





Abstract
Chronic kidney disease (CKD) is defined as an alteration of kidney structure and/or function lasting for >3 months [1]. CKD affects 10% of the general adult population and is responsible for large healthcare costs [2]. Since the end of the last century, the role of hypoxia in CKD progression has controversially been discussed. To date, there is evidence of the presence of hypoxia in late-stage renal disease, but we lack time-course evidence, stage correlation and also spatial co-localization with fibrotic lesions to ensure its causative role. The classical view of hypoxia in CKD progression is that it is caused by peritubular capillary alterations, renal anaemia and increased oxygen consumption regardless of the primary injury. In this classical view, hypoxia is assumed to further induce pro-fibrotic and pro-inflammatory responses, as well as oxidative stress, leading to CKD worsening as part of a vicious circle. However, recent investigations tend to question this paradigm, and both the presence of hypoxia and its role in CKD progression are still not clearly demonstrated. Hypoxia-inducible factor (HIF) is the main transcriptional regulator of the hypoxia response. Genetic HIF modulation leads to variable effects on CKD progression in different murine models. In contrast, pharmacological modulation of the HIF pathway [i.e. by HIF hydroxylase inhibitors (HIs)] appears to be generally protective against fibrosis progression experimentally. We here review the existing literature on the role of hypoxia, the HIF pathway and HIF HIs in CKD progression and summarize the evidence that supports or rejects the hypoxia hypothesis, respectively.
THE CHRONIC HYPOXIA HYPOTHESIS
The kidneys display a particular vascularization, creating a steep cortico-medullary pO2 gradient with tissue pO2 as low as 10 mmHg in the medulla [3, 4]. Tubular cells are major contributors to the generation and maintenance of the cortico-medullary pO2 gradient due to their high energy usage fuelled by their constant metabolic demand for endocytosis and transport, among other processes [5]. This particular setting in the kidney likely explains why erythropoietin (EPO) is synthesized by renal peritubular interstitial cells within the cortico-medullary border [6, 7]. Hence, the renal EPO-producing (REP) cells of the kidney are sensors for systemic oxygen supply [8, 9].
Hypoxia occurs when the local tissue/cellular demand exceeds its oxygen supply. Hypoxia is proposed as a common feature of different types of renal diseases [10]. The classical theory about the role of hypoxia in chronic kidney disease (CKD) was first described in 1998 [11]. It was hypothesized that hypoxia was intrinsically linked to the pathophysiology of CKD, as a direct consequence of any glomerular and tubular damage, promoting renal injury. Independent of the initial injury, CKD presents two common features: glomerulosclerosis and tubulointerstitial fibrosis [12]. The rationale for the theory that hypoxia is detrimental in CKD is that the initial glomerular injury induces itself a stagnation of peri-glomerular capillary blood flow [8]. Inflammation and epithelial cell dedifferentiation lead to accumulation of extracellular matrix that disorganizes architecture and hinders the normal peritubular capillary vascularization, as well as decreasing the oxygen supply of the injured tissue area [13]. An imbalance between vasoconstriction and vasodilatation has also been proposed by Fine and Norman [12] as the cause of hypoxia in diseased renal tissue. During late stages of CKD, a large majority of patients develop renal anaemia, which exaggerates the diminution of O2 supply. Besides, there is also an increase in O2 consumption within the kidney due to the metabolic alterations occurring in tubular cells, including the uncoupling of mitochondrial respiration due to oxidative stress [14]. Because of the loss of functional tissue, the remnant nephrons become hypertrophic and hyperfiltrate, potentially leading to an increase in their oxygen consumption [15]. All these factors induce an imbalance between O2 supply and O2 consumption causing hypoxia, especially within the damaged tissue areas.
EVIDENCE FOR HYPOXIA IN CKD
Although of great interest, the role of hypoxia in CKD progression is experimentally not conclusively demonstrated yet. Our understanding of spatial and time-dependent differences in kidney tissue oxygenation is currently limited by the resolution of the available methods.
