





Abstract
Collapsing focal segmental glomerulosclerosis (FSGS) has various underlying etiologies and often leads to renal failure. The impact of biopsy-proven renal comorbidities in promoting collapsing glomerulopathy (CG) has not been systematically evaluated in large comparative studies. Those data are reported here.
Biopsies with the initial diagnosis of CG in native (n = 321) or transplant kidneys (n = 30) were identified in the University of North Carolina nephropathology database (1 January 2011 to 1 January 2016). Two cohorts were defined: ‘sole’ CG without and ‘accompanied’ CG with significant morphologic renal comorbidities. Tip-variant FSGS (T-FSGS) and time-matched biopsies served as control cohorts for comparative analyses.
CG was significantly more common in native (4.4%) and transplant biopsies (4.1%) compared with T-FSGS (0.7 and <0.1%, respectively, difference versus CG P < 0.01). ‘Associated’ disease was significantly more common in CG (native: 151/321; 47.0%, transplant: 21/30; 70%, P < 0.05) versus T-FSGS (native: 14/51; 27.5%, transplant: exceptional; all differences versus CG P < 0.05). In native biopsies with ‘accompanied’ CG but not in control groups, stenosing vasculopathies including thrombotic microangiopathies were significantly more prevalent (P < 0.01). In transplants, the high incidence of ‘accompanied’ CG was linked to de novo diseases, mainly rejection and vascular injury. In native kidneys, membranous glomerulopathies were prevalent in ‘accompanied’ T-FSGS (36%) and CG (14%) (difference versus time-matched controls P < 0.01 and P < 0.05, respectively); they were uncommon in transplants.
CG but not T-FSGS shows a high rate of comorbidities, with prominent vasculopathies presumably driving ‘ischemic’ CG-specific glomerular injury and also the disease course. These findings facilitate future studies into therapy, prognosis and reversibility of ‘accompanied’ CG.
KEY LEARNING POINTS
What is already known about this subject?
collapsing glomerulopathy (CG) is usually believed to be idiopathic/primary or triggered by events such as HIV infections or drug toxicity, such as pamidronate;
CG associated with other kidney diseases, such as systemic lupus erythematous or diabetic nephropathy, has been described in limited series; and
secondary variants of CG arising in association with other biopsy-proven renal diseases are poorly defined.
What this study adds?
this study is the largest and most comprehensive morphology-based comparative analysis of disease associations in CG;
the study shows that CG in contrast to focal segmental glomerulosclerosis tip lesions is significantly more often seen in patients with biopsy-proven renal comorbidities; and
two types of renal injury—obstruction of intra renal vessels with glomerular ischemia and membranous glomerulopathies—are identified as significant promoters of CG in native and transplant kidneys.
What impact this may have on practice or policy?
our data will broaden the understanding of presumed secondary variants CG and can help to stratify patient cohorts in future studies;
our observations will alert clinicians about disease associations in CG with possible implications for patient management such as avoidance of immunosuppressive therapy; and
our findings suggest that outcome in secondary CG is profoundly influenced by the underlying triggering renal comorbidity with pathways leading to non-CG induced end-stage renal disease or in some instances CG resolution.
INTRODUCTION
Focal segmental glomerulosclerosis (FSGS), collapsing variant [i.e. collapsing glomerulosclerosis (CG)] usually presents with severe proteinuria. Therapeutic intervention strategies remain limited, and CG, often believed to be idiopathic/primary, frequently progresses to chronic renal failure and end-stage renal disease [1–3]. CG was first described decades ago, predominantly in patients with AIDS, and referred to as ‘HIV-associated nephropathy’ (HIVAN) with tubulocystic dilatation, carrying a poor prognosis [4]. It has a strong association with trypanolytic variants of the APOL1 gene found primarily in patients of African descent [5, 6]. CG was also found in association with some other diseases, such as parvovirus or CMV infections, diabetes mellitus (DM), systemic lupus erythematous (SLE) or ‘chronic allograft nephropathy’ [7–11]. CG in patients with coronavirus disease 2019 (COVID-19) through direct viral infection of podocytes has been suggested by some authors [12], but this pathway has subsequently been disputed by others [13, 14]. However, comorbidities in CG have mainly been explored in case reports and small case series; a large-scale systematic comparative review has not been conducted. What renal comorbidities are prevalent in CG and can potentially promote the onset of glomerular tuft collapse? Such comparative biopsy-based study results are reported here. Our findings will further our understanding of currently incompletely characterized secondary variants of CG.
MATERIALS AND METHODS
This study was approved by the local University of North Carolina (UNC) ethics review board. The UNC database was searched for a diagnosis of CG in native and transplant kidney biopsies referred to the UNC Division of Nephropathology between 1 January 2011 and 1 January 2016; cases with insufficient biopsy material were excluded. A diagnosis of CG was made by one of three expert nephropathologists (H.K. Singh, J.C. Jennette and V.N.) according to Columbia FSGS classification criteria [3, 15], i.e. presence of at least one glomerulus with segmental or global tuft collapse and hyperplasia of overlying podocytes typically containing protein resorption droplets. In cases with associated renal diseases that may present with crescents, CG was differentiated from crescents by the absence of leukocytes or fibrinoid necrosis, presence of tuft collapse and prominence of resorption droplets in epithelial cells.
All available demographic and clinical data provided at time of biopsy were reviewed and coded renal comorbidities detected at time of histologic work-up used for comparative studies. No clinical follow-up data were available.
