAs have in Brassicaceae remains to be explored.
While 21-nt pre-meiotic phasiRNAs cause post-transcriptional regulation, 24-nt meiotic phasiRNAs are proposed to induce DNA methylation (Jiang et al., 2020; Zhang et al., 2021). In maize meiotic anthers, 24-nt PHAS loci are highly methylated at CHH contexts and that methylation is depleted in dcl5 mutant anthers, indicating that 24-nt phasiRNAs induce methylation in cis (Zhang et al., 2021). Whether 24-nt phasiRNAs can also trigger methylation in trans to impact gene expression is less clear. In rice, some 24-nt phasiRNAs match to the promoter of CKI1, a casein kinase, and this promoter has panicle-specific CHH methylation (Yu et al., 2021). However, it is unknown if this methylation is directed by 24-nt phasiRNAs or Pol IV-dependent siRNAs produced from the methylated PHAS locus in parallel. Investigating if 24-nt meiotic phasiRNAs target genes in trans and the rules for this targeting will be an important area for future research.
Although their mechanism of action is unknown, it is clear that 24-nt phasiRNAs are central to male fertility during stress conditions. Maize dcl5, which is defective in 24-nt phasiRNAs production, exhibits short anthers with defective tapetal cells when grown at higher temperatures, suggesting that 24-nt phasiRNAs might play a role in conferring male fertility during heat stress (Teng et al., 2020). Loss of the transcription factors necessary for PHAS and DCL5 expression also causes male sterility in maize (Nan et al., 2022). Loss of 21-nt reproductive phasiRNAs also causes environment-dependent sterility, indicating that both size classes of reproductive phasiRNAs influence fertility in the face of abiotic stress (Zhai et al., 2015b; Araki et al., 2020; Yadava et al., 2021). Because many transposons respond to environmental signals, it might be that reproductive phasiRNAs are required to transcriptionally or post-transcriptionally silence such transposons, and therefore phenotypes due to loss of reproductive phasiRNAs are only visible in conditions when the transposons are active.
easiRNAs
While 24-nt nurse cell siRNAs and 24-nt phasiRNAs are produced from the tapetum during meiosis and may regulate protein-coding genes, a different class of siRNA is produced from the male gametophyte and proposed to control transposons. Epigenetically activated small interfering RNAs (easiRNAs) are 21/22-nt siRNAs that are generated from reactivated transposons in the vegetative cell (Figure 1D; Slotkin et al., 2009; Martínez et al., 2016). In Arabidopsis, the vegetative cell is actively demethylated by demeter (DME) and repressor of silencing 1 (ROS1), related DNA glycosylases that demethylate transposons and some protein-coding genes in the vegetative nucleus (Schoft et al., 2011; Ibarra et al., 2012; Park et al., 2017; Khouider et al., 2021). At the same time, decrease in DNA methylation 1 (DDM1) is lower in the vegetative cell than the sperm cells, further relaxing heterochromatin (Slotkin et al., 2009). Homologous glycosylases demethylate the vegetative cell in rice, indicating that this process is evolutionarily conserved (Kim et al., 2019). Demethylation and relaxation of heterochromatin makes transposons more accessible, and either Pol II or Pol IV transcribe easiRNA precursors in the vegetative nucleus (Slotkin et al., 2009; Borges et al., 2018). Like PHAS transcripts, these precursors are recognized by 21- or 22-nt miRNAs, which mediate cleavage with the aid of AGO1 (Creasey et al., 2014; Borges et al., 2018). The cleaved transposon transcripts are then converted into double-stranded RNA (dsRNA) by an RDR, although it is unclear whether RDR2 or RDR6 are involved in this process (Creasey et al., 2014; Martinez et al., 2018; Wang et al., 2020). The resulting dsRNAs are processed by DCL4 and DCL2, producing 21- and 22-nt easiRNAs, respectively.