Current techniques allowing the assessment of hypoxia in tissues are pimonidazole staining, microelectrode-dependent measurements, analyses of hypoxia-inducible factor (HIF)-α stabilization/HIF transcriptional activity and bioimaging techniques such as Blood oxygenation level dependent - Magnetic resonance Imaging (BOLD-MRI) [16]. Immunohistochemical detection of pimonidazole-bound proteins in hypoxic cells allows spatial detection of hypoxia, but is non-quantitative, has a low sensitivity (staining only occurs at pO2 <10 mmHg) and cannot be used in living animals [17]. Microelectrodes allow efficient quantitative oxygen detection but they can only be used post-decapsulation in anesthetized animals, which leads to numerous biases, and the obtained values do not always reflect intra-tissue pO2 levels. The analysis of HIF-α stabilization/HIF transcriptional activity is also indirect and potentially biased by any non-hypoxic regulation of HIF during CKD, such as the transcriptional regulation of HIF-1α by nuclear factor (NF)-κB [18, 19]. BOLD-MRI has the advantage of being performed in clinical settings, but it is a rather indirect marker of hypoxia as it measures de-oxy haemoglobin (Hb) [20]. Other non-invasive imaging techniques include Positron Emission Tomography-computed tomography (PET-CT) with nitroimidazole-family tracers [21] or two-photon phosphorescence lifetime microscopy (2PLM) of oxygen, allowing quantitative pO2 measurement in living animals [22]. Although promising, PET-CT with 18F-fluoromisonidazole can lack reproducibility and correlation with other methods assessing hypoxia [21]. 2PLM is limited by the performance of the probe used [23]. Altogether, no gold standard or easy to use method is currently available, likely explaining some of the discrepancies in the different analyses of hypoxia in CKD.
As the techniques to measure tissue pO2 in clinical settings are rather limited, few data exist in humans regarding hypoxia in kidney tissue during CKD. BOLD-MRI clinical studies have offered an interesting insight, showing that low cortical oxygenation may be predictive of renal function decline [24–26]; however, no direct correlation between CKD stage and BOLD-MRI was found in other studies [27, 28]. In animal models, studies with oxygen-detecting microelectrodes found hypoxia throughout the cortex in a rat model of polycystic kidney disease [29]. Hypoxia was also detected by the same method in streptozotocin-induced rat diabetic nephropathy [30], even at early stages [31]. In the rat subtotal nephrectomy model, hypoxia assessed by biosensors was found at late stages [32]. However, we lack evidence regarding co-localization of hypoxia with fibrotic lesions or remnant nephrons as well as time courses of tissue pO2 during CKD progression, which are needed to assess the pathophysiological role of hypoxia. A recent time course study using microelectrodes in an animal model of adenine-induced CKD showed that hypoxia was present at late stages of the disease, but not at early and intermediate stages, indicating that hypoxia may not play a key role in the pathogenesis of CKD progression in the adenine model [33]. Indeed, although areas of hypoxia are detected in CKD, the global effect may not be as clear as previously stated, since overall tubular workload decreased in parallel to oxygen supply in renal disease [33, 34]. Finally, in an experimental model of diabetic kidney disease, repeated exposition to a hypoxic environment rather improved the course of the disease [35].
Altogether, the currently available evidence points towards the presence of hypoxia in late-stage renal disease. The presence and the role of hypoxia during the development and progression of renal disease are in contrast much less clear and will need further investigation.
THE HIF SYSTEM AND ITS TARGETS
In tissues, the main regulator of the cellular transcriptional adaptive response to hypoxia is the HIF pathway (Figure 1). In 2019, William G. Kaelin Jr, Sir Peter J. Ratcliffe and Gregg L. Semenza were awarded the Nobel Prize in Physiology or Medicine for their seminal discoveries in cellular oxygen sensing and cellular adaptations to hypoxia via the HIF pathway [36]. Briefly, HIFs are heterodimeric complexes made of three different oxygen-dependently regulated α subunits (HIF-1α to -3α) and one constitutional β subunit (HIF-β) that can bind as a heterodimer to the transcriptional coactivators C-AMP Response Element-binding protein (CREB-binding protein)/binding protein p300 (CBP/p300) in the nucleus, enhancing transcription of downstream genes [37]. HIF-dependent regulation of gene expression leads to the adaption of cells and tissues to hypoxia. There are likely >1000 different HIF target genes, including EPO, transferrin and vascular endothelial growth factor (VEGF). HIF also switches the cellular metabolism towards anaerobic energy production through increased transcription of glycolytic enzymes and the inhibition of oxidative metabolism by increased expression of pyruvate dehydrogenase kinase 1 [37–39].