The analysis was based on diagnoses made in index biopsies (first diagnostic biopsy with CG); repeat biopsies were excluded from all cohorts. (i) For study purposes two major morphologic CG groups were defined based on biopsy findings in native or allograft biopsies: (a) ‘sole’ CG: biopsies with CG but no other significant or diagnostically relevant morphologic kidney disease. Included in this 'sole' CG group were biopsies with mild-to-moderate arterio(nephro)sclerosis (AS) and biopsies with mild to moderate mesangial expansion and minor mesangial immunoglobulin A (IgA) deposits. Per definition glomerulonephritides with endo- or extra-capillary proliferations were excluded. (b) ‘Accompanied’ CG: biopsies with CG and other diagnostically relevant pathology diagnoses. In this cohort, severe AS was defined as arterial intimal fibroelastosis of greater thickness than the medial smooth muscle layer and/or arteriolosclerosis with subendothelial circumferential hyaline deposits or stenosing hyaline deposits. (ii) ‘Control native renal biopsy groups’ (same collection approach as outlined for CG): (a) control Cohort 1: tip-variant FSGS (T-FSGS) with biopsies showing according to the Columbia FSGS classification scheme at least one segmental area of sclerosis/intra-capillary foam cells near the glomerular urinary pole; per definition CG was not present [3, 15]. This group represents a well-defined FSGS category. (b) Control Cohort 2: biopsy cohort post exclusion of cases with CG and T-FSGS (referred to as ‘time-matched control group’).
Statistical comparisons were performed using Chi-square tests and standard Student's t-tests with α = 0.050.
RESULTS
There were 351 patients with a histologic diagnosis of CG in the total cohort of 8056 patients whose index biopsy was examined at UNC over a period of 5 years (incidence: 4.4%). The median patient age was 42 years (range 2–82) with a male to female ratio of 1.2:1. There was no significant difference between the incidence of CG in the native cohort (321/7318; 4.4%) compared with the transplant kidney cohort (30/738; 4.1%). In the study period 51 patients presented with T-FSGS (control Cohort 1; incidence 0.7%); 37/51 (73%) patients were classified as ‘sole’ T-FSGS, constituting a significantly higher proportion when compared with the CG group (P < 0.01). T-FSGS was exceptionally rare in renal allograft recipients with only one diagnosed case (1/738; 0.1%). The male to female ratio in T-FSGS patients was 1.6:1, and the median age 49 years (range 14–87). Control Cohort 2, i.e. the group of ‘time-matched’ native control biopsies, consisted of 7318 patients.
Native renal biopsies: CG and disease entities
About half of native cases were classified as ‘sole' CG (170/351; 53%). The remaining patients (151/351; 47%) fell into the category of ‘accompanied' CG (Tables 1 and 2) with various renal disease associations. Among those, thrombotic microangiopathies (TMAs) were significantly more common in CG, either in the entire study set (Table 1) or in a sub-analysis of ‘accompanied’ cases only (Table 2 and Figure 1). Significant associations were, however, not limited to vascular injury due to TMA but seen in a whole spectrum of renal diseases all characterized by severe stenosing vasculopathies, such as hypertension-induced arterio-, and arteriolo-sclerosis often prominent in afferent arterioles (CG 26% versus T-FSGS 6%, P < 0.01; Table 3). In a sub-analysis of biopsies limited to ‘accompanied’ CG, the tight association between severe vasculopathies and glomerular tuft collapse was further underscored ('accompanied' CG with severe vasculopathies: 56%; versus time-matched control Cohort 2, 31%, P < 0.01; Table 2). Of note: TMAs were not seen in biopsies also carrying a diagnosis of T-FSGS. In comparison with severe vasculopathies, nonvascular renal comorbidities were uncommon in CG with the only exception of membranous glomerulopathies (MGNs) showing a higher prevalence in CG as well as in control Cohort 1, T-FSGS. Differences reached statistical significance in the subgroup of ‘accompanied’ cases; MGN in time-matched controls: 9% versus ‘accompanied’ CG: 14%, P < 0.05, versus ‘accompanied’ T-FSGS: 36%, P < 0.01 (Table 2). Cases of MGN diagnosed in the setting of CG were in 50% (10/20) phospholipase A2 receptor (PLA2R)-antibody positive. In the cohorts, no differences in prevalence were noted for lupus nephritis or IgA glomerulopathies. Other diagnoses, summarized in the category ‘other non-vascular diseases’ were uncommon in CG, including anti-neutrophil cytoplasmic autoantibody (ANCA) glomerulonephritis, fibrillary glomerulopathy, athero-embolic disease, thin glomerular basement membrane (GBM) disease, acute interstitial nephritis, cryoglobulin associated glomerulonephritis, sarcoidosis and C3 glomerulonephritis.
Comorbidities in CG and FSGS tip-lesion: a comparative analysis with time-matched control Cohort 2
| Category | CG (%); P-value | Control Cohort 2 (%) | Tip-lesion FSGS control Cohort 1 (%); P-value |
|---|---|---|---|
| Total number of cases | 321 | 7318 | 51 |
| ‘Accompanied' CG, n (%) | 151 (47) | NA | 14 (27) |
| Vascular diseases, n (%) | 84 (26); 0.06 | 2267 (31) | 3 (6); <0.01 |
| Severe AS | 38 (12); 0.37 | 757(10) | 2 (4); 0.13 |
| Diabetic nephropathy | 20 (6); 0.01 | 951 (13) | 1 (2); <0.05 |
| Severe AS + diabetic nephropathy | 12 (4); 0.26 | 372 (5) | 0 |
| TMA | 14 (4.3); <0.05 | 187 (2.6) | 0 |
| Non-vascular diseases, n (%) | 67 (21); <0.01 | 5051 (69) | 11 (22); <0.01 |
| MGN | 21 (7); 0.20 | 622 (9) | 5 (10); 0.74 |
| Lupus nephropathy | 20 (6); 0.07 | 663(9) | 1 (2); 0.08 |
| Class II | 2 | – | – |
| Class III | 1 | – | – |
| Class IV | 7 | – | – |
| Class V | 4 | – | – |
| Class V + III or IV | 6 | – | – |
| IgA nephropathy (proliferative) | 4 (1); <0.01 | 320 (4) | 0 |
| Other nonvascular diseases | 22 (7); <0.01 | 3446 (47) | 5 (10); <0.01 |
| Category | CG (%); P-value | Control Cohort 2 (%) | Tip-lesion FSGS control Cohort 1 (%); P-value |
|---|---|---|---|
| Total number of cases | 321 | 7318 | 51 |
| ‘Accompanied' CG, n (%) | 151 (47) | NA | 14 (27) |
| Vascular diseases, n (%) | 84 (26); 0.06 | 2267 (31) | 3 (6); <0.01 |
| Severe AS | 38 (12); 0.37 | 757(10) | 2 (4); 0.13 |
| Diabetic nephropathy | 20 (6); 0.01 | 951 (13) | 1 (2); <0.05 |
| Severe AS + diabetic nephropathy | 12 (4); 0.26 | 372 (5) | 0 |
| TMA | 14 (4.3); <0.05 | 187 (2.6) | 0 |
| Non-vascular diseases, n (%) | 67 (21); <0.01 | 5051 (69) | 11 (22); <0.01 |
| MGN | 21 (7); 0.20 | 622 (9) | 5 (10); 0.74 |
| Lupus nephropathy | 20 (6); 0.07 | 663(9) | 1 (2); 0.08 |
| Class II | 2 | – | – |
| Class III | 1 | – | – |
| Class IV | 7 | – | – |
| Class V | 4 | – | – |
| Class V + III or IV | 6 | – | – |
| IgA nephropathy (proliferative) | 4 (1); <0.01 | 320 (4) | 0 |
| Other nonvascular diseases | 22 (7); <0.01 | 3446 (47) | 5 (10); <0.01 |
Cases of CG and FSGS tip-lesion (total study cohorts) are compared with the time-matched control group two of native renal biopsies. ‘Vascular diseases’ refers to the sum of the four diagnoses listed in this table. ‘Other diseases’ refers to all other ‘nonvascular’ diagnoses. All n (%) refer to the total number of cases in each category and all P-values refer to comparisons to the time-matched control group, when applicable (bold indicates statistically significantly higher incidence of P-value; normal values indicate statistically lower incidence or not significant P-values).
