





Abstract
Imatinib mesylate has transformed the treatment for chronic myeloid leukemia (CML). The vast majority of patients obtain hematologic remission, with a low probability of progression of disease. Yet imatinib rarely cures CML, and current recommendations dictate lifelong treatment with imatinib. In this review we analyze the biology behind the failure of imatinib to fully eradicate CML. We review evidence that indicates that the leukemic stem cell for CML is inherently resistant to imatinib, and that imatinib treatment itself may enhance this resistance.
Introduction
The development of imatinib mesylate has transformed the treatment of chronic myeloid leukemia (CML), and has become the paradigm for targeted therapy [1]. CML is caused by abnormal signaling through the BCR-ABL fusion protein, a product of the t(9,22) Philadelphia chromosome [2]. Imatinib binds with high affinity and specificity to BCR-ABL, blocking its kinase domain and inhibiting the proliferation of CML progenitors. Imatinib is active in all phases of CML, and patients in chronic phase treated with imatinib have a >85% 5-year survival rate [3].
Despite its impressive response rate, imatinib is rarely curative. In the landmark International Randomized Study of Interferon and STI571 (IRIS) trial, 96% of patients with chronic-phase CML continued to have minimal residual disease throughout 5 years of follow-up [3]. The majority of patients maintained on imatinib displayed a three-log decrease in the burden of BCR-ABL–expressing cells, but only 4% achieved polymerase chain reaction (PCR) negativity. Yet those who continued to maintain low levels of leukemic cells had a minimal rate of progression. Other studies have reported the incidence of obtaining at least one negative PCR result to be higher, up to 32% [4–6]. However, it is faulty to equate PCR negativity with cure. Multiple case reports indicate that the majority of patients who discontinue imatinib, even after obtaining PCR negativity, relapse [7, 8]. In the largest analysis of patients with sustained PCR negativity who then stopped imatinib, Mahon et al. [9] reported a relapse in 19 of 34 patients followed for at least 6 months. Hence, current dogma dictates that patients need to take imatinib for life [10].
In this review, we explore the potential basis for the failure of imatinib to cure CML.
CML Stem Cells
The hypothesis that a rare subpopulation of leukemic cells retains the potential to self-renew as well as generates malignant progeny was substantiated by the discoveries of Dick and colleagues over a decade ago [11]. Those investigators found that a subpopulation of leukemic cells with the phenotype CD34+CD38− was capable of serial transplantation of acute myeloid leukemia (AML) in a murine model. Other subpopulations of leukemic cells, which made up the majority of the AML, were unable to serve as leukemia-initiating cells (LICs) in the xenotropic model. LICs do not necessarily represent malignant transformation of hematopoietic stem cells; in many model systems, introduction of leukemic oncogenes into more mature progenitors results in acquisition of characteristics of “stemness,” including expression of many genes associated with self-renewal (Bmi-1, Hedgehog, telomerase, HOX, Notch) [12]. LICs are rare, representing between one per 1,000,000 and one per 10,000 leukemic cells. The molecular characterization of these cells was covered in recent reviews and is not detailed here [13].
Following in the footsteps of Dick's early work on AML, cancer-imitating cells have since been identified as playing a role in many malignancies, including CML [14–16]. The stem cell hypothesis has served well in explaining many clinical observations, in particular the feature that many cancers can be adequately debulked by chemotherapies or radiation, but most recur. Cancer stem cells seem to be more resistant to conventional therapy, and so are able to survive in small numbers and repopulate the malignancy, as Dick demonstrated in the murine AML xenotransplantation model.
The cancer stem cell hypothesis holds particular relevance for CML, because it explains many of the clinical characteristics of the disease. The differentiation capacity of the putative stem cell clearly explains the finding of mature BCR-ABL–expressing myeloid progenitors in chronic- phase CML. Furthermore, since the seminal work of Fialkow in the 1960s, it has been known that the same malignant clone of CML can develop into either a myeloid or lymphoid blast crisis [17, 18]. Recent work by Jamieson and colleagues indicates that the majority of proliferating cells in chronic-phase CML represent a proliferation of daughter cells with characteristics of immature progenitors [14, 19]; the acquisition of mutations—often involving the Wnt pathway—changes the proliferating daughter population into one with a phenotype of granulocyte/macrophage progenitors, heralding the development of blast crisis.