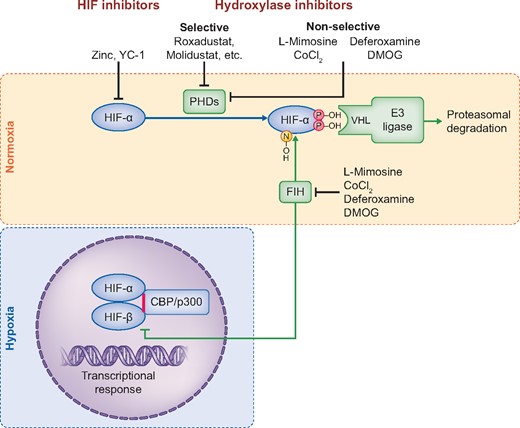
Regulation of HIF transcriptional activity. Under normoxia, HIF-α is hydroxylated by PHD enzymes, resulting in the recognition of HIF-α by an ubiquitin–E3 ligase complex and subsequent proteasomal degradation. Asparagine hydroxylation by FIH prevents the recruitment of the transcriptional coactivators/histone acetyltransferases p300/CBP, decreasing HIF transactivation activity. Under hypoxia, HIF-α is stabilized and translocates into the nucleus, forming an heterodimer with HIF-β. Once activated by CBP/p300, this structure will bind to hypoxia-response elements and enhance the expression of target genes. The HIF pathway can be modulated through the inhibition of the HIF hydroxylases by different compounds, such as L-mimosine, CoCl2, DMOG or deferoxamine. More selective HIF HIs are contained within the new class of –dustat inhibitors, which were recently developed for the treatment of anaemia. All HIF HIs will increase HIF-α. Zinc and the compound YC-1 were reported to be HIF-α inhibitors.
HIF-1α and HIF-2α, the most studied isoforms, differ regarding their tissue expression and regulated target genes. HIF-1β is ubiquitously expressed. HIF-1α is found in all organs, whereas HIF-2α is mainly expressed in lung and heart, as well as endotheliums. In kidneys, HIF-1α is confined to renal tubules, whereas HIF-2α is expressed in endothelial [40] and REP cells [7]. HIF-1α is more dedicated to the regulation of genes involved in glycolytic metabolism [41] and HIF-2α regulates the gene expression of EPO and genes important for iron metabolism [42]. HIF-3α is less well understood but might act as a negative regulator [43].
The oxygen-dependent regulation of the HIF pathway occurs through the enzymatic activity of three prolyl-4-hydroxylase domain (PHD1–3) enzymes and the asparagine hydroxylase factor inhibiting HIF (FIH) [44] (Figure 2). PHDs hydroxylate HIF-α isoforms which in turn affects HIF-α protein stability. Prolyl hydroxylation of HIF-α promotes its proteasomal degradation by increasing its binding affinity to the von Hippel Lindau (VHL) protein, which is part of an E3 ubiquitin ligase that targets HIF-α for proteasomal degradation by ubiquitination [45]. FIH-dependent asparagine hydroxylation of HIF-α prevents the interaction of the HIF heterodimer with CBP/p300, decreasing HIF transactivation activity for selected target genes.
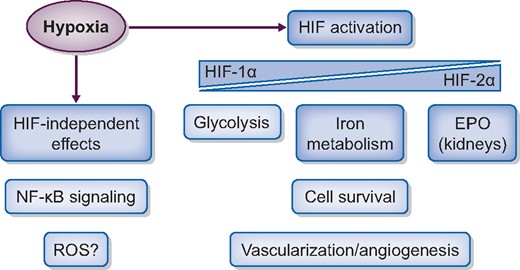
The effects of hypoxia. Hypoxia activates the HIF, which will trigger a transcriptional adaptive response regulating metabolism, increasing EPO and iron metabolism, vascularization and angiogenesis. Hypoxia can also lead to HIF-independent responses, for example through regulation of the NF-κB pathway. Besides the HIF-mediated response, there is also a hypoxia-dependent regulation of gene expression independent of HIF.
