





Abstract
KIT has been suggested to be a potential therapeutic target for malignant melanoma. We evaluated the antitumor activity and safety of the KIT inhibitor nilotinib in metastatic melanoma patients harboring KIT gene mutations or amplifications.
We conducted a phase II multicenter trial of nilotinib in metastatic malignant melanoma with KIT mutations or amplifications. Patients received 400 mg oral nilotinib twice daily. The primary endpoint was response rate, and if seven or more responders were observed from the cumulative 36 patients, nilotinib would be considered worthy of further testing in this study population.
Between October 2009 and June 2013, 176 patients underwent molecular screening for KIT gene aberrations, and 42 patients harboring KIT gene mutations and/or amplification were enrolled in the study. Overall, 25 (59.5%), 15 (35.7%), and 2 (4.8%) patients had KIT mutations, KIT amplifications, and both KIT mutations and amplification, respectively. Of the 42 enrolled patients, 1 patient achieved complete response, 6 patients achieved partial response, and 17 patients achieved stable disease, resulting in an overall response rate of 16.7% (95% confidence interval [CI]: 5.4%−28.0%) and a disease control rate of 57.1% (95% CI: 42.1%−72.1%). The median duration of response was 34 weeks (range: 5–55 weeks). Of the 7 responders, 6 patients had KIT mutations (exon 11: 5 patients; exon 17: 1 patient), and 1 patient had KIT amplification only.
Although this study did not meet its primary endpoint of response rate, nilotinib showed durable response in a subset of metastatic melanoma patients with specific KIT mutations.
KIT aberration can be detected in a subset of metastatic melanoma patients. This phase II trial showed that nilotinib demonstrates durable response in a subset of patients with KIT mutations. The safety profile was very tolerable. This study suggests that a KIT inhibitor may benefit a small subset of metastatic melanoma patients with KIT mutations.
Introduction
Recent research in the fields of tumor biology and immunology has led to the development of new targeted and immunotherapeutic agents for the treatment of melanoma. The cytotoxic T lymphocyte-associated antigen 4 antibody ipilimumab and blocking of programmed cell death 1 (PD1) and its ligand PDL1 with nivolumab or pembrolizumab have demonstrated clinical efficacy in patients with metastatic melanoma [1–4]. The BRAF inhibitor vemurafenib showed an objective response rate of 48% and survival benefit in melanoma patients harboring BRAF V600E mutation [5, 6]. More recently, a phase III trial demonstrated that dabrafenib plus trametinib improved median progression-free survival (PFS) in melanoma patients with BRAF V600E or V600K mutations compared with dabrafenib alone (9.3 vs. 8.8 months, p = .03) [7]. However, the incidence of BRAF mutations is relatively low in acral and mucosal melanomas, the prevalent subtype in Asians [8, 9].
Intracellular signaling through KIT plays a critical role in melanocyte development [10–12]. Its role as an oncogene and therapeutic target in melanoma has recently emerged. KIT is an established therapeutic target in cancers with KIT-activating mutations, such as gastrointestinal stromal tumors (GISTs). KIT mutations and amplifications have also been identified in melanoma. Curtin et al. found that BRAF and NRAS mutations were infrequent in mucosal and acral melanomas and those resulting from chronic sun-induced damage (CSD), whereas KIT mutations and/or increased copy number were prevalent in mucosal (39%), acral (36%), and CSD (28%) melanomas [13]. In our previous study, increased KIT copy number and KIT mutations were identified in approximately 30% and 7.6% of Chinese melanoma patients, respectively [8]. KIT mutations were detected in 20.7% of CSD, 11.9% of acral, and 9.6% of mucosal melanomas in a Chinese patient cohort (n = 502) [9].