The cancer stem cell hypothesis is central to the question of why imatinib alone does not cure CML. Attainment of PCR negativity is far from the norm[3–6, 20]. Withdrawal of imatinib in PCR− patients often results in relapse [7–9]. It is not clear if this represents suppression of the BCR-ABL+ clone to levels below the limit of detection (at best, one in one million cells), and hence growth of a suppressed, but not fully ablated, population of CML cells, or the persistence of imatinib-resistant LICs. Mathematical modeling of repopulation kinetics is most consistent with expansion of an imatinib-resistant LIC [21, 22]. Indeed, Oravecz-Wilson et al. [23] demonstrated the persistence of LICs in an imatinib-responsive myeloproliferative mouse system (though the model system focused on cells expressing imatinib-sensitive Huntington Interacting Protein 1 (HIP1)/platelet-derived growth factor receptor, not BCR-ABL). Although CML progeny cells may become resistant to imatinib through selection of mutated clones, there is evidence to suggest that not only may LICs be inherently resistant but imatinib itself may induce resistance.
CML Stem Cells Express High Levels of BCR-ABL
Jamieson et al. [19] analyzed the levels of expression of BCR-ABL transcripts in bone marrow progenitor cells isolated from patients with CML. In their analysis of the chronic phase, they found that levels of BCR-ABL mRNA were fourfold higher in the population enriched for the LIC population than in either common myeloid progenitors, granulocyte–macrophage progenitors, or megakaryocyte–erythroid progenitors. Indeed, these levels were even higher than those observed in mature progenitors from samples obtained from imatinib-resistant chronic-phase patients. This analysis indicates that the CML LIC population expresses levels of BCR-ABL that could engender resistance to imatinib.
CML Stem Cells Have Elevated Levels of Membrane Transporter Proteins
CML stem cells have been shown to express the ATP-dependent transporter cassette protein ABCG2, which could decrease the accumulation of imatinib intracellularly, to inhibit imatinib activity in CML LICs [24]. Indeed, BCR-ABL may regulate the expression of ABCG2 through its effects on AKT kinase [25]. However, it is controversial whether ABCG2 truly regulates imatinib transport. Brendel et al. [26] demonstrated that ectopic expression of ABCG2 in K562 cells leads to lower levels of intracellular radiolabeled imatinib. Yet Jordanides showed that, although imatinib serves as an inhibitor of ABCG2, it might not itself be a substrate of ABCG2 [27]. The significance of ABCG2 in regulating the activity of imatinib thus remains unclear.
Imatinib Itself Increases CML Stem Cell Homing to the Bone Marrow Microenvironment
The CXC chemokine receptor 4 (CXCR4) protein is a cell-surface adhesion molecule important for stromal interaction [28]. Upregulation increases the propensity of cells to home to the bone marrow microenvironment. CML stem cells express levels of CXCR4 that are similar to those of other myeloid cells. However, exposure to imatinib increases the expression of CXCR4 in CML stem cells [29]. The implication of this finding is that CML stem cells exposed to imatinib are more likely to home to the marrow microenvironment. The bone marrow microenvironment is relatively hypoxic. Hypoxia enhances the survival of quiescent CML LICs. Nondividing LICs are relatively resistant to imatinib [29]. Indeed, Giuntoli et al. [30] have shown that hypoxic culturing of CML LICs leads to the development of imatinib resistance.