Comorbidities in CG and FSGS tip-lesion: a comparative analysis with time-matched control Cohort 2
| Category | CG (%); P-value | Control Cohort 2 (%) | Tip-lesion FSGS control Cohort 1 (%); P-value |
|---|---|---|---|
| Total number of cases | 321 | 7318 | 51 |
| ‘Accompanied' CG, n (%) | 151 (47) | NA | 14 (27) |
| Vascular diseases, n (%) | 84 (26); 0.06 | 2267 (31) | 3 (6); <0.01 |
| Severe AS | 38 (12); 0.37 | 757(10) | 2 (4); 0.13 |
| Diabetic nephropathy | 20 (6); 0.01 | 951 (13) | 1 (2); <0.05 |
| Severe AS + diabetic nephropathy | 12 (4); 0.26 | 372 (5) | 0 |
| TMA | 14 (4.3); <0.05 | 187 (2.6) | 0 |
| Non-vascular diseases, n (%) | 67 (21); <0.01 | 5051 (69) | 11 (22); <0.01 |
| MGN | 21 (7); 0.20 | 622 (9) | 5 (10); 0.74 |
| Lupus nephropathy | 20 (6); 0.07 | 663(9) | 1 (2); 0.08 |
| Class II | 2 | – | – |
| Class III | 1 | – | – |
| Class IV | 7 | – | – |
| Class V | 4 | – | – |
| Class V + III or IV | 6 | – | – |
| IgA nephropathy (proliferative) | 4 (1); <0.01 | 320 (4) | 0 |
| Other nonvascular diseases | 22 (7); <0.01 | 3446 (47) | 5 (10); <0.01 |
| Category | CG (%); P-value | Control Cohort 2 (%) | Tip-lesion FSGS control Cohort 1 (%); P-value |
|---|---|---|---|
| Total number of cases | 321 | 7318 | 51 |
| ‘Accompanied' CG, n (%) | 151 (47) | NA | 14 (27) |
| Vascular diseases, n (%) | 84 (26); 0.06 | 2267 (31) | 3 (6); <0.01 |
| Severe AS | 38 (12); 0.37 | 757(10) | 2 (4); 0.13 |
| Diabetic nephropathy | 20 (6); 0.01 | 951 (13) | 1 (2); <0.05 |
| Severe AS + diabetic nephropathy | 12 (4); 0.26 | 372 (5) | 0 |
| TMA | 14 (4.3); <0.05 | 187 (2.6) | 0 |
| Non-vascular diseases, n (%) | 67 (21); <0.01 | 5051 (69) | 11 (22); <0.01 |
| MGN | 21 (7); 0.20 | 622 (9) | 5 (10); 0.74 |
| Lupus nephropathy | 20 (6); 0.07 | 663(9) | 1 (2); 0.08 |
| Class II | 2 | – | – |
| Class III | 1 | – | – |
| Class IV | 7 | – | – |
| Class V | 4 | – | – |
| Class V + III or IV | 6 | – | – |
| IgA nephropathy (proliferative) | 4 (1); <0.01 | 320 (4) | 0 |
| Other nonvascular diseases | 22 (7); <0.01 | 3446 (47) | 5 (10); <0.01 |
Cases of CG and FSGS tip-lesion (total study cohorts) are compared with the time-matched control group two of native renal biopsies. ‘Vascular diseases’ refers to the sum of the four diagnoses listed in this table. ‘Other diseases’ refers to all other ‘nonvascular’ diagnoses. All n (%) refer to the total number of cases in each category and all P-values refer to comparisons to the time-matched control group, when applicable (bold indicates statistically significantly higher incidence of P-value; normal values indicate statistically lower incidence or not significant P-values).
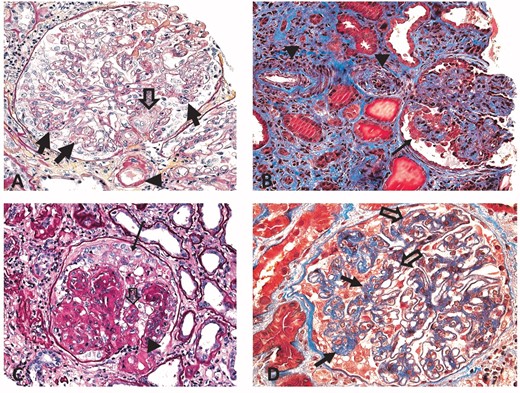
CG associated with other significant renal diseases. (A) TMA. The glomerulus shows segmental tuft collapse (closed arrows). The vascular pole is ‘occluded’ by activated mononuclear cell elements (endothelial cells and/or myofibroblasts; open arrow). The arrowhead points to arteriolar wall changes with intramural hyaline deposits often seen in TMAs. (B) Severe AS. The glomerulus shows segmental tuft collapse (closed arrow). An interlobular artery and pre-arteriole show severe, stenosing hypertension-induced intimal widening including fibroelastosis (arrowheads). Fibrosis, tubular atrophy and few small tubular microcysts are noted in the tubulo-interstitial compartment. (C) Diabetic glomerulopathy. The glomerulus shows segmental podocyte crowding (arrow) and segmental collapse of ‘non-nodular’ tufts (open arrow). The afferent arteriole at the vascular pole demonstrates severe stenosing hyalinosis (arrowhead). (D) MGN. The glomerulus shows global thickening of capillary walls (open arrows) and collapse of a tuft segment (closed arrows). (Typical immunofluorescence and ultrastructural features confirmed a diagnosis of MGN.) Masson trichrome stains, 40× original magnification (A) and (D), 20× (B) and (C).
