





Abstract
Prostate cancer is the most common noncutaneous cancer and the second leading cause of death from cancer in men in most western countries. Advanced prostate cancer is typically sensitive to androgen‐deprivation therapy, but invariably progresses to the castration‐resistant state. Most current prostate cancer treatments are based on cytotoxicity directed against tumor cells via androgen‐deprivation therapy or chemotherapy. Chemotherapy with docetaxel represents the standard first‐line treatment in patients with castration‐resistant prostate cancer (CRPC). Following progression after treatment with docetaxel, cabazitaxel (XRP6258)–prednisone treatment leads to a significantly longer overall survival (OS) time than with mitoxantrone–prednisone. Several other novel agents are currently being evaluated, including sipuleucel‐T, abiraterone acetate, and MDV3100, as well as the radionuclide alpharadin. The cell‐based immunotherapy sipuleucel‐T produces longer OS times in chemotherapy‐naïve patients, whereas the androgen biosynthesis inhibitor abiraterone acetate results in longer OS times following docetaxel. It is envisioned that these agents will change the standard of care for patients with metastatic CRPC. This review focuses on the clinical development of cabazitaxel and abiraterone acetate.
摘要
前列腺癌是最常见的非上皮性癌症,为大多数西方国家男性的第二大癌症死亡原因。晚期前列腺癌通常对雄激素去势治疗敏感,但都不可避免进展为去势抵抗。目前,前列腺癌治疗大多是通过雄激素去势治疗或化疗所产生的细胞毒性直接杀伤肿瘤细胞。多西他赛化疗是去势难治性前列腺癌(CRPC)患者的标准一线治疗。多西他赛化疗后若发生进展,卡巴他赛(XRP6258)联合泼尼松治疗的总生存期(OS)显著长于米托蒽醌与泼尼松联合方案。还有几种新药目前也正在进行临床试验,包括sipuleucel‐T、醋酸阿比特龙、MDV3100及放射性核素alpharadin(氯化镭‐223)。细胞免疫治疗药物sipuleucel‐T可延长未曾化疗患者的OS,而雄激素生物合成抑制剂醋酸阿比特龙可延长多西他赛经治患者的OS。可以预见,上述药物将会改变转移性CRPC患者的治疗标准。本综述将重点阐述卡巴他赛和醋酸阿比特龙的临床开发。
Background
Prostate cancer is the most frequently diagnosed nonskin malignancy in men, with >200,000 cases being diagnosed each year in the U.S. alone [1]. The treatment of localized disease is controversial, given the prolonged natural history of the disease and the tendency toward overtreatment of indolent cancers that pose little risk to men in their lifetime [2, 3]. Nevertheless, both radiotherapy and surgery are curative options for localized prostate cancer. If surgery fails, salvage radiotherapy has the potential to cure some patients. If initial radiotherapy fails, surgical therapies occasionally can provide benefit. However, once radiotherapy and surgery have both failed, no curative options are currently available.
Treatment of metastatic disease and treatment of prostate cancers that have progressed despite radiation and/or surgical therapies typically involve androgen deprivation. Few comparative studies have been performed on the optimal timing of androgen‐deprivation therapy (ADT), making this a particularly controversial topic in men with prostate‐specific antigen (PSA)‐only disease. The timing of ADT in patients who have experienced a rise in PSA after definitive local therapy is the subject of much discussion. ADT can be achieved by surgical or medical castration, but it is not curative. Castration resistance (cancer progression despite castrate levels of serum testosterone) develops in virtually all patients, and current treatment options are relatively limited in this setting, although it is important to note that many men, particularly those with PSA‐only disease, die as a result of other causes before they ever develop castration resistance.
Docetaxel in Metastatic Castration‐Resistant Prostate Cancer
Until recently, only docetaxel had been shown to produce longer survival times in patients with metastatic castration‐resistant prostate cancer (mCRPC) [4, 5]. The survival benefit is relatively limited (median survival time extended by approximately 2–3 months in large, controlled trials) [4, 5], and for patients progressing after docetaxel there has been no clear standard of care. Various palliative therapies are available but none has led to longer survival times.
Treatment Options Post‐Docetaxel
What to do after docetaxel fails in a patient with mCRPC is the subject of much discussion and research. Part of this discussion has focused on the appropriate control groups to use in phase III trials because the standard of care is not defined. Recent clinical trials have included prednisone alone, placebo, mitoxantrone, and a generically defined “standard of care” as control treatments. In the clinic, the post‐docetaxel treatment setting is difficult because, until recently, no therapy has been shown to be effective and because the patient's life expectancy is limited. Furthermore, this patient population is often elderly with a variety of comorbidities.
The first major study to address post‐docetaxel treatment of mCRPC patients was the Satraplatin and Prednisone Against Refractory Cancer (SPARC) trial [6], which evaluated satraplatin, an orally administered platinum agent, in a total of 950 patients, ∼50% of whom had received docetaxel before trial entry. It should be noted that SPARC trial accrual began before docetaxel was approved by the U.S. Food and Drug Administration (FDA) for prostate cancer. The trial showed a modestly longer median progression‐free survival (PFS) interval for satraplatin plus prednisone (11.1 weeks versus 9.7 weeks), but no significant difference in the overall survival (OS) times. The median OS duration was 14.1 months for both the satraplatin–prednisone and placebo–prednisone arms.