For this subset of melanoma patients, several phase II trials on imatinib have been reported [14–16]. In these studies, response rates ranged from 20%−30%, and exon 11 and 13 mutations were more predictive of imatinib response compared with exon 9, 17, and 18 mutations. We previously conducted and reported a preliminary analysis of this phase II trial [17]. In that report, 2 of 9 evaluable patients achieved partial response, and both showed durable response to KIT inhibitors. A decrease in tumor size from baseline was observed in four of nine patients [17]. Nilotinib is a novel phenylaminopyrimidine derivative with potent activity against the tyrosine kinases BCR-ABL, KIT, discoidin domain receptor, and platelet-derived growth factor receptor. It was approved in 2007 for the treatment of imatinib-resistant and accelerated-phase chronic myeloid leukemia (CML). In 2010, nilotinib received accelerated approval from the U.S. Food and Drug Administration for the treatment of newly diagnosed Philadelphia chromosome-positive CML following a randomized phase III trial demonstrating its superiority over imatinib [18, 19]. Based on these findings, we conducted a phase II clinical trial of nilotinib in patients with metastatic melanoma harboring KIT mutations or amplifications.
Patients and Methods
Study Design
This was an open-label, phase II multicenter study of the Korean Cancer Study Group conducted at nine centers. Patients were enrolled between October 2009 and June 2013. The protocol was approved by the institutional review board of each participating center, and the trial was conducted in accordance with the principles of the Declaration of Helsinki. All patients provided written informed consent before enrollment. Novartis (Seoul, Republic of Korea, https://www.novartis.com) provided nilotinib but was not involved in patient accrual, data analysis, or manuscript preparation. The trial was registered at ClinicalTrials.gov (01099514). We screened all melanoma patients who would be eligible for clinical trial enrollment, and the study was not enriched for acral or mucosal melanoma.
Patient Selection
Eligibility criteria included histologically confirmed melanoma, unresectable stage III or IV melanoma, presence of at least one measurable lesion according to Response Evaluation Criteria in Solid Tumors (RECIST 1.1), Eastern Cooperative Oncology Group (ECOG) performance status of 0−2, normal organ function, and no evidence of progression and normal neurologic function within 8 weeks of enrollment in patients with brain metastasis. Patients were eligible regardless of prior therapy including surgery, radiotherapy, immunotherapy, and cytotoxic chemotherapy. Patients with KIT gene alterations were defined as those with KIT mutations in exons 9, 11, 13, 17, or 18 or KIT gene amplification (>3 gene copies by quantitative polymerase chain reaction), as previously described [8, 20]. Patients who had undergone prior treatment with KIT inhibitors were not eligible for enrollment.
Study Treatment and Dose Modification
Patients received 400 mg oral nilotinib twice daily in 4-week treatment cycles until disease progression or unacceptable toxicity. Toxicities were evaluated according to the National Cancer Institute's Common Toxicity Criteria version 3.0. Grade ≥2 nonhematologic and grade ≥3 hematologic toxicities were managed by holding the drug until resolution to grade ≤1 and then resuming without a dose reduction. If the patient experienced a second toxicity, the drug was reduced to 600 mg once daily. Grade 3 or 4 hepatic toxicities were managed through dose interruption followed by dose reduction to 600 mg once daily.
Response Evaluation
The primary endpoint was response rate, and secondary endpoints were OS, PFS, and toxicity. Radiographic imaging and tumor assessments were performed every 8 weeks based on RECIST 1.1 criteria [21]. Patients with documented disease progression were followed up for survival status every 2 months until death. Evaluable patients were defined as those who had undergone at least one computed tomography (CT) evaluation for nilotinib treatment response. All complete responses (CRs) and partial responses (PRs) were confirmed by repeated CT scan evaluation per RECIST 1.1.
Statistical Considerations
A Simon's two-stage phase II optimal design was used to assess the primary endpoint of response according to the following parameters: reference response rate of 10%; experimental response rate of 30%; and type I and II error rates of 5% and 10%, respectively. Eighteen patients were treated at the first stage, and the study would be stopped early if fewer than three patients responded. Otherwise, the study would proceed to the second stage, in which an additional 18 patients would be treated. If seven or more responders were observed from the cumulative 36 patients, nilotinib would be considered worthy of further testing in this study population. This study was planned to accrue a maximum of 40 patients to account for an attrition rate of up to 10% due to ineligibility or dropout.
Standard descriptive and analytical methods were used to describe the patient population and their baseline characteristics and clinical outcomes. PFS and overall survival (OS) were estimated using the Kaplan-Meier method, and the log-rank test was used to compare cumulative survival in the two groups (patients with clinical benefit vs. progressive disease [PD]). Statistical data were analyzed using SPSS version 18 (IBM Corp., Armonk, NY, http://www.ibm.com).