Sub-analysis: ‘accompanied’ CG and FSGS tip cohorts compared with time-matched control Cohort 2
| Category | ‘Accompanied', CG (%); P-value | Control Cohort 2 (%) | ‘Accompanied' tip-lesion FSGS control Cohort 1 (%); P-value |
|---|---|---|---|
| Number of cases | 151 | 7318 | 14 |
| Vascular diseases, n (%) | 84 (56); <0.01 | 2267 (31) | 3 (21); 0.44 |
| Severe AS | 38 (25); <0.01 | 757(10) | 2 (14); 0.63 |
| Diabetic nephropathy | 20 (13); 0.95 | 951 (13) | 1 (7); 0.51 |
| Severe AS + diabetic nephropathy | 12 (8); 0.11 | 372 (5) | 0 |
| TMA | 14 (9); <0.01 | 187 (3) | 0 |
| Nonvascular diseases, n (%) | 67 (44); <0.01 | 5051 (69) | 11 (79); 0.44 |
| MGN | 21 (14); <0.05 | 622 (9) | 5 (36); <0.01 |
| Lupus nephropathy | 20 (13); 0.07 | 663 (9) | 1 (7); 0.80 |
| Class II | 2 | – | – |
| Class III | 1 | – | – |
| Class IV | 7 | – | – |
| Class V | 4 | – | – |
| Class V + III or IV | 6 | – | – |
| IgA nephropathy (proliferative) | 4 (3); 0.30 | 320 (4) | 0 |
| Other nonvascular diseases | 22 (15); <0.01 | 3446 (47) | 5 (36); 0.45 |
| Category | ‘Accompanied', CG (%); P-value | Control Cohort 2 (%) | ‘Accompanied' tip-lesion FSGS control Cohort 1 (%); P-value |
|---|---|---|---|
| Number of cases | 151 | 7318 | 14 |
| Vascular diseases, n (%) | 84 (56); <0.01 | 2267 (31) | 3 (21); 0.44 |
| Severe AS | 38 (25); <0.01 | 757(10) | 2 (14); 0.63 |
| Diabetic nephropathy | 20 (13); 0.95 | 951 (13) | 1 (7); 0.51 |
| Severe AS + diabetic nephropathy | 12 (8); 0.11 | 372 (5) | 0 |
| TMA | 14 (9); <0.01 | 187 (3) | 0 |
| Nonvascular diseases, n (%) | 67 (44); <0.01 | 5051 (69) | 11 (79); 0.44 |
| MGN | 21 (14); <0.05 | 622 (9) | 5 (36); <0.01 |
| Lupus nephropathy | 20 (13); 0.07 | 663 (9) | 1 (7); 0.80 |
| Class II | 2 | – | – |
| Class III | 1 | – | – |
| Class IV | 7 | – | – |
| Class V | 4 | – | – |
| Class V + III or IV | 6 | – | – |
| IgA nephropathy (proliferative) | 4 (3); 0.30 | 320 (4) | 0 |
| Other nonvascular diseases | 22 (15); <0.01 | 3446 (47) | 5 (36); 0.45 |
The comparative analysis is limited to the ‘accompanied’ disease cohorts of CG and tip-lesion FSGS. ‘Vascular diseases’ refers to the sum of the four diagnoses listed in this table. ‘Other diseases’ refer to all other ‘non-vascular’ diagnoses. All N/percentages refer to ‘accompanied’ cases of CG and tip-lesion FSGS and all P-values refer to comparison of those cases to the time-matched control cohort (bold values indicate statistically significant higher incidence; normal values indicate statistically lower incidence P-value).