There is evidence that the cellular hypoxia response is not only mediated by the HIF pathway. HIF-independent gene regulation has been shown under hypoxia [46–49], and hypoxia sensitivity was reported for almost 20 transcription factors [50]. Interestingly, it was demonstrated that hypoxia and inflammation mutually regulate each other through complex mechanisms [51, 52]. Furthermore, the HIF-α hydroxylases have likely substrate proteins outside the HIF pathway [50, 53–55]. Of note, these substrates include several proteins of the NF-κB signalling pathway [56]. NF-κB is the master regulator of the cellular transcriptional response to pro-inflammatory stimuli. Therefore, a regulation of the NF-κB pathway by the HIF-α hydroxylases could take part in the interaction of hypoxia and CKD progression. However, the relevance of these regulations and the interplay of hypoxia and inflammation specifically for CKD progression is not well understood. Oxidative stress and reactive oxygen species (ROS) have previously been implicated in the progression of CKD [57, 58]. While it is well established that intracellular ROS increase during the reoxygenation period following hypoxia (as it is e.g. occurring during ischaemia–reperfusion injury), it is more controversially discussed whether ROS also increase during hypoxia [59–61]. The differences in ROS detection may be due to differences in basal cell metabolism, and the hypoxia degree as well as its duration [62]. Overall, it is likely that hypoxia-mediated regulations contribute to CKD progression independent of HIF, but the detailed mechanisms need further analyses.
IS HIF ACTIVATION BENEFICIAL OR DETRIMENTAL IN CKD?
The contribution of the HIF pathway is an important question in various diseases, including CKD. However, the available methods to assess the pathway have several limitations. As the regulation of HIF-α does not occur at a transcriptional level, mRNA analyses of HIF-α usually cannot offer an indication about the activation of the HIF pathway. The analyses of the mRNA levels of HIF target genes can be used to gain some insights into the activity of the HIF pathway, but it can be confounded by additional gene expression regulations, e.g. through other transcription factors. Protein analyses of HIF pathway components or down-stream targets are limited by the quality of the existing antibodies. In addition, because HIF-α protein regulation occurs very fast [63], animal euthanasia and tissue collection can introduce a bias by changing the tissue pO2.
The HIF pathway has been extensively studied in AKI. HIF activation is universally observed in ischaemic models [64] and genetic ablation of the HIF pathway, either HIF-1α or HIF-2α, induced an aggravation of the renal injury [64–67]. Systemic activation of the HIF pathway using HIF hydroxylase inhibitors (HIs) before the injury prevented AKI episodes [66–70]. Similarly, global HIF-1α overexpression was protective [71, 72]. Altogether, HIF-α deletion is detrimental in AKI, whereas pre-stimulation of the HIF pathway genetically or pharmacologically prevents AKI episodes, mostly in ischaemia–reperfusion.
In contrast to AKI, there is more debate on the regulation of the HIF pathway in CKD. Some clinical evidence shows an increase of HIF-1α protein levels in lupus nephritis [73] and its target genes in nephrosclerosis [74]. HIF-1α mRNA has even been proposed as a urinary biomarker for CKD [75]. More recent experimental evidence is rather in favour of spatially limited HIF activation [33, 34], or even a downregulation of the pathway [76] and its target genes such as VEGF in CKD [77]. The cause of the inadequate response of the HIF pathway described in some experimental settings, such as diabetic nephropathy, is unclear. Glucose may play a direct inhibitory role on the HIF pathway [78–80]. Some authors have proposed that HIF upregulation could be suboptimal in CKD [76, 78], potentially accelerating the pathophysiological process. Finally, HIs allow renal anaemia correction through HIF stimulation in CKD and are now well validated in different experimental models and clinical trials. Their stimulatory effect on EPO production may further indicate a suboptimal activation of the HIF pathway in CKD (at least of HIF-2α), which can be sufficiently increased pharmacologically to re-stimulate EPO synthesis.