Results
Molecular Screening
A total of 176 patients consented to undergo molecular screening for KIT gene aberrations. Among 142 patients who had available tissue for KIT mutation testing, 30 (21.1%) had KIT mutations. KIT amplification was detected in 29 of 118 patients (24.6%). Two patients had both KIT mutation and amplification. Of the 59 patients with KIT mutations or amplification, 57 met the eligibility criteria. Fifteen patients were excluded because of other ongoing treatment, patient refusal, poor performance status due to rapidly progressive disease, loss to follow-up, and progressive brain metastasis. A total of 42 were enrolled in the study (Fig. 1).
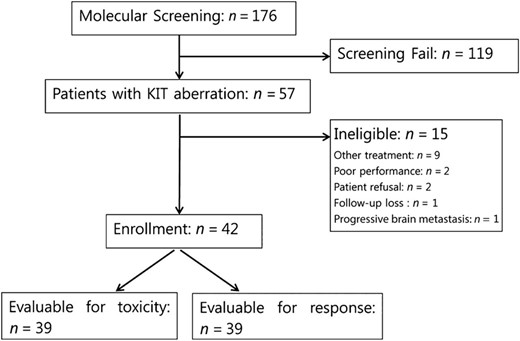
CONSORT diagram of patient enrollment.
Patient Characteristics
Patient characteristics are shown in Table 1. Of the 42 enrolled patients, 21 (50%) had acral melanoma, 12 (28.6%) had mucosal melanoma, and 9 (21.4%) had cutaneous melanoma. The patient population consisted of 21 men and 21 women with a median age of 56 years (range: 28−81 years). Most patients (95.2%) had good performance status (ECOG of 0 or 1). All patients had metastatic disease at the time of study enrollment, and 15 patients had elevated lactate dehydrogenase level at baseline. The most common metastatic site was the lung (61.9%) followed by the distant lymph nodes (54.8%), liver (40.5%), bone (19.0%), brain (11.9%), peritoneum (11.9%), and pleura (9.5%). Overall, 28 patients (66.7%) were previously treated with at least one cytotoxic chemotherapy regimen, and no one had prior immunotherapy such as ipilimumab, nivolumab, or pembrolizumab. A total of 191 4-week cycles of nilotinib were administered to the study participants. The median number of cycles per patient was 3 (range: 1−19 cycles).
Patient characteristics
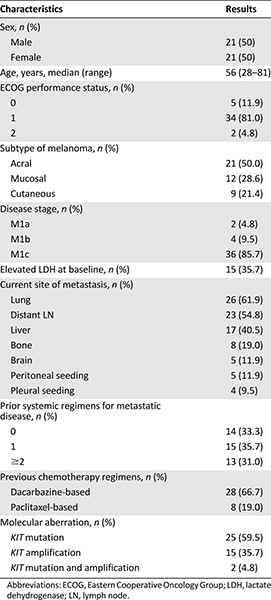
Patient characteristics
Adverse Events
Nilotinib safety was assessed in 40 of the 42 patients (laboratory evaluation was done in 39 patients). Two patients lost to follow-up were not included in the safety assessment. The toxicity profiles of the patients are shown in Table 2. Nilotinib showed a very favorable toxicity profile. Grade 4 toxicities were not observed. Grade 3 leukopenia, anemia, fatigue, skin rash, and lipase elevation were observed in 1 patient, and grade 3 jaundice and liver enzyme elevation were observed in 2 patients. Treatment was delayed in 10 cycles (5.2%) due to jaundice (n = 2), liver enzyme elevation (n = 2), cytopenia (n = 2), lipase elevation (n = 1), skin rash (n = 1), nausea (n = 1), and unknown cause (n = 1). Nine patients underwent a 25% dose reduction because of grade 3 (leukopenia, liver enzyme elevation, jaundice, and lipase elevation) and grade 2 (headache, fatigue, and nausea) toxicities. Nilotinib treatment was terminated in 2 patients because of grade 3 and grade 2 jaundice. The most commonly reported adverse events were anemia (n = 29), skin rash (n = 19), liver enzyme elevation (n = 16), jaundice (n = 17), anorexia (n = 13), fatigue (n = 13), and nausea (n = 12).