Sub-analysis: ‘accompanied’ CG and FSGS tip cohorts compared with time-matched control Cohort 2
| Category | ‘Accompanied', CG (%); P-value | Control Cohort 2 (%) | ‘Accompanied' tip-lesion FSGS control Cohort 1 (%); P-value |
|---|---|---|---|
| Number of cases | 151 | 7318 | 14 |
| Vascular diseases, n (%) | 84 (56); <0.01 | 2267 (31) | 3 (21); 0.44 |
| Severe AS | 38 (25); <0.01 | 757(10) | 2 (14); 0.63 |
| Diabetic nephropathy | 20 (13); 0.95 | 951 (13) | 1 (7); 0.51 |
| Severe AS + diabetic nephropathy | 12 (8); 0.11 | 372 (5) | 0 |
| TMA | 14 (9); <0.01 | 187 (3) | 0 |
| Nonvascular diseases, n (%) | 67 (44); <0.01 | 5051 (69) | 11 (79); 0.44 |
| MGN | 21 (14); <0.05 | 622 (9) | 5 (36); <0.01 |
| Lupus nephropathy | 20 (13); 0.07 | 663 (9) | 1 (7); 0.80 |
| Class II | 2 | – | – |
| Class III | 1 | – | – |
| Class IV | 7 | – | – |
| Class V | 4 | – | – |
| Class V + III or IV | 6 | – | – |
| IgA nephropathy (proliferative) | 4 (3); 0.30 | 320 (4) | 0 |
| Other nonvascular diseases | 22 (15); <0.01 | 3446 (47) | 5 (36); 0.45 |
| Category | ‘Accompanied', CG (%); P-value | Control Cohort 2 (%) | ‘Accompanied' tip-lesion FSGS control Cohort 1 (%); P-value |
|---|---|---|---|
| Number of cases | 151 | 7318 | 14 |
| Vascular diseases, n (%) | 84 (56); <0.01 | 2267 (31) | 3 (21); 0.44 |
| Severe AS | 38 (25); <0.01 | 757(10) | 2 (14); 0.63 |
| Diabetic nephropathy | 20 (13); 0.95 | 951 (13) | 1 (7); 0.51 |
| Severe AS + diabetic nephropathy | 12 (8); 0.11 | 372 (5) | 0 |
| TMA | 14 (9); <0.01 | 187 (3) | 0 |
| Nonvascular diseases, n (%) | 67 (44); <0.01 | 5051 (69) | 11 (79); 0.44 |
| MGN | 21 (14); <0.05 | 622 (9) | 5 (36); <0.01 |
| Lupus nephropathy | 20 (13); 0.07 | 663 (9) | 1 (7); 0.80 |
| Class II | 2 | – | – |
| Class III | 1 | – | – |
| Class IV | 7 | – | – |
| Class V | 4 | – | – |
| Class V + III or IV | 6 | – | – |
| IgA nephropathy (proliferative) | 4 (3); 0.30 | 320 (4) | 0 |
| Other nonvascular diseases | 22 (15); <0.01 | 3446 (47) | 5 (36); 0.45 |
The comparative analysis is limited to the ‘accompanied’ disease cohorts of CG and tip-lesion FSGS. ‘Vascular diseases’ refers to the sum of the four diagnoses listed in this table. ‘Other diseases’ refer to all other ‘non-vascular’ diagnoses. All N/percentages refer to ‘accompanied’ cases of CG and tip-lesion FSGS and all P-values refer to comparison of those cases to the time-matched control cohort (bold values indicate statistically significant higher incidence; normal values indicate statistically lower incidence P-value).
Comorbidities in CG and tip-lesion FSGS: a comparative analysis
| Category | CG (%) | Tip-lesion FSGS (%) | P-value |
|---|---|---|---|
| Total number of cases | 321 | 51 | |
| ‘Sole', n (%) | 170 (53) | 37 (73) | <0.01 |
| ‘Accompanied', n (%) | 151 (47) | 14 (28) | |
| Vascular diseases,a n (%) | 84 (26) | 3 (6) | <0.01 |
| Severe AS | 38 (12) | 2 (4) | 0.09 |
| Diabetic nephropathy | 20 (6) | 1 (2) | 0.22 |
| Severe AS + diabetic nephropathy | 12 (4) | 0 | NA |
| TMA | 14 (4) | 0 | NA |
| Nonvascular diseases,a n (%) | 67 (21) | 11 (22) | 0.94 |
| MGN | 21 (7) | 5 (10) | 0.45 |
| Lupus nephropathy | 20 (6) | 1 (2) | 0.22 |
| IgA nephropathy (proliferative) | 4 (1) | 0 | NA |
| Other nonvascular diseases | 22 (7) | 5 (10) | 0.16 |
| Category | CG (%) | Tip-lesion FSGS (%) | P-value |
|---|---|---|---|
| Total number of cases | 321 | 51 | |
| ‘Sole', n (%) | 170 (53) | 37 (73) | <0.01 |
| ‘Accompanied', n (%) | 151 (47) | 14 (28) | |
| Vascular diseases,a n (%) | 84 (26) | 3 (6) | <0.01 |
| Severe AS | 38 (12) | 2 (4) | 0.09 |
| Diabetic nephropathy | 20 (6) | 1 (2) | 0.22 |
| Severe AS + diabetic nephropathy | 12 (4) | 0 | NA |
| TMA | 14 (4) | 0 | NA |
| Nonvascular diseases,a n (%) | 67 (21) | 11 (22) | 0.94 |
| MGN | 21 (7) | 5 (10) | 0.45 |
| Lupus nephropathy | 20 (6) | 1 (2) | 0.22 |
| IgA nephropathy (proliferative) | 4 (1) | 0 | NA |
| Other nonvascular diseases | 22 (7) | 5 (10) | 0.16 |
Evaluated in the total study cohorts of CG (n = 321) and tip-lesion FSGS (n = 51).
‘Vascular diseases’ refers to the sum of the four diagnoses listed in this table. ‘Other diseases’ refers to all other ‘nonvascular’ diagnoses (bold indicates statistically significant higher incidence of P-value).
Comorbidities in CG and tip-lesion FSGS: a comparative analysis
| Category | CG (%) | Tip-lesion FSGS (%) | P-value |
|---|---|---|---|
| Total number of cases | 321 | 51 | |
| ‘Sole', n (%) | 170 (53) | 37 (73) | <0.01 |
| ‘Accompanied', n (%) | 151 (47) | 14 (28) | |
| Vascular diseases,a n (%) | 84 (26) | 3 (6) | <0.01 |
| Severe AS | 38 (12) | 2 (4) | 0.09 |
| Diabetic nephropathy | 20 (6) | 1 (2) | 0.22 |
| Severe AS + diabetic nephropathy | 12 (4) | 0 | NA |
| TMA | 14 (4) | 0 | NA |
| Nonvascular diseases,a n (%) | 67 (21) | 11 (22) | 0.94 |
| MGN | 21 (7) | 5 (10) | 0.45 |
| Lupus nephropathy | 20 (6) | 1 (2) | 0.22 |
| IgA nephropathy (proliferative) | 4 (1) | 0 | NA |
| Other nonvascular diseases | 22 (7) | 5 (10) | 0.16 |
| Category | CG (%) | Tip-lesion FSGS (%) | P-value |
|---|---|---|---|
| Total number of cases | 321 | 51 | |
| ‘Sole', n (%) | 170 (53) | 37 (73) | <0.01 |
| ‘Accompanied', n (%) | 151 (47) | 14 (28) | |
| Vascular diseases,a n (%) | 84 (26) | 3 (6) | <0.01 |
| Severe AS | 38 (12) | 2 (4) | 0.09 |
| Diabetic nephropathy | 20 (6) | 1 (2) | 0.22 |
| Severe AS + diabetic nephropathy | 12 (4) | 0 | NA |
| TMA | 14 (4) | 0 | NA |
| Nonvascular diseases,a n (%) | 67 (21) | 11 (22) | 0.94 |
| MGN | 21 (7) | 5 (10) | 0.45 |
| Lupus nephropathy | 20 (6) | 1 (2) | 0.22 |
| IgA nephropathy (proliferative) | 4 (1) | 0 | NA |
| Other nonvascular diseases | 22 (7) | 5 (10) | 0.16 |
Evaluated in the total study cohorts of CG (n = 321) and tip-lesion FSGS (n = 51).