Rechallenge with docetaxel was investigated as an alternative strategy for patients progressing on docetaxel in several small retrospective studies. Results of a study conducted in France suggested that reintroduction of docetaxel at relapse following an initial response to docetaxel provided benefit in some patients [7]. A PSA decrease ≥50% was achieved in 24 of 50 patients (48%) and the median OS time after docetaxel reintroduction was 16 months. Docetaxel retreatment was reported to be well tolerated, with 6% of patients reporting grade 3 or 4 hematologic toxicities. Large, randomized, controlled trials are necessary in order to evaluate this strategy definitively.
Two new agents have been evaluated in the post‐docetaxel CRPC setting. One of these, cabazitaxel (XRP6258/RPR116258A/TXD258), recently showed a longer OS time than with mitoxantrone in a phase III trial and has been approved in the U.S. and Europe for the treatment of men with mCRPC whose disease has progressed after docetaxel. The other, abiraterone acetate, also showed an OS advantage in a phase III trial of 1,195 patients and was recently approved by the U.S. FDA. These two agents are the focus of this review. A brief discussion of other agents currently being evaluated in phase III trials in the post‐docetaxel CRPC setting is also included, because they represent the next generation of options for this difficult‐to‐treat patient population.
Cabazitaxel
Background
The taxanes paclitaxel and docetaxel were originally derived from taxol, a compound extracted from the Pacific Yew Taxus brevifolia [8]. Taxanes are important anticancer agents with activity in a variety of solid tumors, including prostate cancer. Taxanes alter tubulin polymerization dynamics, which impacts microtubule disassembly and can lead to arrested cell division during mitosis [9]. Taxanes may also impact interphase tubulin functions [10] and inhibit androgen receptor (AR) nuclear localization and signaling [11, 12].
Pharmacology and Development
It is well recognized that prostate cancer develops resistance to current taxane‐based regimens. Some patients never respond to taxanes whereas others respond and then progress [13]. In the Southwest Oncology Group 9916 trial, the group receiving docetaxel and estramustine every 3 weeks had a median time to progression (TTP) of 6.3 months and a median OS time of 17.5 months [4]. In the TAX327 trial, the median OS duration was 18.9 months for docetaxel, 75 mg/m2 every 3 weeks [5]. These are the standard dose and schedule currently approved by the U.S. FDA and European Medicines Agency. Multiple mechanisms of taxane resistance have been described, including overexpression of various transmembrane molecular transporters, such as the bile salt export pump (sister gene of P‐glycoprotein) [14] and the multidrug‐resistance pump [15], although the clinical relevance of these mechanisms is unknown.
Efforts have been made to generate novel taxanes with antitumor activity in cancers resistant to approved agents. Cabazitaxel is one such compound with antitumor activity in cell lines resistant to chemotherapy, including docetaxel [16–18]. Cabazitaxel is a taxoid showing cytotoxic activity in cold‐induced depolymerization assays (similar to docetaxel or paclitaxel) [16, 17]. Modifications in the chemical structure of cabazitaxel (Fig. 1) are associated with equipotency versus docetaxel in several cancer cell lines [17], but superior potency in a variety of docetaxel‐resistant cell lines [16, 17]. In cell lines with acquired resistance to doxorubicin, vincristine, vinblastine, paclitaxel, or docetaxel, cabazitaxel was over five times more active than docetaxel [17]. Cabazitaxel has a broad spectrum of antitumor efficacy in tumor models of murine and human origin [16, 19] and is also active in vivo against docetaxel‐resistant tumor models including B16/TXT [16, 20]. Unlike docetaxel, cabazitaxel crosses the blood–brain barrier and is active against early‐stage glioblastoma [21].
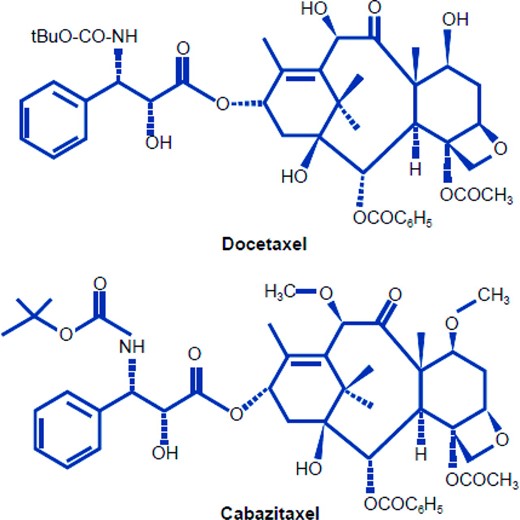
Chemical structure of cabazitaxel and docetaxel.
For solubility reasons, cabazitaxel is formulated in polysorbate 80, and premedication may be required to prevent hypersensitivity reactions, although they appear to occur less frequently than with docetaxel. Dexamethasone is administered i.v. 30 minutes before the administration of cabazitaxel, rather than in multiple doses orally starting the day before treatment, as for docetaxel. Premedication with an i.v. antihistamine and an H2 receptor antagonist is also recommended with cabazitaxel [22]. In the TROPIC trial, the overall incidence of events classified as allergic conditions was low and they were mostly grade 1 or 2 (2% in the cabazitaxel group compared with 1% in the mitoxantrone group). All hypersensitivity events were either grade 1 or grade 2, except for one patient in the cabazitaxel group who experienced serious (grade 4) anaphylactic shock, which occurred 18 days post‐treatment and was considered unrelated to study drug, and was attributed to a nut and fish (food) allergy.