There is evidence that vegetative cell-derived easiRNAs move intercellularly and function within sperm cells. Purified sperm cells contain abundant 21-nt retrotransposon siRNAs but lack the precursor transposon transcripts, suggesting that easiRNAs are not produced in sperm cells (Slotkin et al., 2009). Similarly, loss of DME in vegetative cells is associated with reduced CHH methylation in sperm cells (Ibarra et al., 2012), suggesting that siRNAs that are produced in vegetative cells act in sperm cells. Movement of siRNAs between the vegetative cell and sperm cells was directly demonstrated by producing siRNAs in the vegetative cell from a truncated green fluorescent protein (GFP) sequence. These siRNAs could silence a full-length GFP reporter that was specifically expressed in sperm cells, demonstrating their non-cell autonomous function (Martínez et al., 2016) Similarly, the sperm cell-specific GFP reporter was silenced when its 3′ untranslated region contained binding sites for endogenous easiRNAs, and this silencing was eliminated when the 2b protein, which sequesters siRNAs, was expressed in vegetative cells (Martínez et al., 2016). Together, these experiments demonstrate that easiRNAs produced in the vegetative cell can move into sperm cells and silence gene expression there. Although it is unclear whether easiRNAs trigger DNA methylation or post-transcriptional gene silencing, they are proposed to silence transposons to prevent new insertions that would be passed to the next generation (Slotkin et al., 2009). EasiRNAs are also implicated in parental dosage balance in the endosperm after fertilization (discussed further below) (Borges et al., 2018; Martinez et al., 2018).
Female germ line
Because they are buried in maternal somatic tissue (Figure 2A), it is more difficult to study siRNAs and the impact of RdDM on female germ line development. Genetic evidence demonstrates that proteins associated with RdDM are involved in specifying a single megaspore mother cell (MMC) and thereby initiating the female germ line (Figure 2B) (Olmedo-Monfil et al., 2010; Mendes et al., 2020). In Arabidopsis, loss of RDR2, DCL3, AGO9, or DRM results in multiple MMC-like cells per ovule (Olmedo-Monfil et al., 2010; Mendes et al., 2020). Because AGO9 accumulates specifically in somatic cells surrounding the MMC, these phenotypes suggest that RdDM acts in somatic cells to restrict female germ line identity. Multiple MMC-like cells arise due to ectopic expression of sporocyteless/nozzle (SPL/NZZ), which has an increased expression domain in ago9 and drm1 drm2 mutants (Mendes et al., 2020). The requirement for DRM function suggests that SPL/NZZ regulation is through transcriptional gene silencing. Trans-acting siRNAs, which cause post-transcriptional gene silencing, are also required to limit female germ line initiation (Su et al., 2017, 2020), suggesting that multiple small RNA pathways interact at this critical point of development.

Female reproductive development and the action of maternal 24-nt reproductive siRNAs. A, Female germ line development. Within each ovule, a single MMC undergoes meiosis to generate four haploid megaspores. Three of these degenerate, leaving a single functional megaspore. The megaspore goes through three rounds of nuclear division before cytokinesis to generate a 7-celled female gametophyte (antipodal cells not shown). The female gametophyte contains a binucleate central cell and a haploid egg cell that are ready for fertilization. The female germ line is surrounded by somatic cells throughout this development. B, Before meiosis, Pol IV, RDR2, and DCL3 produce 24-nt siRNAs, which interact with AGO9 to mediate DNA methylation and transcriptional gene silencing of SPL/NZZ via DRM1 and/or DRM2 (DRM). Regulation of SPL/NZZ is required for the specification of a single MMC. C, In somatic cells of a mature ovule, CLSY3 and CLSY4 direct Pol IV to transcribe siren loci. These transcripts are converted into double stranded by RDR2 and further processed into 24-nt siren siRNAs by DCL3. siren siRNAs induce methylation at protein coding genes in somatic cells and are proposed to move intercellularly, causing methylation in the gametophyte.
Consistent with RdDM’s proposed role in maintaining somatic cell identity (i.e. repressing germ line identity), AGO9 accumulates in somatic cells surrounding the haploid megaspore and the developing female gametophyte. However, AGO9 is also required to silence transposons in the developing ovules (Olmedo-Monfil et al., 2010). This observation is similar to intercellular movement of nurse cell siRNAs from the tapetum to the microspores suggesting siRNA movement might occur from the soma to germ line during both male and female reproductive development.
Like the tapetum, somatic tissue surrounding the female germ line expresses abundant 24-nt siRNAs from a modest number of loci (Rodrigues et al., 2013; Grover et al., 2020; Zhou et al., 2022a). Known as siren loci due to their original description as siRNAs in endosperm, these siRNAs are produced by RNA Pol IV in ovules before fertilization and from the maternal somatic seed coat after fertilization (Figure 2C) (Rodrigues et al., 2013; Grover et al., 2020). Loci expressing siren siRNAs are present in both monocots and dicots, but there is little conservation of individual loci, even between species within the same family (Grover et al., 2020).