The current literature on the role of HIF in experimental models of CKD progression using genetic modifications for altering HIF-1α and/or HIF-2α levels is summarized in Table 1. At baseline, without any renal injury, inducing a stable tubular overexpression of HIF-1α and HIF-2α by VHL deletion is sufficient to induce a rapidly progressive fibrosis [81]. A genetic endothelial-specific knockout of PHD2 (leading to constitutive HIF-1α and HIF-2α stabilization) also induced fibrosis and elevated serum creatinine at baseline [82]. Similarly, genetic HIF-2α overexpression in tubules increased renal fibrosis and dysfunction and induced cysts without specific induction of renal disease [83]. In ageing mice, tubular overexpression of HIF-1 α accelerated renal damage [84]. Thus, genetic HIF stabilization/overexpression in the kidney appears to be detrimental.
Experimental studies of the effect of HIF in rodent models of CKD: genetic modulation of the HIF pathway
| Genetic modification | Experimental model | Effect | Reference |
|---|---|---|---|
| Global HIF overexpression | UUO | Suppressed inflammation and fibrogenesis | [85] |
| Genetic HIF-2α overexpression | Adenine-induced nephropathy | At early stages, aggravated histological lesions without impairment of renal function; at late stages, improved renal function and fibrosis | [88] |
| Genetic ablation of endothelial HIF-1α | Angiotensin-II-induced renal injury | Decreased microalbuminuria and fibrosis | [91] |
| Genetic ablation of epithelial HIF-1α | UUO | Decreased tubulointerstitial fibrosis | [94] |
| Genetic PHD2 knockout | Baseline | Increased renal fibrosis, dysfunction and cysts | [83] |
| Baseline | Elevation of serum creatinine, increased interstitial fibrosis | [82] | |
| HIF-1α shRNA | Angiotensin-II-induced renal injury | Decreased albuminuria and glomerular damage | [89] |
| Unilateral clamping of the left renal artery for 3 weeks | Attenuated glomerular injury index and fibrotic markers | [90] | |
| Inducing stable expression of HIF-1/2 via Vhl knock-out in podocytes | Baseline | Induces rapidly progressing glomerulonephritis | [81] |
| Inducing stable expression of HIF-1/2 via Vhl knock-out | UUO | Increased fibrosis | [95] |
| 5/6 nephrectomy | Increased fibrosis | [95] | |
| Tubular HIF-1α overexpression | Baseline | Accelerated renal damage in ageing mice | [84] |
| Systemic HIF-1α knock-down | Streptozotocin-induced diabetic nephropathy in mice | Increased glomerular and podocytes damage | [87] |
| Systemic HIF-1α knock-out | Streptozotocin-induced diabetic nephropathy in mice | Increased histological damage, fibrosis and albuminuria | [86] |
| Mouse podocyte ablation model | Decreased glomerulosclerosis and col-1α accumulation | [93] | |
| Polycystic disease induced by Pkd deletion | In the severe phenotype, attenuated cyst growth; no effect in the mild phenotype | [96] | |
| Systemic Hif1α + Hif2α deletion | UUO | Aggravated fibrosis and inflammatory cell infiltration | [67] |
| None | MRL/lpr lupus nephritis mouse model | Decreased proteinuria and tubulointerstitial injury | [92] |
| Genetic modification | Experimental model | Effect | Reference |
|---|---|---|---|
| Global HIF overexpression | UUO | Suppressed inflammation and fibrogenesis | [85] |
| Genetic HIF-2α overexpression | Adenine-induced nephropathy | At early stages, aggravated histological lesions without impairment of renal function; at late stages, improved renal function and fibrosis | [88] |
| Genetic ablation of endothelial HIF-1α | Angiotensin-II-induced renal injury | Decreased microalbuminuria and fibrosis | [91] |
| Genetic ablation of epithelial HIF-1α | UUO | Decreased tubulointerstitial fibrosis | [94] |
| Genetic PHD2 knockout | Baseline | Increased renal fibrosis, dysfunction and cysts | [83] |