Phase I Study
In a dose‐ranging phase I study, 25 patients with advanced solid tumors were treated with cabazitaxel every 3 weeks [23]. In total, 102 courses were administered at four dose levels in the range of 10–25 mg/m2. The main dose‐limiting toxicity was neutropenia; one patient experienced febrile neutropenia and two others had prolonged grade 4 neutropenia at the 25‐mg/m2 dose. Other toxicities were reported to be generally mild to moderate and included nausea, vomiting, diarrhea, neurotoxicity, and fatigue. Partial responses were observed in two patients with metastatic prostate cancer including a patient with docetaxel‐refractory disease; one unconfirmed partial response and two minor responses were also recorded.
Pharmacokinetic analyses in the phase I study [23] revealed a proportional relationship between cabazitaxel dose and the area under the plasma versus concentration curve from 0 to 48 hours (AUC0–48h) and the maximal plasma concentration. The decline in the cabazitaxel plasma concentration was triphasic, with mean half‐life (t1/2) values of 2.6 minutes, 1.3 hours, and 77.3 hours in the first, second, and third phases, respectively. Clearance rates averaged 53.5 L/hour. The clearance and AUC(0–48h) values did not change with repeated treatment. Interpatient variability in pharmacokinetic values was relatively low, although there was higher variability in terminal (third‐phase) t1/2 values. The pharmacokinetic profile of cabazitaxel within the dose ranges studied was generally similar to that of docetaxel (triphasic elimination, proportional dose) [24].
Cabazitaxel is metabolized primarily by liver cytochrome P450 enzymes CYP3A4 and CYP3A5, and to a lesser extent via CYP2C8 [22]. The parent drug is the primary form found in the circulation [22]. Cabazitaxel is mainly eliminated in the feces (∼76% of the dose), whereas renal elimination accounts for only 3.7% of the dose [22]. Because cabazitaxel is mainly metabolized via CYP3A, substances that induce or inhibit this enzyme may affect cabazitaxel pharmacokinetics.
Phase II Data
One cabazitaxel phase II study was performed in patients with metastatic breast cancer resistant to prior taxanes [25]. Patients were divided into those progressing after adjuvant therapy and those progressing after first‐ or second‐line therapy and were treated with cabazitaxel at a dose of 20 mg/m2 every 3 weeks. Grade 3 or 4 neutropenia was reported in 52 of 71 patients (73%); 20 patients (28%) with good tolerability after cycle 1 received cabazitaxel at a dose of 25 mg/m2 in cycle 2. Two complete and eight partial responses were noted. The response rate for patients progressing after neoadjuvant or adjuvant treatment was 14%, and for those progressing after first‐ or second‐line therapy the response rate was 12%. The median TTP was 2.7 months (95% confidence interval [CI], 1.45–4.07 months) and the median OS time was 12.3 months (9.49–15.05 months).
Phase III Trial: TROPIC
Based on clinical data from phase I trials demonstrating antitumor activity in docetaxel‐pretreated patients, including those with docetaxel‐refractory prostate cancer, a decision was made to evaluate the efficacy of cabazitaxel in a phase III trial (TROPIC) in patients with mCRPC progressing after docetaxel‐based therapy [26]. A summary of the design of the TROPIC trial is shown in Table 1. Additional entry criteria for TROPIC included evidence of cancer progression by the Response Evaluation Criteria in Solid Tumors (RECIST) or PSA criteria either during or after docetaxel therapy, no prior treatment with mitoxantrone, an Eastern Cooperative Oncology Group (ECOG) performance status score of 0–2, adequate organ function, a cardiac ejection fraction >50%, no brain or leptomeningeal metastases, and no concurrent or planned treatment with strong inhibitors of CYP3A4 or CYP3A5. Inclusion and exclusion criteria were not based on the response to prior docetaxel.
Phase III randomized trials with cabazitaxel and abiraterone acetate
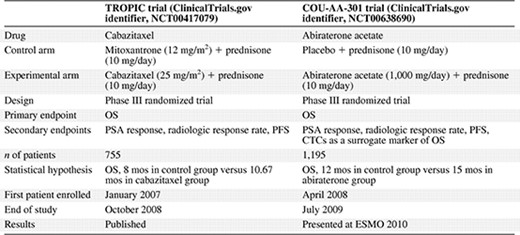
Phase III randomized trials with cabazitaxel and abiraterone acetate
The TROPIC study was initiated in January 2007 and the last patient was enrolled in October 2008. In total, 755 patients were randomized 1:1 to cabazitaxel or mitoxantrone. The primary endpoint was OS and the predefined statistical plan required 511 events in an intent‐to‐treat analysis to detect a 25% difference in the hazard rate between the two arms with 90% power at a two‐sided 5% α level. An assumption was made that survival would be 8.0 months in the control group and 10.7 months in the experimental group [26]. Based on current knowledge of the survival rates from the SPARC trial [6], this may have been an overly pessimistic estimate.