Like nurse cell siRNAs, Pol IV transcription of siren loci is CLSY3- and CLSY4-dependent, with the former having a dominant role (Zhou et al., 2022a). However, there is only a small overlap between loci producing these siRNAs (12 shared loci among 797 nurse cell and 68 siren loci), suggesting that control of siRNA expression in somatic cells surrounding the male and female germ lines is distinct in some way. While they are largely produced from different loci, siren siRNAs also induce methylation in trans at protein coding genes and cause altered gene expression at a subset of target genes (Burgess et al., 2022). However, it is not yet known whether this methylation occurs in the germ line or is restricted to maternal somatic cells. Live cell imaging of methylation in Arabidopsis revealed that global CHH methylation in the egg cell requires Pol V but not Pol IV, indicating that siRNAs might be made elsewhere and transported into the egg cell to induce methylation (Ingouff et al., 2017). Consistent with this hypothesis, rice egg cells and ovaries have highly similar siRNA patterns. Both cell types accumulate abundant siRNAs from a small number of discreet loci, suggesting that siren siRNAs might move from the maternal soma to the gametophyte in rice (Li et al., 2020). While these observations hint that siren siRNAs might be transported from the maternal soma to the germ line, direct evidence in support of this hypothesis is lacking.
Seed development
Double fertilization in angiosperms results in two fertilization products, the embryo and the endosperm, that carry the same genetic information but differ in ploidy (Figure 3A). The embryo grows into the next generation and therefore maintaining epigenetic control of transposons is critical in this tissue. In contrast, transposon control is less important in the ephemeral endosperm, which will ultimately be consumed by the embryo. However, recent research indicates that RdDM also plays a role during endosperm development by regulating expression of dosage-sensitive growth factors (Lu et al., 2012; Kirkbride et al., 2019).
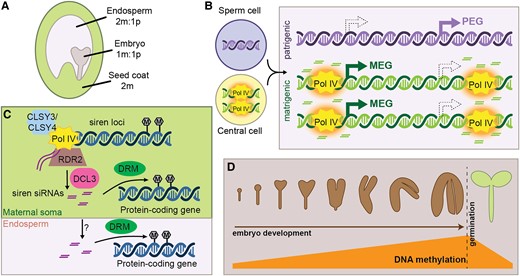
24-nt siRNA production during seed development. A, The developing seed is composed of three tissues, with distinct genetic complements. m, maternal or matrigenic; p, patrigenic. B, Pol IV activity in the central cell is hypothesized to establish an epigenetic state that is maintained on matrigenic chromosomes in the endosperm (orange halo). This epigenetic state causes allele-specific siRNA production in the endosperm and might also influence imprinted gene expression. However, whether methylation caused by allele-specific siRNAs reinforces allele-specific gene expression is unclear. (MEG, PEG; filled arrows depict the expressed state while hollow arrows with dashed outline show the non-expressed allele). C, Siren siRNAs are produced in the immature seed coat and trigger DNA methylation at protein-coding genes via DRM proteins. Siren siRNAs might also move intercellularly, resulting in maternally specific accumulation of siRNAs in the endosperm. Siren siRNA methylation of protein-coding genes in the endosperm might influence seed development. D, During embryo development, canonical RdDM is upregulated at many transposons, resulting in hypermethylation of the genome in mature embryos. This methylation is rapidly lost upon germination.
Endosperm: balancing parental contributions
Because the endosperm forms from fertilization of the diploid central cell by a haploid sperm cell, it is triploid with a 2:1 matrigenic:patrigenic ratio. (We use “matrigenic” and “patrigenic” to distinguish the maternally or paternally transmitted alleles in the zygote from the true maternal and paternal alleles in the sporophyte and gametophytes (Queller and Strassmann, 2002).) Maintenance of this 2 matrigenic:1 patrigenic dosage is important for successful endosperm and seed development, and an imbalance in parental dosage is a key mechanism of reproductive isolation following polyploidization (Müntzing, 1933; Scott et al., 1998). The Parental Conflict Hypothesis describes the fact that matrigenic alleles benefit by sharing resources with their maternal half-siblings, while patrigenic alleles benefit by extracting maximum resources from the maternal sporophyte (Haig and Westoby, 1989). This conflict might be resolved by the evolution of genomic imprinting, where the expression of a gene depends on its parent of origin. Parental Conflict Hypothesis therefore predicts that matrigenically expressed genes (MEGs) tend to restrict growth while patrigenically expressed genes (PEGs) generally enhance growth, and balance between MEGs and PEGs is necessary for successful endosperm development. Expression or activity of imprinted genes might vary in different species (independent of polyploidization), creating an imbalance in effective parental dosage (also known as the Endosperm Balance Number) (Johnston et al., 1980).