‘Vascular diseases’ refers to the sum of the four diagnoses listed in this table. ‘Other diseases’ refers to all other ‘nonvascular’ diagnoses (bold indicates statistically significant higher incidence of P-value).
Native renal biopsies: CG and clinical data
Data on ethnicity was available for 283 patients (88%); 203 (71.7%) were identified as African Americans (AAs), 55 (19.4%) as Caucasians, and the remaining patients as Latinos, Middle-Easterners, Asians or native Americans. In CG, the proportion of AA patients was almost identical in the ‘sole' CG category (95/151; 63%) versus the ‘associated’ CG category (108/170; 64%). Vice versa, AA patients were in ∼50% categorized as ‘sole’ CG (108/203; 53.2%); similar to patients of Caucasian ethnicity ('sole' CG in 28/55; 50.9%).
Nephrotic-range proteinuria was reported in 179/235 patients (75.7%) with similar frequencies in ‘sole’ and ‘accompanied’ CG (99/127; 78.0% and 80/108; 74.1%, respectively; difference not significant). Serum creatinine levels were provided for most patients (304/322; 94.4%); they were generally elevated with highly variable titers: mean 4.97 mg/dL (standard deviation 4.01). ‘Sole’ CG in contrast to ‘accompanied’ CG showed a trend toward lower mean serum creatinine levels (4.62 versus 5.11 mg/dL, difference not significant).
Thirty-one patients were reported as being HIV positive (9.6%), with 21/31 (67.7%) being classified as ‘sole’ CG; no patient was suffering from AIDS. Eighteen patients (5.6%) tested positive for hepatitis C virus (HCV) (8/18; 44% ‘sole’ CG). Only one patient presented with a documented parvovirus infection. Two patients had a history of cocaine (one ‘sole’ and one with severe AS), one of heroin and one of Opana abuse; the latter presented with a TMA.
Transplant biopsies
A diagnosis of CG was made in 30 renal allograft recipients over a 5-year period (30/738; 4.1%), with a high proportion of ‘accompanied’ cases (21/30; 70% versus 152/322; 47% in native biopsies, P < 0.05). The time between transplantation and the first diagnosis was available for 27/30 patients: mean 76.4 months (range 1–206); time to diagnosis of ‘sole’ CG 65.6 months (mean) and of ‘accompanied' CG 81.0 months (difference not statistically significant; Table 4). In cases presenting as ‘sole’ CG, FSGS was reported as the underlying native disease in 4/9 (44.4%), including one case of recurrent CG. In the cohort of ‘accompanied’ CG, fewer patients reportedly suffered from FSGS as underlying native renal disease (4/21; 19%); however, compared with the ‘sole’ CG group, the difference was not significant (Table 4). In the cohort of ‘accompanied’ CG, 3/21 (14.3%) cases showed TMA, none of which had documented concurrent rejection. Four cases showed severe AS and CG, one with additional diabetic nephropathy. Rejection-related cases (9/21; 42.9%) included antibody-mediated rejection (ABMR) (3/21; 14.3%), T cell-mediated rejection (TCMR) (4/21; 19.0%) and mixed ABMR and TCMR (2/21; 9.5%). One case showed endarteritis (1/21; 4.7%). Thus, various forms of vascular obstruction and flow compromise presumably occurred in TMA, severe AS, ABMR and Banff Type 2 rejection with endarteritis (13/21; 61.9%). In the total transplant cohort, only one case of T-FSGS and one of MGN were diagnosed. None of the transplant patients was known to be HIV positive; one was reportedly positive for HCV.
CG in kidney transplants
| Category | ‘Sole’ CG (%) | ‘Accompanied’ CG (%); P-value |
|---|---|---|
| CG in renal allografts (n = 30) | ||
| Number of cases, n (%) | 9/30 (30) | 21/30 (70) |
| Average time from transplantation to diagnosis (months) | 66 | 81; 0.57 |
| Number of cases with FSGS as primary underlying native renal disease | 4/9 (44) | 4/21 (19); 0.15 |
| Number of C4d-positive cases | 0 | 4/21 (19) |
| Number of cases with TMA | 0 PD | 3/21 (14) |
| Number of cases with severe AS and/or DM | 0 PD | 4/21 (19) |
| Number of cases with either acute TCMR, ABMR or mixed rejection (excluding borderline) | 0 PD | 9/21 (43) |
| Number of cases with transplant glomerulopathy | 0 PD | 5/21 (24) |
| Number of cases with CNI toxicity | 0 PD | 2/21 (9) |
| Cases with >1 type of comorbidity | 0 PD | 7/21 (33) |
| Category | ‘Sole’ CG (%) | ‘Accompanied’ CG (%); P-value |
|---|---|---|
| CG in renal allografts (n = 30) | ||
| Number of cases, n (%) | 9/30 (30) | 21/30 (70) |
| Average time from transplantation to diagnosis (months) | 66 | 81; 0.57 |
| Number of cases with FSGS as primary underlying native renal disease | 4/9 (44) | 4/21 (19); 0.15 |
| Number of C4d-positive cases | 0 | 4/21 (19) |
| Number of cases with TMA | 0 PD | 3/21 (14) |
| Number of cases with severe AS and/or DM | 0 PD | 4/21 (19) |
| Number of cases with either acute TCMR, ABMR or mixed rejection (excluding borderline) | 0 PD | 9/21 (43) |
| Number of cases with transplant glomerulopathy | 0 PD | 5/21 (24) |
| Number of cases with CNI toxicity | 0 PD | 2/21 (9) |
| Cases with >1 type of comorbidity | 0 PD | 7/21 (33) |
In renal allografts, the vast majority of cases fall into the ‘accompanied’ CG category with often more than one renal comorbidity. Cases with more than one comorbidity include the following combinations: TCMR + transplant glomerulopathy [1], TCMR + DM [1], recurrent IgA nephropathy or MGN + chronic/active rejection [4] and a case of polyomavirus infection with chronic/active rejection. PD, per definition; CNI, calcineurin inhibitor.