Secondary endpoints included PSA response (for patients with a baseline PSA level ≥20 ng/mL) and PSA progression (≥25% increase from the nadir in PSA nonresponders or ≥50% increase from the nadir in PSA responders). Tumor response (RECIST) was assessed by cross‐sectional imaging (computed tomography or magnetic resonance imaging) every other treatment cycle. Bone scans were performed at the beginning and end of the study treatments, and when clinically indicated. PFS was defined as the time between randomization and the first date of progression, as measured by PSA progression, tumor progression, pain progression, or death [26].
Cabazitaxel was administered i.v. at a dose of 25 mg/m2, with the control group receiving i.v. mitoxantrone at a dose of 12 mg/m2. Patients in both arms received oral prednisone, 10 mg/day. A maximum of 10 cycles of therapy was allowed because of concerns about cardiac toxicity with mitoxantrone. Of note, the TAX327 trial comparing mitoxantrone with docetaxel also limited the maximum number of cycles to 10 [5]. Treatment was discontinued because of disease progression, because of unacceptable toxicity, or after 10 cycles. Given prior data from the SPARC trial [6], life expectancy in this cohort of patients was expected to be ∼14 months.
In an intention‐to‐treat analysis, the median OS time was 15.1 months in patients who received cabazitaxel, compared with 12.7 months in those who received mitoxantrone (Fig. 2) [26]. Patients in the cabazitaxel group had a hazard ratio for death of 0.70 (95% CI, 0.59–0.83; p < .0001 by stratified log‐rank test) relative to those who received mitoxantrone. The PFS interval, tumor response rate, PSA response rate, and TTP were also better with cabazitaxel than with mitoxantrone (Table 2). The most frequent, clinically significant grade ≥3 toxicities were neutropenia (cabazitaxel, 81.7%; mitoxantrone, 58.0%) and diarrhea (cabazitaxel, 6.2%; mitoxantrone, 0.3%); the rates of febrile neutropenia were 7.5% and 1.3%, respectively (Table 2).
![Overall survival with cabazitaxel (TROPIC) [26].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/oncolo/16/11/10.1634_theoncologist.2010-0412/5/m_oncolo_16_11_1487_f2.jpeg?Expires=1748302147&Signature=3Nuj0lrioAaSRWbTPn3Il3Q5OO-u~T6BSv4679HCPpi1bQzjBdK6jVbJZ-bgpnjuCwA3YfHSwkzYY14tyvZphvYDAC3YAgkVZT3OFIDs~dcXTYfU1ooC9d-VV0rpZegb0OTcySVbh0nZIibArtFPgsBKQJ7CMaPHBTwdsxRx5twboAn58rEaM9GiW3zFGEpAIyo~bBgJqGyCVndHPLxI5gZ03CwQk4V7S7oAEHoyrHednzYpajdmRhd79TeieZMVG3EAZo4AdFRQuiDJkaWRPY-bh9BdvQd4FJAzmiYv-H0SnZ3BLCiXXkjU~x433W9YcvreZ4qPVD212jRSJMFGnQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Overall survival with cabazitaxel (TROPIC) [26].
Abbreviation: CI, confidence interval.
Reprinted from de Bono JS, Oudard S, Ozguroglu M et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration‐resistant prostate cancer progressing after docetaxel treatment: A randomised open‐label trial. Lancet 2010;376:1147–1154, Copyright 2010, with permission from Elsevier.
Summary of efficacy results and adverse events from the TROPIC trial
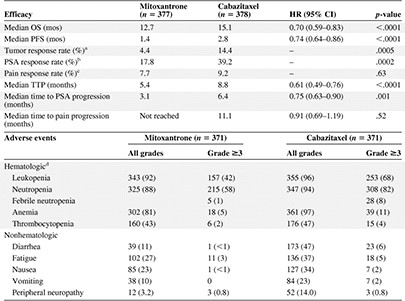
Summary of efficacy results and adverse events from the TROPIC trial
The overall rate of death within 30 days of drug infusion was 4.9% for patients treated with cabazitaxel and 2.4% for patients treated with mitoxantrone. Neutropenia and associated complications were the most frequent cause of death in the cabazitaxel group (1.9% of treated patients). Potential cardiac issues were associated with deaths in 1.3% of patients in the cabazitaxel group [26].
Trials have not yet defined the optimal strategy for the management of treatment‐induced neutropenia and related risks in this population. At this time, the FDA‐approved cabazitaxel label recommends considering the use of G‐CSF as primary prophylaxis for men with high‐risk clinical features known to predispose them to complications from prolonged neutropenia (i.e., age >65 years, poor performance status, serious comorbidities) [22]. Additional studies to evaluate whether or not pharmacogenomic predictors of drug disposition and neutropenic sepsis could be used clinically to personalize drug dosing are now warranted. Other strategies to deal with neutropenic sepsis risk include dose reductions from 25 mg/m2 to 20 mg/m2 in patients with grade 4 neutropenia following their first course of treatment and the routine use of prophylactic growth factors to abrogate myelosuppression. The dose of cabazitaxel in the post‐docetaxel setting is also being investigated in a current phase III trial comparing the 20 mg/m2 and 25 mg/m2 doses (PROSELICA trial; ClinicalTrials.gov identifier, NCT01308580).