There is accumulating evidence that RdDM influences endosperm development by mediating expression differences between the matrigenic and patrigenic genomes. Compared to the embryo, endosperm is enriched for siRNAs mapping to genes (Erdmann et al., 2017) and thousands of genes are misexpressed in endosperm lacking Pol IV (Satyaki and Gehring, 2022). Imprinted genes are associated with parentally biased siRNA loci (Rodrigues et al., 2013; Xin et al., 2014; Pignatta et al., 2015; Erdmann et al., 2017), leading to the hypothesis that allele-specific siRNAs help repress transcription at the non-expressed allele (Figure 3B). However, it is not clear how siRNAs might differentially act on the homologous alleles. Additionally, in maize, siRNAs are frequently associated with the expressed allele (Xin et al., 2014), suggesting that allele-specific expression of siRNAs and genes might be driven in parallel by a single parent-specific epigenetic state. There is evidence that RdDM is required for establishing such an epigenetic state before fertilization, particularly in the female germ line. Over 4,500 genes are misexpressed in heterozygous endosperm produced from Pol IV mutant mothers, and these genes are enriched for MEGs and PEGs (Satyaki and Gehring, 2022). Similarly, matrigenically biased siRNA regions are overwhelmingly downregulated in endosperm from Pol IV mutant mothers (Satyaki and Gehring, 2022). In one example, loss of Pol IV in mothers results in loss of maternal siRNAs at the Agamous-like transcription factor AGL91 (a PEG) and derepression of the matrigenic AGL91 allele (Kirkbride et al., 2019). Together, these observations suggest that RdDM might establish a heritable epigenetic state before fertilization that persists and impacts siRNA and gene expression after fertilization. However, identification of such epigenetic states has remained elusive (Satyaki and Gehring, 2022). Alternatively, rather than establishing a heritable epigenetic state (or in addition to such establishment), siRNAs might be produced in the maternal somatic tissue, and transported into endosperm where they accumulate and regulate gene expression (Grover et al., 2020., discussed further below).
Further support for the hypothesis that RdDM establishes epigenetic states in gametes comes from the observation that loss of RdDM components such as Pol IV in tetraploid pollen donors allows successful interploidy hybridization with diploid mothers (Erdmann et al., 2017; Martinez et al., 2018; Satyaki and Gehring, 2019), while loss of maternal Pol IV exacerbates seed lethality in such paternal excess crosses (Satyaki and Gehring, 2022). These observations suggest that Pol IV-dependent siRNAs, or the methylation marks they establish, increase the effective dosage of both matrigenic and patrigenic genomes. The nature of effective dosage is unknown, but might be related to expression of MEGs and PEGs (Brandvain and Haig, 2018; Raunsgard et al., 2018). Consistent with this hypothesis, disruption of several PEGs restores viability to endosperm with excess patrigenic genomes (Kradolfer et al., 2013; Wolff et al., 2015) and many PEGs are upregulated in paternal-excess endosperm beyond the two-fold that is expected due to increased copy number (Martinez et al., 2018; Satyaki and Gehring, 2019). Upregulation of these PEGs might be due to loss of CHH methylation at nearby transposons in paternal excess endosperm, a process that unexpectedly requires Pol IV activity in pollen (Martinez et al., 2018). This counterintuitive observation is reconciled by hypothesizing that Pol IV-transcripts are cleaved by alternative DCL proteins to produce 21/22-nt easiRNAs in the male germ line, and that these shorter siRNAs compete with Pol IV-dependent 24-nt siRNAs (Borges et al., 2018; Martinez et al., 2018; Panda et al., 2020; Wang et al., 2020). Under this model, the additional genomes in tetraploid fathers produce an excess of 21/22-nt siRNAs, which overwhelm maternal or matrigenically derived 24-nt siRNAs, causing demethylation, misexpression of PEGs, and endosperm failure (Borges et al., 2018; Martinez et al., 2018). However, whether 21/22-nt siRNAs produced in pollen are transmitted to the zygote where they might encounter maternally derived 24-nt siRNAs is unclear (discussed in “Outstanding Questions”).
In addition, there is a substantial debate regarding the nature of 24-nt siRNAs in endosperm, with various publications reporting either an extensive matrigenic bias (Mosher et al., 2009; Grover et al., 2020) or limited parental bias (Rodrigues et al., 2013; Xin et al., 2014; Erdmann et al., 2017; Satyaki and Gehring, 2022). The different conclusions are correlated with differing approaches for measuring siRNAs: assessing the many thousands of individual siRNA loci identifies only limited examples of either matrigenic or patrigenic bias, while considering the siRNA population as a whole yields substantial matrigenic bias. This discrepancy is likely due to accumulation of maternal-specific siren siRNAs in endosperm (Grover et al., 2020). Although they are produced from only 1% to 2% of all loci, siren siRNAs are highly expressed and form a substantial fraction of the total siRNA population. Given their abundant expression in maternal somatic tissue, the presence of siren siRNAs in endosperm samples could result from contamination of endosperm with maternal tissue before sequencing (Schon and Nodine, 2017). However, these maternal siRNAs are also found in laser microdissected samples, which are unlikely to contain maternal contamination (Grover et al., 2020). This observation suggests that matrigenic expression (or transport from the maternal soma, as discussed below) is limited in the number of loci but extensive in the impact on siRNA transcriptome.