Toxicity profile

Toxicity profile
Response and Survival
Tumor response and follow-up data are shown in Figure 2. Three patients discontinued nilotinib before computed tomography response evaluation because of pneumonia (n = 2) and consent withdrawal before study initiation (n = 1). In the intent-to-treat population (n = 42), 1 patient showed complete response, 6 patients showed partial response, and 17 patients showed stable disease (SD), resulting in an overall response rate (ORR) of 16.7% (95% confidence interval [CI]: 5.4%−28.0%) and a disease control rate of 57.1% (95% CI: 42.1%−72.1%). The ORR was significantly greater than the hypothesized null value of 10%, although the lower boundary of the 95% CI (5.4%) for the observed response rate in the intent-to treat population is below the 10% null response rate expected.
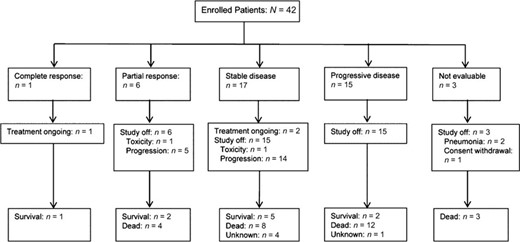
Treatment response to nilotinib.
KIT mutation and amplification status, treatment response and duration, and survival status of individual patients are presented in Table 3. The median duration of response was 34 weeks (range: 5–55 weeks). Among the 7 responders, 6 harbored KIT mutations (exon 11 mutations: 5 patients; exon 17 mutations: 1 patient) and 1 had KIT amplification (KIT copy number: 264) without KIT mutation. In addition, 3 of 5 patients with exon 11 mutations had L576P mutations. Although the ORR was higher in patients with than without KIT mutations, this difference was not significant (22.2% vs. 6.7%; p = .390).
Molecular aberration and treatment response
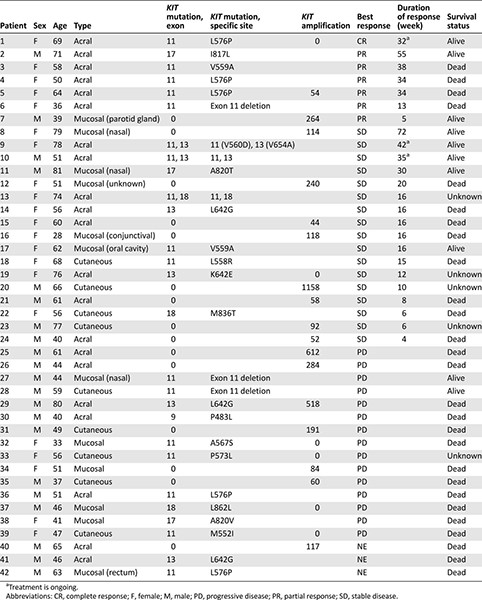
Molecular aberration and treatment response
At the time of the analysis, 3 patients were still receiving ongoing treatment, 39 patients had terminated treatment (disease progression: 34 patients [87.2%]; toxicity: 2 patients [5.1%]; and patient refusal: 1 patient [2.6%]), and 2 patients (5.1%) were lost to follow-up. After a median follow-up duration of 57 weeks (95% CI: 35.7–78.3 weeks), the median PFS for the clinical benefit group (CR/PR/SD) versus the PD group was 34 weeks (95% CI: 10.1–57.9 weeks) versus 7 weeks (95% CI: 5.3–8.7 weeks), respectively (p < .001). Median OS was also longer in the clinical benefit group (74 weeks; 95% CI: 48.0–100.0 weeks) than the PD group (29 weeks; 95% CI: 15.6–42.4 weeks; p < .001) (Fig. 3).