Abiraterone Acetate
Background
The treatment of patients with advanced or high‐risk prostate cancer has been based on androgen deprivation, with the objective of diminishing testicular testosterone production, either by bilateral orchiectomy or with the use of luteinizing‐hormone releasing hormone (LHRH) agonists. Despite continuing ADT, the disease eventually progresses [27]. Preclinical and clinical studies indicate that the AR continues to drive the proliferation of CRPC cells. In CRPC, the AR axis remains active with continued activation of downstream genes [28]. Abiraterone acetate is an oral, selective, irreversible, inhibitor of CYP17 that inhibits androgen and estrogen synthesis (Fig. 3). In patients with CRPC resistant to LHRH analogs, abiraterone acetate has shown impressive antitumor activity [29, 30].
![Abiraterone acetate mode of action and chemical structure. (A): The cytochrome P450 enzyme CYP17 and steroids. Androgen biosynthesis pathway. The physiological effects of abiraterone acetate on steroidogenesis are indicated by arrows next to each steroid precursor. Abiraterone acetate inhibits 17α‐hydroxylase (blunt arrow), causing a decline in serum cortisol and a consequent rise in adrenocorticotrophic hormone (ACTH) (broken arrow). This, in turn, results in a rise in deoxycorticosterone and corticosterone by approximately 10‐ and 40‐fold, respectively. The elevated deoxycorticosterone levels result in the expected toxicities of secondary mineralocorticoid syndrome. Abiraterone acetate also inhibits C17,20‐lyase (blunt arrow), resulting in significant declines in dehydroepiandrosterone (DHEA), androstenedione, and testosterone. Aldosterone levels fall as a result of suppression of the renin–angiotensin pathway by high levels of deoxycorticosterone. However, there is a fourfold increase in 11‐deoxycortisol, which may be a result of the higher ACTH levels driving the partial reversal of the activity of 17α‐hydroxylase but not C17,20‐lyase. (B): Chemical structures of abiraterone acetate and abiraterone [29].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/oncolo/16/11/10.1634_theoncologist.2010-0412/5/m_oncolo_16_11_1487_f3.jpeg?Expires=1748302147&Signature=bxdLYrDda3fZAC80I4fj~AUzMpblgQmDiqaUBcato1IQ2i3PoqfqjcQKJYcJXHHhUgdqJto47ORuMy7xHUOs2sBKt1sa4nQAFV40JzDL8KDRsYvKjc2daVSdtyqshMO-KNIomFBI5SkHURqbmNcRjYUpDwCnJm-QKyhVhgkKn8yNWJDuJMwuH~IeOur6bXfheQbep0eAu~QME5M2K7U5Qo62CVcpCy6vUSNEivXaneBExZjcWXR9VAz3nO9tjKZmSP4wvNM23bJVuDsJXP8eZ6X9c7fWkpONHqaJBLPIvEE3bIkSw71MfvaoS-MblBcB4QnxaCYgspvoVlzMkEmzmA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Abiraterone acetate mode of action and chemical structure. (A): The cytochrome P450 enzyme CYP17 and steroids. Androgen biosynthesis pathway. The physiological effects of abiraterone acetate on steroidogenesis are indicated by arrows next to each steroid precursor. Abiraterone acetate inhibits 17α‐hydroxylase (blunt arrow), causing a decline in serum cortisol and a consequent rise in adrenocorticotrophic hormone (ACTH) (broken arrow). This, in turn, results in a rise in deoxycorticosterone and corticosterone by approximately 10‐ and 40‐fold, respectively. The elevated deoxycorticosterone levels result in the expected toxicities of secondary mineralocorticoid syndrome. Abiraterone acetate also inhibits C17,20‐lyase (blunt arrow), resulting in significant declines in dehydroepiandrosterone (DHEA), androstenedione, and testosterone. Aldosterone levels fall as a result of suppression of the renin–angiotensin pathway by high levels of deoxycorticosterone. However, there is a fourfold increase in 11‐deoxycortisol, which may be a result of the higher ACTH levels driving the partial reversal of the activity of 17α‐hydroxylase but not C17,20‐lyase. (B): Chemical structures of abiraterone acetate and abiraterone [29].
Reprinted from Ang JE, Olmos D, de Bono JS. CYP17 blockade by abiraterone: Further evidence for frequent continued hormone‐dependence in castration‐resistant prostate cancer. Br J Cancer 2009;100:671–675, Copyright 2009, with permission from Macmillan Publishers Ltd.
Pharmacology and Development
Several preclinical and clinical studies have shown that, despite being characterized as “hormone refractory,” that is, relapsing after initial hormone ablation [31], prostate cancers continue to be AR driven [32, 33]. There are several known mechanisms of resistance to ADT, most of which result in greater AR signaling. These include intracrine steroid synthesis; amplification of the AR gene; constitutive, ligand‐independent activation of AR; AR mutations that decrease AR specificity and increase AR promiscuity; and ligand‐independent AR activation by protein kinases or other effectors [34, 35].
The importance of continued AR signaling in CRPC supported the investigation of the inhibition of CYP17 by abiraterone to block extragonadal sources of steroid and to block intratumoral androgen and estrogen synthesis [32]. This strategy should impact CRPC driven by ligand‐dependent AR signaling [28, 36, 37]. CYP17 is a key enzyme in the production of androgens and estrogens by the adrenal glands and tumor tissue (Fig. 3). In patients with congenital CYP17 deficiency, lower production of cortisol, androgens, and estrogens leads to absent sexual development [38]. Glucocorticoid generation is maintained in these patients through the synthesis of corticosterone, which explains why they do not develop adrenal insufficiency. However, CYP17 blockade results in high adrenocorticotrophic hormone (ACTH) levels and a syndrome of secondary mineralocorticoid excess. This can be managed with mineralocorticoid antagonists or low doses of glucocorticoids.