Intercellular RdDM in seed development
While siRNAs from most loci accumulate normally in Pol IV heterozygous endosperm, some loci differentially accumulate siRNAs in Pol IV heterozygous endosperm produced from Pol IV mutant mothers or fathers (Satyaki and Gehring, 2022). Maternal Pol IV mutations have a much stronger impact on endosperm siRNA accumulation and most matrigenically biased siRNA loci require maternal Pol IV (Satyaki and Gehring, 2022). One hypothesis to explain these observations is that RdDM establishes a heritable epigenetic state in the central cell, and this state causes siRNAs production from matrigenic alleles after fertilization (Mosher, 2010). Alternatively, maternal-specific siRNAs might result from the fact that the endosperm is surrounded by, and intimately connected with, maternal sporophytic cells. As discussed above, the tapetum (male sporophytic cells surrounding the developing male germ line) produces reproductive phasiRNA and Pol IV-dependent 24-nt siRNAs that move intercellularly into the male germ line. It is reasonable to propose that maternal sporophytic cells function similarly and continue transporting siRNAs into the endosperm throughout seed development (Figure 3C) (Grover et al., 2020). Post-fertilization movement of siRNA provides a unique opportunity for the maternal genome to respond to endosperm cues (e.g. speed of growth and development), and potentially influence gene expression to maximize embryo success. Maternal siren siRNAs have already been demonstrated to impact gene expression in Brassica rapa ovules (Burgess et al., 2022), although it is not known whether they also impact gene expression in endosperm, nor which gene expression changes are developmentally significant. Such a system would explain why maternal sporophytic, but not gametophytic, RdDM is required for successful seed development in B. rapa (Grover et al., 2018) and Capsella rubella (Wang et al., 2020).
Although they are hypothesized to trigger methylation in the endosperm, there is no evidence of siRNA movement from maternal soma to developing embryos. Firstly, siren siRNAs are biparental in embryos, indicating that there is not a meaningful accumulation of maternally derived siRNAs in embryos (Grover et al., 2020). Secondly, there is little difference in methylation between rdr2 embryos whether they develop on rdr2 mutant mothers who lack siRNA production or rdr2/RDR2 heterozygous mother who produce somatic siRNAs (Chakraborty et al., 2021).
In addition to movement from the soma to the filial tissues, siRNAs could move between the endosperm and embryo. Just as DME-mediated demethylation of the vegetative cell causes production of siRNAs that trigger methylation in sperm cells (Ibarra et al., 2012) (discussed above), it is possible that a the demethylated endosperm provides siRNAs to the developing embryo. This hypothesis was raised based on the observed correlation between sites that are demethylated in endosperm and hypermethylated in embryos (Hsieh et al., 2009; Mosher and Melnyk, 2010; Bauer and Fischer, 2011; Bouyer et al., 2017). However, this correlation is not seen in B. rapa (Chakraborty et al., 2021). Furthermore, soybean somatic embryos are hypermethylated, suggesting that the signals for methylation are intrinsic to the embryogenesis program, rather than driven by demethylation of neighboring tissues (Ji et al., 2019). Although such movement is a compelling hypothesis, over a decade of research has not provided evidence for siRNA movement between endosperm and embryo, which are symplastically isolated shortly after fertilization (Lafon-Placette and Köhler, 2014). On the whole, evidence suggests that RdDM in embryos is entirely cell autonomous.
Embryo hypermethylation
During embryogenesis, the genome becomes heavily methylated at CHH sites (An et al., 2017; Bouyer et al., 2017; Kawakatsu et al., 2017; Lin et al., 2017; Narsai et al., 2017; Ji et al., 2019; Rajkumar et al., 2020; Chakraborty et al., 2021). Indeed, mature embryos in dry seed are one of the most highly methylated plant tissues (Kawakatsu et al., 2017). Embryo hypermethylation occurs predominantly on transposons and requires RDR2 and DRM, indicating that it occurs through RdDM (Figure 3D; Lin et al., 2017; Chakraborty et al., 2021), although siRNA-independent methylation via CMT2 is also implicated in this process (Kawakatsu et al., 2017; Papareddy et al., 2020). In rice, a resetting of 24-nt siRNA production is detected in single-celled zygotes, indicating an immediate transition to an embryogenic pattern of RdDM (Li et al., 2022). Detailed analysis of siRNAs and DNA methylation throughout Arabidopsis embryogenesis indicates that heterochromatin decondensation in the embryo allows 24-nt siRNA production and subsequent CHH methylation (Papareddy et al., 2020), further supporting a model of cell-autonomous embryo methylation.