CG in kidney transplants
| Category | ‘Sole’ CG (%) | ‘Accompanied’ CG (%); P-value |
|---|---|---|
| CG in renal allografts (n = 30) | ||
| Number of cases, n (%) | 9/30 (30) | 21/30 (70) |
| Average time from transplantation to diagnosis (months) | 66 | 81; 0.57 |
| Number of cases with FSGS as primary underlying native renal disease | 4/9 (44) | 4/21 (19); 0.15 |
| Number of C4d-positive cases | 0 | 4/21 (19) |
| Number of cases with TMA | 0 PD | 3/21 (14) |
| Number of cases with severe AS and/or DM | 0 PD | 4/21 (19) |
| Number of cases with either acute TCMR, ABMR or mixed rejection (excluding borderline) | 0 PD | 9/21 (43) |
| Number of cases with transplant glomerulopathy | 0 PD | 5/21 (24) |
| Number of cases with CNI toxicity | 0 PD | 2/21 (9) |
| Cases with >1 type of comorbidity | 0 PD | 7/21 (33) |
| Category | ‘Sole’ CG (%) | ‘Accompanied’ CG (%); P-value |
|---|---|---|
| CG in renal allografts (n = 30) | ||
| Number of cases, n (%) | 9/30 (30) | 21/30 (70) |
| Average time from transplantation to diagnosis (months) | 66 | 81; 0.57 |
| Number of cases with FSGS as primary underlying native renal disease | 4/9 (44) | 4/21 (19); 0.15 |
| Number of C4d-positive cases | 0 | 4/21 (19) |
| Number of cases with TMA | 0 PD | 3/21 (14) |
| Number of cases with severe AS and/or DM | 0 PD | 4/21 (19) |
| Number of cases with either acute TCMR, ABMR or mixed rejection (excluding borderline) | 0 PD | 9/21 (43) |
| Number of cases with transplant glomerulopathy | 0 PD | 5/21 (24) |
| Number of cases with CNI toxicity | 0 PD | 2/21 (9) |
| Cases with >1 type of comorbidity | 0 PD | 7/21 (33) |
In renal allografts, the vast majority of cases fall into the ‘accompanied’ CG category with often more than one renal comorbidity. Cases with more than one comorbidity include the following combinations: TCMR + transplant glomerulopathy [1], TCMR + DM [1], recurrent IgA nephropathy or MGN + chronic/active rejection [4] and a case of polyomavirus infection with chronic/active rejection. PD, per definition; CNI, calcineurin inhibitor.
DISCUSSION
Here we report data from a large comparative biopsy-based study in order to shed light on secondary variants of CG. Coded biopsy diagnoses from a single nephropathology referral center were analyzed and comparative studies conducted with time-matched and T-FSGS cohorts. A diagnosis of CG was rendered in 4% of all native renal biopsies, approximately half of which were associated with other significant renal lesions, termed here the ‘accompanied’ CG cohort. ‘Accompanied’ CG was significantly more often seen than ‘accompanied’ T-FSGS, especially in renal allografts, pointing toward specific CG triggering comorbidities. Those were identified as severe stenosing vasculopathies, e.g. stenosing arterio- or arteriolo-sclerosis and TMAs. TMA was less frequently diagnosed in time-matched control biopsies and absent in T-FSGS. In transplants, CG was diagnosed in 4% of all biopsies with predominance of ‘accompanied’ cases. More than 60% of renal comorbidities in allografts directly or indirectly affected blood flow, such as rejection, TMA, severe AS or calcineurin inhibitor-induced toxic arteriolopathies; thus overall findings in transplants were similar to those made in native renal biopsies. Interestingly, FSGS as underlying native renal disease was uncommon in the cohort of ‘accompanied’ allograft CG, further supporting the notion of specific comorbidities as triggering events for secondary variants CG.
How can we best interpret these findings? In CG accompanied stenosis is found in intra-parenchymal small arteries, i.e. interlobular arteries and afferent arterioles, resulting in impaired glomerular blood flow and ischemic injury in podocytes and endothelial cells. Glomerular ischemia presumably alters trans-GBM cross signaling, vascular endothelial growth factor (VEGF) feedback loops and leads to tuft collapse and crowding of podocytes. Similar phenomena seem to trigger CG in renal transplants. Interestingly, histologic small vessel changes can range from chronic, i.e. severe arterial intimal fibroelastosis and/or arteriolar hyalinosis, to acute, i.e. transplant endarteritis or intimal swelling in TMA. Since only a relatively small subgroup of patients with intra-parenchymal small vessel obstruction present with concurrent CG, we hypothesize that a sudden, significant impairment of glomerular perfusion is needed to promote CG. Such a decrease of glomerular blood flow can be caused by an acute disease process, e.g. a TMA, or acute chronic vascular changes, e.g. vascular sclerosis plus superimposed drug-induced vasoconstriction or dehydration. Anecdotal personal experience seems to further support this notion, i.e. CG-like changes are seen in glomeruli located in transitional zones of ischemic renal infarcts. Transient episodes of CG can be found during severe acute ABMR with endothelial cell injury that resolves under anti-rejection therapy (V.N., unpublished personal observations). The intriguing concept of potential CG resolution in cases with reversible sudden impairment of glomerular blood flow and endothelial injury is currently poorly understood and requires targeted future studies. In contrast to the sudden decrease of flow, a more protracted glomerular flow impairment results in shrinkage of glomerular tufts and wrinkling of capillary walls with largely unaltered podocyte morphology. Also other diseases, such as atheroembolization or sickle cell crisis affecting blood flow, follow the same pathophysiologic chain of events and can lead to secondary variants of ‘accompanied’ CG as previously reported in a few anecdotal reports [16, 17]. Likely due to the small number of recorded cases in the UNC nephropathology database, such disease associations remained statistically undetected in this study. Interestingly, sole intra-glomerular events, such as glomerulonephritides with endocapillary proliferations, tuft necrosis or crescent formation, seem to be less powerful CG promoters. In comparison, in T-FSGS, severe vasculopathies were uncommon findings; no case of a TMA was recorded. Thus, in the right window of opportunity, a specific type of ischemic glomerular and podocyte injury occurs that triggers CG rather than other forms of podocytopathies, such as T-FSGS.