Abiraterone acetate (CB 7630; 3β‐acetoxy‐17‐(3‐pyridyl)androsta‐5,16‐diene) was designed and first synthesized at The Institute of Cancer Research in Sutton, U.K. [39–41] as an androgen and estrogen synthesis inhibitor. Abiraterone acetate is the 3‐acetate prodrug of abiraterone; the acetate salt is more soluble than the parent compound and rapidly converted to abiraterone following absorption (Fig. 3). By irreversibly inhibiting CYP17, also known as 17α‐hydroxylase or C17,20‐lyase, abiraterone inhibits both adrenal and intratumoral androgen synthesis. Preclinical studies showed that abiraterone acetate reduced the volume of androgen‐dependent organs, including the ventral prostate, seminal vesicles, and testes, significantly more than ketoconazole.
Phase I Studies
A first‐in‐man phase I study showed that treatment with abiraterone acetate resulted in acceptable safety and tolerability, but the agent was only administered for a maximum of 12 days [42]. That study demonstrated that it is possible to suppress testosterone levels to the castrate range in men with intact gonadal function at an abiraterone acetate dose of 800 mg. Antitumor activity was not evaluated, but the study provided proof of principle that the drug could block CYP17, thus warranting a further clinical trial in patients with CRPC.
A phase I/II trial (COU‐AA‐001) in patients with chemotherapy‐naïve CRPC was designed to evaluate the safety and efficacy of continuous abiraterone acetate administered once daily as a capsule formulation; the dose was escalated from 250 mg/day to 2,000 mg/day [43]. Abiraterone acetate had an acceptable safety profile and antitumor activity at all evaluated dose levels. The most frequent side effects were related to a secondary mineralocorticoid excess syndrome, with hypertension, hypokalemia, and lower‐limb edema. These side effects were managed with the mineralocorticoid receptor antagonist eplerenone. Spironolactone was avoided because it activates the AR [44]. Abiraterone acetate treatment induced increases in ACTH and steroids upstream of CYP17, and decreases in serum testosterone, androgenic steroids, and estradiol. No patient developed adrenocortical insufficiency, as anticipated from the natural history of congenital syndromes of CYP17 deficiency. Antitumor activity was observed at all doses, with declines in PSA, radiologic partial responses, and improvement in symptoms. In that study, 66% of treated patients had a ≥30% decline in PSA levels; 38% showed a partial response (RECIST) or reduction in analgesic use. This first phase I trial in chemotherapy‐ and ketoconazole‐naïve patients with CRPC confirmed that CYP17 blockade by abiraterone acetate has an acceptable safety profile and antitumor activity in CRPC patients. Moreover, patients received abiraterone acetate in that study in an extension protocol for up to 48 months.
A second phase I/II study (COU‐AA‐002) [45], evaluating the safety and tolerability of a tablet formulation of abiraterone acetate at doses in the range of 250–1,000 mg, also found an acceptable safety profile for further development. Consistent with abiraterone acetate's mechanism of action, hypertension, hypokalemia, and lower extremity edema were the most commonly observed drug‐related adverse events (AEs); these were all manageable with mineralocorticoid antagonists or low‐dose steroids. Adrenal metabolite analysis showed inhibition of CYP17 even at low abiraterone doses and an ACTH‐driven compensatory increase in levels of corticosterone and deoxycorticosterone. Data from dose‐finding studies indicated that when pharmacokinetic, adrenal CYP17 inhibition, and efficacy signals were taken into consideration, the 1,000‐mg dose offered consistent pharmacologic effects without additional side effects. Therefore, this dose was chosen for further efficacy and safety evaluation in phase II and III studies.
Phase II Data
After the very promising phase I results, several phase II studies were conducted to evaluate the efficacy and toxicity of abiraterone acetate in both chemotherapy‐naïve and taxane‐resistant CRPC patients [45–51] (Table 3).
Phase I/II trials with abiraterone acetate

Phase I/II trials with abiraterone acetate
In docetaxel‐naïve patients, the PSA response rate was 60%–80% [46, 50]. Following expansion at the 1,000‐mg dose, the COU‐AA‐001 study enrolled additional patients to further evaluate antitumor activity in patients with chemotherapy‐naïve CRPC [46]. Chemotherapy‐naïve men (n = 54) with CRPC resistant to multiple hormonal therapies were treated in this two‐stage phase I/II study. Declines in PSA ≥30%, ≥50%, and ≥90% were observed in 43 (80%), 38 (70%), and 14 (26%) patients, respectively. Independent, blinded, radiologic evaluation reported disease regression by the RECIST in 37% of patients (nine of 24 patients with measurable disease). Decreases in circulating tumor cell (CTC) counts, normalization of lactate dehydrogenase, and improved symptoms with a reduction in analgesic use were commonly documented. Similar response rates were seen in the COU‐AA‐002 trial, a parallel phase I/II study investigating the tablet as opposed to the capsule formulation [45, 50].