While control of transposons is important in embryos, it is not clear that embryo hypermethylation is to defend the genome against mobile elements. Firstly, the meristematic cells where transposon control is most important make up only a small fraction of cells in the mature embryo, indicating that most of the methylation is present on somatic tissues of the embryo (Papareddy et al., 2020). Secondly, methylation is lost within two days following germination, despite the fact that transposon control is still important during early growth and development (Bouyer et al., 2017; Kawakatsu et al., 2017; Narsai et al., 2017). And finally, CHH hypermethylation is dispensable for embryo development in Arabidopsis and B. rapa (Lin et al., 2017; Chakraborty et al., 2021). There are several theories for the function of this methylation, including maintaining a transcriptional quiescent state during dormancy (Kawakatsu et al., 2017), a side-effect of chromatin decondensation during rapid protein synthesis or cell-cycle arrest (Papareddy et al., 2020; Papareddy and Nodine, 2021). Ancient barley grains from archeological sites indicate that methylated cytosines spontaneously deaminate to thymine in dormant seeds (Smith et al., 2014), and therefore hypermethylation might be an opportunity to induce mutations in transposons while the seed awaits germination.
Trans-chromosomal methylation
Most angiosperms are outcrossing and thus the embryo results from fusion of genomes that vary both genetically and epigenetically. While gene conversion is rare, epigenetic alteration of one allele by its homologous pair is much more frequent. In its simplest form, this process is called trans-chromosomal methylation (TCM) or trans-chromosomal demethylation (TCdM) and results in DNA methylation in an F1 hybrid that diverges from the average of the two parental types (Figure 4; Greaves et al., 2012a). Loci undergoing TCM and TCdM are associated with abundant siRNAs in parental genomes, suggesting that siRNAs move between homologous chromosomes to trigger methylation at the paired allele (Chodavarapu et al., 2012; Shen et al., 2012; Greaves et al., 2012b; Zhang et al., 2016; Cao et al., 2022). Analysis of allele-specific siRNAs demonstrates that the allele gaining methylation does not necessarily begin producing its own siRNAs (Zhang et al., 2016), further indicating that siRNAs are responsible for inter-allele communication, not just perpetuation of the newly methylated state.
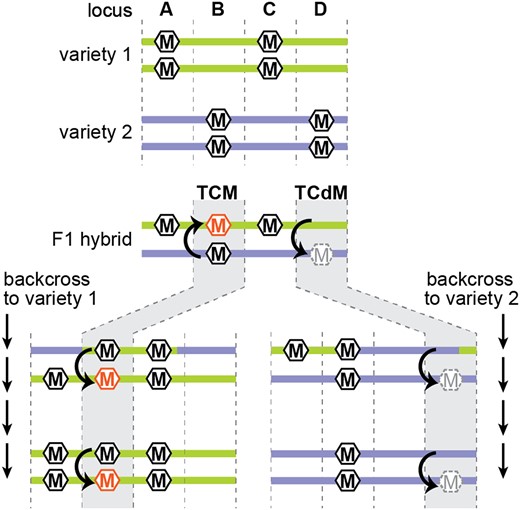
Trans-chromosomal methylation and demethylation. Four hypothetical loci (A–D) that are differentially methylated between two varieties. At loci A and C, there is no allelic interaction, and methylation depends on the genetic background (green is methylated, purple is unmethylated). At locus B, TCM triggers methylation on the previously unmethylated allele (orange hexagon). In repeated backcrosses to the unmethylated background, the newly methylated allele continues to induce methylation of naive alleles. At locus D, TCdM results in demethylation of the high methylation alleles (grey dashed hexagon). This unmethylated state continues to cause TCdM in subsequent generations of backcrossing to the methylated allele.