A Model
These observations suggest a model in which CML LICs, relatively resistant to imatinib as a result of their inherent quiescence, relatively high levels of BCR-ABL, and perhaps high levels of ABCG2 transporter, respond to imatinib by upregulating CXCR4, which increases their homing to a hypoxic marrow microenvironment where their quiescence and imatinib resistance are further enhanced. (See Figure 1.) This model predicts that imatinib treatment only increases the inherent resistance of CML LICs. Thus, imatinib, which so successfully targets imatinib-sensitive daughter cells, is not expected to adequately target CML LICs, and hence never fully cure CML.
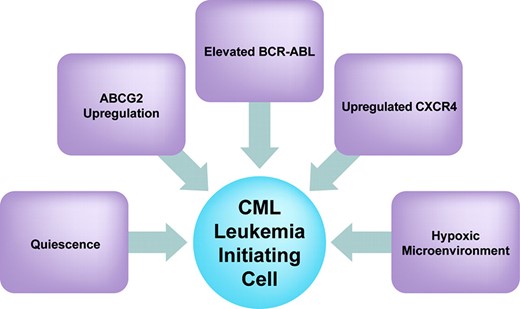
Factors that may play a role in determining the inherent resistance of CML leukemia-initiating cells to imatinib.
Abbreviations: ABCG2, ATP-dependent transporter cassette protein G2; CML, chronic myeloid leukemia; CXCR4, CXC chemokine receptor 4.
Future Strategies
Few studies of second- or third-generation tyrosine kinase inhibitors, or other novel agents, have focused on the LIC. Most strategies that address LIC resistance to imatinib propose other approaches in conjunction with imatinib. Being quiescent, CML LICs are relatively resistant to most chemotherapies [31]. Indeed, Jorgensen and colleagues [32, 33] found insensitivity to cytosine arabinoside (Ara-C), Ly294002 (a phosphatidylinositol 3-kinase inhibitor), 17-N-Allylamino-17-demethoxygeldanamycin (17-AAG, a heat shock protein-90 inhibitor), or lonafarnib (a farnesyl transferase inhibitor). The addition of imatinib had little effect on cells treated with Ara-C, LY294002, or 17-AAG, but 5 μM imatinib (a concentration achievable in vivo), given in concert with lonafarnib, decreased the growth of CML LICs.
Several groups have explored the strategy of overcoming the quiescence of CML stem cells with growth factors. Holtz et al. [34] found that GM-CSF and G-CSF enhanced CML LIC proliferation. Growth factor treatment significantly reduced the number of viable, nondividing cells after imatinib exposure. Stimulation with growth factors before imatinib treatment further reduced the number of residual nondividing CML CD34+ cells.
For decades, interferon was a cornerstone of CML treatment, but the role of immune modulators as an adjunct to imatinib has not been extensively explored. In a retrospective analysis, Rousselot et al. [8] found that the small number of patients who achieved BCR-ABL PCR negativity with imatinib and continued to be PCR− after discontinuation of imatinib, all had histories of previous treatment with interferon. Preliminary analysis of an ongoing study of 50 patients, however, has not fully substantiated an advantage to interferon pretreatment [9].
Targeting specific genes that are overexpressed in CML stem cells is also a potential strategy. The Wnt pathway is critical for CML stem cell growth: murine models demonstrate that inhibition of the Wnt signaling pathway enhances survival in a murine model of CML [35]. Similarly, expression of the PML tumor suppressor gene, implicated in the pathogenesis of acute promyelocytic leukemia, is critical to the survival of CML cells: Pandolfi's group has shown that targeting PML with arsenic decreases CML cell survival [36]. Targeting the nuclear factor (NF)-κB pathway, also important for CML stem cell growth, has offered an attractive strategy through the use of parthenolide and other NF-κB inhibitors [37–40].
Acknowledgments
The author would like to thank the University of Pittsburgh Cancer Institute Writing Group and Richard Steinman, M.D., Ph.D., for constructive suggestions.
References
Author notes
Disclosures: Robert L. Redner: None.
The content of this article has been reviewed by independent peer reviewers to ensure that it is balanced, objective, and free from commercial bias. No financial relationships relevant to the content of this article have been disclosed by the author or independent peer reviewers.

{kind=link}