Two phase II studies have also been conducted in patients with CRPC who had received prior docetaxel. In one phase II study (COU‐AA‐003), 47 patients were treated with abiraterone acetate at a dose of 1,000 mg/day; 18 started the study on a stable dose of steroids to maintain performance status [49]. Declines in PSA ≥30%, ≥50%, and ≥90% were observed in 32 (68%), 24 (51%) and seven (15%) patients, respectively. Moreover, of the 30 patients evaluable by the RECIST, eight (27%) had a partial response. In this heavily pretreated population, more than half of the patients had a PSA response and more than two thirds had stable disease or a partial response.
The other phase II study (COU‐AA‐004) [47] evaluated abiraterone acetate at a dose of 1,000 mg/day with prednisone (5 mg twice daily) in 58 men with mCRPC who had experienced treatment failure on docetaxel‐based chemotherapy; 27 of the patients had also previously received ketoconazole. PSA declines ≥50% were confirmed in 21 (36%) patients—14 of 31 (45%) ketoconazole‐naïve patients and seven of 27 (26%) ketoconazole‐pretreated patients. Partial tumor responses were observed in four of 22 (18%) patients, with improved ECOG performance status scores in 28% of patients. The vast majority of AEs related to abiraterone treatment were grade 1–2, with the most common being fatigue, nausea, and vomiting. There was one case of grade 3 fatigue and no grade 4 events were observed [47].
To date, no relationship has been reported between response to abiraterone acetate and true progression on prior docetaxel chemotherapy or stopping docetaxel for another reason, such as toxicity. The drug has been well tolerated in the post‐docetaxel setting with toxicities similar to those seen in docetaxel‐naïve patients.
Post‐Docetaxel Phase III Evaluation
The high degree of antitumor activity seen with abiraterone acetate in combination with steroids in patients with CRPC, together with a favorable toxicity profile, supported the study of this regimen in phase III trials. A large, multicenter, randomized, double‐blind, placebo‐controlled phase III trial assessing abiraterone acetate and prednisone was initiated in April 2008 in patients with mCRPC who had failed docetaxel‐based chemotherapy (ClinicalTrials.gov identifier, NCT00638690) (Table 1) and completed accrual in July 2009. The primary endpoint of that trial was OS. The trial enrolled 1,195 patients, randomized 2:1 to receive abiraterone acetate plus prednisolone (or prednisone) or placebo plus prednisolone (or prednisone). In the trial, abiraterone led to a longer OS time at the time of an interim analysis, by 3.9 months compared with placebo [52], with a hazard ratio of 0.646 (95% CI, 0.543–0.768; p < .0001) (Table 4). Abiraterone was well tolerated; however, some AEs were deemed of special interest, including those associated with elevated mineralocorticoid levels resulting from CYP17 blockade (fluid retention and edema, hypokalemia, and hypertension), as well as cardiac disorders and liver function test abnormalities. These AEs were more common in the abiraterone acetate group than in the placebo group (55% versus 43%; p < .001) [52]. There was no significant difference in the rate of cardiac events between the abiraterone acetate group and the placebo group (13% versus 11%; p = .14) [52]. Tachycardia and atrial fibrillation were the most common cardiac events, occurring in 3% and 2% of patients in the abiraterone group, respectively. All tachycardia events were grade 1 or 2, whereas all atrial fibrillation events were grade ≤3. There were not significantly more cardiac deaths in the abiraterone acetate group than in the placebo group (1.1% versus 1.3%, respectively) [52]. No individual grade 4 AEs occurred in ≥2% of patients in either treatment group.
Abiraterone phase III trial results
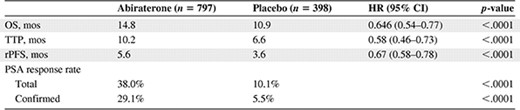
Abiraterone phase III trial results
As a consequence of these findings, the data monitoring committee informed the sponsor to notify participants in the placebo arm about the option to cross over to abiraterone. All secondary endpoints, including time to PSA progression, time to radiologic progression, and the PSA response rate, favored the abiraterone arm. However, clinicians must be aware of the potential for life‐threatening side effects with abiraterone, such as fatal arrhythmias resulting from hypokalemia, and careful monitoring of electrolyte and liver function tests are essential during abiraterone therapy.
This large, phase III trial will also prospectively assess whether treatment‐induced changes in CTC counts can serve as a surrogate endpoint for OS in CRPC patients. Preliminary studies indicate that CTCs represent an extremely promising source of tumor tissue for biomarker studies and that CTC counts are prognostic of OS in breast, colon, and prostate cancer patients, suggesting that this approach may even be superior to radiologic or PSA evaluation [53, 54].
Docetaxel‐Naïve Phase III Evaluation
A placebo‐controlled, randomized, phase III study with the aim of assessing whether or not abiraterone improves outcomes in the docetaxel‐naïve setting has now completed accrual (ClinicalTrials.gov identifier, NCT00887198). More than 1,000 patients were randomized 1:1 to abiraterone acetate plus prednisolone or placebo plus prednisolone, with coprimary endpoints of the radiologic‐free survival and OS times.
Biomarkers of Abiraterone Antitumor Activity
The antitumor activity of abiraterone acetate is partially explained by the suppression of serum androstenedione and dehydroepiandrosterone (DHEA), in addition to testosterone and estradiol. Patients with higher levels of androstenedione and DHEA at baseline had a better response rate than patients with lower levels [55]. Resistance to abiraterone may also be related to greater production of steroids upstream of CYP17. To reverse resistance to abiraterone administered in the absence of therapeutic steroids but with a mineralocorticoid receptor antagonist, low‐dose steroids were successfully used to decrease production of ACTH and upstream steroids at disease progression on abiraterone alone. Low‐dose steroids inhibit the ACTH feedback loop and upstream steroid precursor synthesis, which can activate promiscuous ARs [55].