On the whole, TCM is more common than TCdM, which might result when siRNAs produced from one allele divide their function between homologous alleles, resulting in an siRNA concentration that is insufficient to maintain methylation at either allele (Chodavarapu et al., 2012; Greaves et al., 2016). There is also some evidence that TCdM is more likely to occur when the alleles are more genetically dissimilar, suggesting that siRNAs might migrate to the opposite allele, but be unable to trigger methylation due to nucleotide mismatches (Zhang et al., 2016). Several recent papers demonstrate that 24-nt siRNAs can function despite mismatches between the siRNA and target site (Fei et al., 2021; Long et al., 2021; Burgess et al., 2022), but it is not clear whether mis-matched siRNA operate as efficiently as perfectly matching siRNAs.
In some cases, TCM and TCdM create heritable epigenetic states, or epialleles. In crosses between two maize varieties, hundreds of TCM or TCdM events were stably maintained through six generations of backcrossing and three generations of selfing (Cao et al., 2022). Similarly, interspecific hybrids between maize and teosinte or between tomato and its wild relative Solanum pennellii also induce heritable methylation changes (Shivaprasad et al., 2012; Gouil and Baulcombe, 2018; Cao et al., 2022). The stability of these changes indicates that the converted allele gains the ability to convert a naïve allele, a process called paramutation (Hollick, 2017). For example, an allele that experiences TCM becomes hypermethylated and gains the ability to transmit that high-methylation state to a low-methylation allele in the next generation (Figure 4). Heritable epialleles are associated with changes in chromatin accessibility, histone modifications, and accumulation of siRNAs (Shivaprasad et al., 2012; Hollick, 2017; Gouil and Baulcombe, 2018; Cao et al., 2022), indicating that gaining or losing the ability to produce siRNAs through changes in chromatin state might underlie the heritability some TCM and TCdM events. However, other TCM and TCdM events might be paramutagenic without the use of siRNAs (Martinho et al., 2022).
Outstanding questions
Research on small RNA-mediated DNA methylation during reproduction is an important and growing area of research. Despite many recent advances, a number of critical questions remain unanswered.
How does pol IV create tissue-specific expression of a subset of 24-nt siRNA loci?
While Pol II transcription of PHAS loci provides a clear mechanism for tissue-specific expression of reproductive phasiRNAs, it is unknown how expression of Pol IV-dependent siRNAs is regulated. Reproductive-specific expression of Pol IV-dependent siRNAs from some loci was first noted in 2009 and was hypothesized to result from DME demethylation in the maternal gametophyte (Mosher et al., 2009; Mosher, 2010). However, ectopic overexpression of DME was insufficient to induce siRNA expression and loss of DME homologs in maize is associated with increased siRNA expression (Mosher et al., 2011; Gent et al., 2022). Subsequent research suggests that chromatin decondensation might allow Pol IV access to chromatin in specific developmental states (Fu et al., 2018; Papareddy et al., 2020; Choi et al., 2021), however how decondensation might be controlled in a locus-specific manner is unclear. Both siren siRNAs in the female gametophyte and nurse cell siRNAs during male germ line development require CLSY3 (Long et al., 2021; Zhou et al., 2022a), hinting that these chromatin remodelers, which are expressed in tissue-specific patterns, might promote site-specific decondensation to allow Pol IV access. Alternatively, decondensation and nucleosome remodeling might work together to uncover binding sites for Pol IV recruitment. Siren siRNAs in Arabidopsis and B. rapa are enriched for sequence motifs (Burgess et al., 2022; Zhou et al., 2022a), although it remains to be determined whether these motifs are functionally significant in triggering siRNA expression.
How do siRNAs move in reproductive tissues?
Although there is increasing evidence for intercellular movement of siRNAs during reproductive development, the route of this transport is unclear. SiRNAs can move from cell to cell in vegetative tissue, presumably through plasmodesmata (Melnyk et al., 2011; Liu and Chen, 2018). During male reproductive development, there are many plasmodesmata connecting the tapetum to the microspore mother cell, but these are occluded during meiosis by callose deposition (Sager and Lee, 2014), suggesting that symplastic movement of 24-nt reproductive phasiRNAs or nurse cell siRNAs must conclude prior to the completion of meiosis. Similarly, plasmodesmata at connecting the megagametophyte to maternal somatic cells appear to be occluded by callose at the four nucleus stage (Thijssen, 2003) and the integuments are symplastically isolated during embryo development (Stadler et al., 2005). Lack of plasmodesmata between maternal somatic cells and the endosperm might serve to protect the endosperm and embryo from chemical intermediates released when the integuments undergo programmed cell death to create the seed coat (Sager and Lee, 2014).
Small RNAs are also found in membrane-bound extracellular vesicles (Baldrich et al., 2019; Ruf et al., 2022), providing a plausible mechanism for apoplastic movement of siRNAs. Interestingly, this pathway can transport RNA from one organism to another (Cai et al., 2018), which evokes the movement from the maternal soma to genetically distinct filial tissues. However, at this time we have only a limited understanding of intercellular small RNA transport via extracellular vesicles.