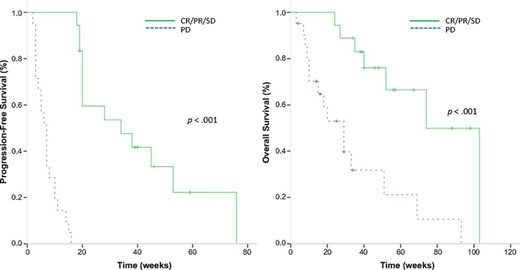
Kaplan-Meier curves of patients with KIT gene aberrations. (A): Progression-free survival (median: 34 vs. 7 weeks; p < .001). (B): Overall survival (median 74 vs. 29 weeks; p < .001).
Abbreviations: CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Discussion
This is our final report on the nilotinib trial in metastatic melanoma patients with KIT mutations or amplifications. We previously reported the preliminary analysis of nilotinib treatment in the first 11 patients [17]. Overall, 1 patient showed complete response, 6 patients showed partial response, and 17 patients showed stable disease, resulting in an ORR of 16.7% (7 of 42 patients) and a disease control rate of 57.1%. Although the ORR was not impressively high, durable responses up to 55 weeks were observed in the 7 responders at the time of analysis.
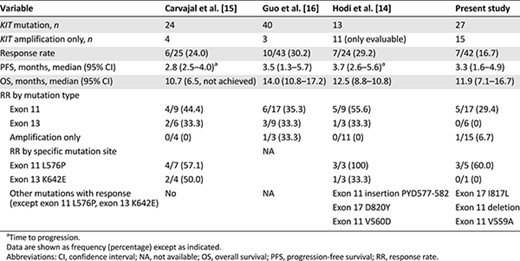
Several studies have demonstrated the value of imatinib in a selected group of patients with metastatic melanoma with KIT alterations [14–16] (Table 4). With careful patient selection, Guo et al. [16] were able to demonstrate significant benefits in a group of patients with metastatic melanoma with either KIT mutations or KIT amplification treated with imatinib. This single-arm, phase II trial enrolled a total of 43 patients, and the overall response rate was 22.0% with 9 PRs. Among 9 responders, all have KIT mutations except 1, who had KIT amplification without mutation. Similarly, Cavajal et al. [15] evaluated 25 patients who harbored the KIT mutation and/or amplification and demonstrated an ORR of 24.0%. The relatively low response rate obtained in this phase II study can be explained by several factors. More than two-thirds of the patient cohort received prior cytotoxic therapies. In addition, approximately 80% of the patient population had acral or mucosal melanoma, subtypes known to exhibit more aggressive clinical behavior [22, 23]. These factors may have affected the response rate to nilotinib.
Correlation of molecular aberration and treatment response
Correlation of molecular aberration and treatment response
Because of their greater potency, the second-generation KIT inhibitors nilotinib and dasatinib are under intense clinical investigation for the treatment of metastatic melanoma with KIT alterations. In particular, ECOG is currently conducting a phase II study of dasatinib in metastatic melanoma patients harboring KIT alterations (ECOG 2607, 00700882). The Tasigna Efficacy in Advanced Melanoma (TEAM) trial is a phase II, single-arm study to assess the efficacy of nilotinib in metastatic melanoma patients (01028222). Patient accrual has been completed for the TEAM trial, and the trial results will be reported soon. These trials will be valuable in further establishing the efficacy and safety of KIT inhibitors in KIT-mutated melanoma. A phase II trial of nilotinib was recently published in the salvage setting following progression to prior KIT inhibition [24]. In a subset of patients, nilotinib achieved durable stabilization.