Interestingly, another renal comorbidity, i.e. primary or secondary MGN, was prevalent in native kidney biopsies not only in CG but also in T-FSGS; in comparison, MGN was uncommon in renal allografts. MGNs characterized by immune complex-type deposits and architectural alterations along the GBM seem to enhance tuft ‘stickiness’ with adhesion formation near urinary poles and formation of T-FSGS and in some cases also CG. Thus, MGNs are less potent CG triggering events.
In our study population, HIV infection was only reported in a small minority of subjects; no patient was known to suffer from AIDS and classical HIVAN was not seen.
Previous limited studies looking into the prevalence of CG in selected renal diseases reported it in 5% of patients with DM and severe AS [10]. The authors speculated that vascular narrowing was involved in the pathogenesis with ischemia-induced over-expression of VEGF in podocytes. Another recently published selected case series reported a high concurrence of CG and TMA (19/53; 35.8%) [18]. It should be noted that the cohort in this last study was almost entirely Caucasian and so it is unlikely that APOL1 polymorphism played a significant role in the development of CG in these patients. Although comparative analyses with control cohorts were not conducted, these studies nevertheless show a strong association between CG and intra-renal vascular obstruction. In addition, autoimmune conditions have been associated with CG, and it is hypothesized that ‘perturbations’ of the immune system might trigger podocytopathies and hyperplasia [11, 19, 20]. An additional factor, increased incidence of APOL1 gene risk variants serving as a potential predisposing factor in the development of CG features in the setting of SLE has been reported, as many of them are AA patients [21].
However, in our study, we were unable to establish a significant association of CG and biopsy-proven SLE nephritis. Our observations raise the possibility of either a coincidental concurrence of SLE and CG or of glomerular blood flow impairment as a triggering event in some cases of SLE nephritis. Several case reports described presumably secondary variants CG in the context of other diseases, such as leukemia, malaria and sickle cell anemia [16, 22–24]. Three cases of CG in patients with active tuberculosis have been reported, two of them showed clinical remission of renal symptoms post anti-tuberculosis treatment [25, 26]. It is tempting to speculate that ischemia and low flow might have contributed to secondary CG in some of these patients.
In previous studies, the incidence of CG in allografts was reported to be 3.5%, with 22% of affected patients presenting with concomitant acute cellular rejection and 56% with severe arteriolar hyalinosis [8]. Santoriello et al. recently reported a series of 38 cases; they associated CG with rejection (in 60% of patients), CMV/EBV/Parvovirus19 infections, vaso-occlusive disease and donors of African descent. Interestingly, in Santoriello et al.’s study APOL1 high-risk status was only one factor associated with CG. The authors noted remittance of CG in 39% of renal transplant recipients [9]. In a small case series of transplant nephrectomies, Nadasdy et al. showed zonal detection of CG in areas with occlusive vascular injury [27]. Our current observations are in line with these previous reports.
Compared with other ethnic groups, patients of African descent more often show APOL1 G1 and G2 gene polymorphism and associated increased risk for the development of various chronic kidney diseases including CG [5, 21, 28–30]. Although in our UNC data base information on APOL1 gene polymorphism is not available, most likely APOL1 risk constellations also play a causative role in our patient cohort. However, since the ratio of cases that presented as ‘accompanied’ CG was similar between AA and Caucasian patients, we speculate that APOL1 gene polymorphism is likely only one underlying precondition that can promote CG. Recently, Santoriello et al. reported similar observations [9].
Our report has limitations. In the UNC nephropathology registry, clinical data collection solely depends on selected information provided at time of biopsy. Information on APOL1 genotypes and risk status is not available, and data on concurrent viral infections such as CMV or Parvovirus 19 is sketchy. Specific information on outcome is not recorded mainly due to the large referral base with biopsies estimated to derive from a population of ∼10 million, and lack of routine follow-up information [31]. It is also beyond the scope of our observational retrospective study to shed light on specific pathophysiologic events and mechanisms leading from vascular obstruction to glomerular low flow, and podocytopathies with tuft collapse.
In conclusion, our unique morphology-based study evaluated disease associations between CG and other renal comorbidities utilizing recorded data from a single major diagnostic nephropathology referral center. We provide evidence that renal comorbidities are commonly seen in biopsies with CG, in particular, post-kidney transplantation, but not in T-FSGS. Among those, intra-renal small vessel obstruction, likely of sudden onset, with impairment of glomerular blood flow is most tightly associated with CG in native and transplant biopsies. Such association is not prominent in controls including T-FSGS. For the first time, we could also show that in CG as well as in T-FSGS MGNs are significantly more common, representing another pathway leading to secondary forms of glomerular tuft injury. Our observations can form the basis for targeted future studies. Morphologic and clinical features, as well as underlying ischemic events characterizing secondary CG, have to be studied, and the potential for CG reversibility analyzed in systematic outcome studies. Secondary CG due to vascular obstruction: a more benign and potentially reversible ‘CG variant’ with a clinical course driven by the underlying renal comorbidity?
ACKNOWLEDGEMENTS
The authors thank J. Charles Jennette, MD for critical review of the study and for his help with editing the manuscript.
AUTHORS’ CONTRIBUTIONS
F.G. and V.N. are the main authors of this manuscript. H.K.S. assisted with the study design and the manuscript preparation.
CONFLICT OF INTEREST STATEMENT
The authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest or nonfinancial interest in the subject matter or materials discussed in this manuscript. The results presented in this article have not been published previously in whole or part, except in abstract form.
REFERENCES
Author notes
Present address: Francois Gougeon, University of Montreal, Department of Pathology and Cellular Biology, Montreal, QC, Canada


{kind=link}
Comments