Different molecular alterations could be involved in abiraterone resistance. We now have preliminary evidence to suggest that the presence of a rearrangement of the ETS transcription factor gene ERG, specifically the TMPRSS2‐ERG fusion gene, which generates an AR and estrogen receptor‐α driven ETS oncogene, may correlate with a higher likelihood of response to abiraterone [55–57]. Further studies evaluating AR mutations are now required. Several reports indicate that splice variants can result in ligand‐independent AR signaling [58, 59]. In addition, mutated AR can be activated by ligands upstream of CYP17, including deoxycorticosterone and pregnenolone [60], or a constitutively activated AR.
Newer Agents in Development in the Post‐Docetaxel Space
In addition to cabazitaxel and abiraterone, there are many other agents with the potential to prolong survival after docetaxel in patients with mCRPC. Many of these are being evaluated in phase III studies [61], including the anti‐androgen MDV3100 (ClinicalTrials.gov identifiers, NCT00974311, NCT01212991), the CYP17 inhibitor TAK‐700 (ClinicalTrials.gov identifiers, NCT01193257, NCT01193244), and ipilimumab, an anticytotoxic T lymphocyte–associated antigen‐4 antibody (ClinicalTrials.gov identifiers, NCT01057810, NCT00861614). Phase III trials are also being carried out to evaluate the bone targeting α‐emitting isotope radium‐223 (ClinicalTrials.gov identifier, NCT00699751), and OGX‐011 [61] (ClinicalTrials.gov identifiers, NCT01188187, NCT01083615), a targeted therapeutic that sensitizes tumors that have become resistant to conventional cancer therapeutics. Radium‐233 has previously demonstrated positive results in a randomized phase II trial [62] and is currently being evaluated in patients unsuitable for additional chemotherapy in the phase III setting. Recent data indicate a survival benefit with this agent [63]. This placebo‐controlled alpharadin (radium‐223 chloride) randomized, phase III registration trial has been announced to show a survival advantage for this radionuclide with an almost 3‐month survival benefit. At interim analysis, a statistically significant improvement in overall survival in patients receiving alpharadin of 14 months compared with 11.2 months in those receiving placebo (p=.0022; hazard ratio, 0.699) has been described. The full reporting of these data is anticipated and it is envisioned that this agent will become a standard of care in the treatment of advanced prostate cancer. Randomized phase II data have suggested a survival benefit with OGX‐011 [64] used in combination with docetaxel. OGX‐011 is now being investigated in phase III trials [61]. Other exciting agents in development include novel androgen receptor–targeting drugs such as the selective AR degrading agent AZD3514, drugs targeting the PI3K/AKT/TOR kinase axis, and the VEGFR/C‐Met–targeting multikinase inhibitor cabozantinib.
These are some of the most promising agents currently being investigated for the treatment of mCRPC patients. However, one of the greatest challenges in the development of novel agents is the development of analytically validated biomarkers that can be clinically qualified as both predictor assays and surrogates for intermediate endpoints.
Conclusions
Both cabazitaxel and abiraterone acetate clearly have antitumor activity in CRPC patients; they have both led to longer OS times in patients who have progressed on prior docetaxel. Moreover, the cell‐based immunotherapy sipuleucel‐T has been shown to produce longer survival times in the prechemotherapy setting [65]. Additional phase III trials to evaluate the optimal sequence of drug delivery now need to be pursued in order to optimize the benefits of treatment. It is likely that the near future will bring several other new therapeutic possibilities for patients with this disease. Other promising new agents are in development, including the antiandrogen MDV3100. Finally, we envision that predictive biomarkers, such as ETS gene rearrangements, will allow the molecular dissection of the heterogeneity of this disease and that CTC counts could become an approvable clinical trial endpoint for this disease.
(C/A), consulting/advisory relationship; (RF) Research funding; (E) Employment; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder
Author Contributions
Conception/Design: Johann Sebastian de Bono, Oliver Sartor, Ross M. Michels, Christophe Massard
Collection and/or assembly of data: Johann Sebastian de Bono, Oliver Sartor, Ross M. Michels, Christophe Massard
Data analysis and interpretation: Johann Sebastian de Bono, Oliver Sartor, Ross M. Michels, Christophe Massard
Manuscript writing: Johann Sebastian de Bono, Oliver Sartor, Ross M. Michels, Christophe Massard
Final approval of manuscript: Johann Sebastian de Bono, Oliver Sartor, Ross M. Michels, Christophe Massard
The authors take full responsibility for the content of the paper but thank Neil Anderson, Ph.D. (Adelphi Communications Ltd, supported by Sanofi‐Aventis), for his editorial, copyediting, and production assistance.
References
Author notes
Disclosures: Oliver Sartor: ASTRO (C/A); Cougar, Johnson & Johnson (RF); Sanofi (H, RF); Ross M. Michels: None; Christophe Massard: None; Johann Sebastian de Bono: AstraZeneca, Genentech (H); Sanofi (H, C/A); Medivation (H, RF); Johnson & Johnson, Astellas (H, C/A, RF).

{kind=link}
{kind=link}
{kind=link}