To correlate KIT aberrations with treatment response to KIT inhibitors, we compared nilotinib response in our study and imatinib response in previous studies [15, 20, 21] according to mutation type and specific mutation site (Table 4). In our study, 6 of the 7 responders had exon 11 mutations. None of the responders had KIT amplification alone; however, 1 responder had concomitant KIT mutation (exon 11 L576P) and amplification. Regarding mutation site, metastatic melanoma patients with exon 11 L576P mutation had the highest ORR in the present study. Reported imatinib response rates in metastatic melanoma patients harboring exon 11 L576P mutation range from 57.1% to 100% [15, 20]. Another KIT point mutation associated with imatinib response is the K642E exon 13 mutation [15, 20] (Table 4). In our study, this particular mutation was detected in 1 patient who had stable disease for 16 weeks. Other mutations associated with nilotinib response in our study included exon 17 I817L and exon 11 V559A mutations. One patient with exon 17 I817L mutation responded to nilotinib for more than 1 year (>55 weeks). This particular missense mutation had not been previously reported to be associated with imatinib response (COSM1651644). Of the 2 patients with exon 11 V559A mutation, 1 showed partial response for 38 weeks and 1 showed stable disease for 16 weeks with nilotinib. V559A and N822I double KIT mutant melanoma has been shown to have some degree of response to imatinib [25]; therefore, our phase II trial demonstrated that nilotinib can induce durable responses in V559A KIT-mutated melanoma. Currently, four primary double KIT mutations in melanoma have been identified in the literature: N566D (exon 11)/K642E (exon 13) [13], N463S (exon 9)/N655S (exon 13) [15], K642E (exon 13)/N822I (exon 17) [26], and V559A (exon 11)/N822I (exon 17) [25]. In our patient cohort, 1 patient had V560D (exon11)/V654A(exon13) (Table 3) and had stable disease for >42weeks on nilotinib. Exon 13 V654A mutation is known as imatinib-resistant mutation in GIST [27]; however, in our study, long-lasting stabilization of the disease was seen in this particular patient. Nevertheless, a preclinical model to test the specific mutation in relation to nilotinib antitumor activity in melanoma should provide stronger evidence to support our clinical finding. In addition, we could not find definite a correlation between KIT amplification and response to KIT inhibitor. Moreover, we did not observe definite correlation between the level of KIT amplification (i.e., 4 vs. >50 copies), although we reported this in our previous pilot report [17].
In a previous study, nilotinib was very well tolerated, with alopecia, skin rash, and headache being the most common adverse events. In our study, most adverse events were grade 1; however, grade 3 jaundice and liver enzyme elevation were each observed in 2 patients. Our findings are consistent with those of previous studies.
Conclusion
Although this study did not meet the primary endpoint of response rate, it showed that nilotinib is efficacious and safe for the treatment of metastatic melanoma with certain KIT aberrations. Patients with melanomas harboring KIT mutations have a high probability of responding to nilotinib but usually with short duration of response compared with KIT-mutant GIST. Nilotinib compares similarly with previous trials with imatinib in terms of response rate and median PFS in melanoma [15, 16]; however, the data do not allow the conclusion that the exon 17 (I817L) mutation or other mutations should preferably be treated with nilotinib. Single-mutant exon 17 mutations may respond to imatinib, and no preclinical evidence shows that these mutants are really resistant. Based on our trial, it is very important to note that KIT amplification alone is very unlikely to predict a response to nilotinib. Taken together with other KIT inhibitor trials in melanoma [15–17], KIT-amplified melanomas without KIT mutations should be excluded from future KIT inhibitor monotherapy trials. Future studies should characterize the actual sensitivity of the KIT mutations found in melanomas because many are different from those in GIST. Future studies should also perform follow-up biopsies to better understand the nature of resistance. The oncogenic dependency on KIT is less pronounced in melanoma than in GIST, and very little is known about the mechanisms of resistance, which clinically evolve quicker than in GIST.
Acknowledgments
This investigator-initiated trial was supported by Novartis. This work was supported by a grant from the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (HI14C0072, HI14C3418).
Author Contributions
Conception/Design: Jeeyun Lee
Provision of study material or patients: Tae Min Kim, Yu Jung Kim, Kee-Taek Jang, Hyo Jin Lee, Soon Nam Lee, Mi Sun Ahn, In Gyu Hwang, Suee Lee, Moon-Hee Lee
Collection and/or assembly of data: Jeeyun Lee, Tae Min Kim, Yu Jung Kim, Hyo Jin Lee, Soon Nam Lee, Mi Sun Ahn, In Gyu Hwang, Suee Lee, Moon-Hee Lee
Data analysis and interpretation: Su Jin Lee
Manuscript writing: Su Jin Lee, Jeeyun Lee
Final approval of manuscript: Jeeyun Lee
Disclosures
The authors indicated no financial relationships.
References
Author notes
Contributed equally.
Disclosures of potential conflicts of interest may be found at the end of this article.

{kind=link}
{kind=link}
{kind=link}