Another interesting question in the mechanism of small RNA movement is whether small RNAs move alone or in complex with RNA binding proteins. Double-stranded siRNA duplexes produced by DCL cleavage are small enough to transport through plasmodesmata, however without the protection of an RNA binding protein, “naked” siRNAs might be subject to degradation. An RNA-binding partner would also allow siRNAs to be targeted to the plasmodesmata, while unbound siRNAs would presumably move through simple diffusion. AGO proteins, the primary binding partner of siRNAs, are large proteins that are unlikely to pass through plasmodesmata (Brosnan et al., 2019; Jullien et al., 2022), but genomes contain many other RNA-binding proteins that might facilitate movement. Indeed, the specific expression of such proteins might enable tissue-specific siRNA mobility. Extracellular vesicles do not have a size exclusion limit, and several RNA-binding proteins have been identified in vesicles, including AGO1 (He et al., 2021). Discovering the mechanism of small RNA transport will undoubtedly unlock new knowledge about the nature and function of mobile small RNAs.
To what extent do reproductive siRNAs mediate heritable, transgenerational epigenetics?
Paramutagenic events following inter- or intra-specific hybridization demonstrate that epigenetic patterns in an individual are at least partially dependent on inherited epigenetic states (Shivaprasad et al., 2012; Gouil and Baulcombe, 2018; Cao et al., 2022). And because these heritable epialleles are sometimes associated with changes in expression of neighboring genes (Cao et al., 2022), an individual’s phenotype might derive from an epigenetic change induced in its ancestors. Yet most epigenetic polymorphisms do not exhibit trans-chromosomal behavior and it is unknown what properties of a locus are necessary for this chromosomal communication. It is also difficult to reconcile paramutagenic events in the face of extensive embryo hypermethylation during development and subsequent loss of methylation during germination (Bouyer et al., 2017; Kawakatsu et al., 2017; Narsai et al., 2017). Improved techniques for assaying epigenetic patterns, including single cell approaches, should enable a better understanding of how stable epigenetic patterns are transmitted.
Another outstanding question is whether siRNAs that are deposited in gametes from somatic cells influence epigenetic states in the zygote. Nurse cell siRNAs, 24-nt reproductive phasiRNAs, and easiRNAs might be loaded into the sperm cell and delivered to the egg cell upon fertilization. Alternatively, they might establish an epigenetic state on patrigenic alleles that could be transmitted to matrigenic alleles after fertilization. Similarly, it is possible that siren siRNA could accumulate in the egg cell before fertilization, or possibly be transported from maternal soma to zygote after fertilization.
How do small RNAs influence diverse plant reproductive systems?
Angiosperms are the dominant class of plants on Earth and their seeds are critically important for humans. But seed production is not the ancestral state for plants, and many extant lineages either do not make seeds, or do not make them through double fertilization as described here. It will be particularly interesting to investigate the role of small RNAs in reproductive development of bryophytes, whose lifecycle is dominated by the haploid gametophyte. For example, dynamic DNA methylation in Marchantia polymorpha suggests that there might be antherozoid (sperm)-specific RdDM (Schmid et al., 2018).
It will also be interesting to understand the rates of birth, death, and mutation for easiRNAs, reproductive phasiRNAs, and siren loci, which can vary even within families (Bélanger et al., 2020; Grover et al., 2020; Pokhrel et al., 2021). Understanding selection to retain these loci and maintain their ability to recognize their targets might help us understand their biological functions.
Conclusion
Advances in sequencing technology have allowed researchers to discover and catalog the many types of siRNAs produced during plant reproductive development and we are developing an increasing understanding of how these siRNAs function. Although we have a strong grasp of how genetic information (i.e. DNA) is transmitted from parent to offspring, we still have much to learn about how epigenetic modifications are faithfully passed between generations. Epigenetic communication between generations via small RNA movement is also an exciting caveat to the standard rules of inheritance. In addition to these fundamental biological questions, understanding how small RNAs influence fertility and seed development will be critical as we attempt to increase agricultural production while minimizing environmental impacts.
Funding
We gratefully acknowledge support from the National Science Foundation (IOS-1546825 to R.A.M.) and the National Institute of Food and Agriculture (AFRI 2021-67013-33797 to R.A.M.).
References
Author notes
Hiu Tung Chow and Rebecca A. Mosher contributed to writing the article.
The author responsible for the distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://dbpia.nl.go.kr/plcell) is: Rebecca A. Mosher ([email protected]).
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}