| Baseline | Elevation of serum creatinine, increased interstitial fibrosis | [82] | |
| HIF-1α shRNA | Angiotensin-II-induced renal injury | Decreased albuminuria and glomerular damage | [89] |
| Unilateral clamping of the left renal artery for 3 weeks | Attenuated glomerular injury index and fibrotic markers | [90] | |
| Inducing stable expression of HIF-1/2 via Vhl knock-out in podocytes | Baseline | Induces rapidly progressing glomerulonephritis | [81] |
| Inducing stable expression of HIF-1/2 via Vhl knock-out | UUO | Increased fibrosis | [95] |
| 5/6 nephrectomy | Increased fibrosis | [95] | |
| Tubular HIF-1α overexpression | Baseline | Accelerated renal damage in ageing mice | [84] |
| Systemic HIF-1α knock-down | Streptozotocin-induced diabetic nephropathy in mice | Increased glomerular and podocytes damage | [87] |
| Systemic HIF-1α knock-out | Streptozotocin-induced diabetic nephropathy in mice | Increased histological damage, fibrosis and albuminuria | [86] |
| Mouse podocyte ablation model | Decreased glomerulosclerosis and col-1α accumulation | [93] | |
| Polycystic disease induced by Pkd deletion | In the severe phenotype, attenuated cyst growth; no effect in the mild phenotype | [96] | |
| Systemic Hif1α + Hif2α deletion | UUO | Aggravated fibrosis and inflammatory cell infiltration | [67] |
| None | MRL/lpr lupus nephritis mouse model | Decreased proteinuria and tubulointerstitial injury | [92] |
The studies showing a protective role of HIF are highlighted in green and those in favour of a detrimental role are highlighted in orange. Yellow indicates a dual role.
Experimental studies of the effect of HIF in rodent models of CKD: genetic modulation of the HIF pathway
| Genetic modification | Experimental model | Effect | Reference |
|---|---|---|---|
| Global HIF overexpression | UUO | Suppressed inflammation and fibrogenesis | [85] |
| Genetic HIF-2α overexpression | Adenine-induced nephropathy | At early stages, aggravated histological lesions without impairment of renal function; at late stages, improved renal function and fibrosis | [88] |
| Genetic ablation of endothelial HIF-1α | Angiotensin-II-induced renal injury | Decreased microalbuminuria and fibrosis | [91] |
| Genetic ablation of epithelial HIF-1α | UUO | Decreased tubulointerstitial fibrosis | [94] |
| Genetic PHD2 knockout | Baseline | Increased renal fibrosis, dysfunction and cysts | [83] |
| Baseline | Elevation of serum creatinine, increased interstitial fibrosis | [82] | |
| HIF-1α shRNA | Angiotensin-II-induced renal injury | Decreased albuminuria and glomerular damage | [89] |
| Unilateral clamping of the left renal artery for 3 weeks | Attenuated glomerular injury index and fibrotic markers | [90] | |
| Inducing stable expression of HIF-1/2 via Vhl knock-out in podocytes | Baseline | Induces rapidly progressing glomerulonephritis | [81] |
| Inducing stable expression of HIF-1/2 via Vhl knock-out | UUO | Increased fibrosis | [95] |
| 5/6 nephrectomy | Increased fibrosis | [95] | |
| Tubular HIF-1α overexpression | Baseline | Accelerated renal damage in ageing mice | [84] |
| Systemic HIF-1α knock-down | Streptozotocin-induced diabetic nephropathy in mice | Increased glomerular and podocytes damage | [87] |
| Systemic HIF-1α knock-out | Streptozotocin-induced diabetic nephropathy in mice | Increased histological damage, fibrosis and albuminuria | [86] |
| Mouse podocyte ablation model | Decreased glomerulosclerosis and col-1α accumulation | [93] | |
| Polycystic disease induced by Pkd deletion | In the severe phenotype, attenuated cyst growth; no effect in the mild phenotype | [96] | |
| Systemic Hif1α + Hif2α deletion | UUO | Aggravated fibrosis and inflammatory cell infiltration | [67] |
| None | MRL/lpr lupus nephritis mouse model | Decreased proteinuria and tubulointerstitial injury | [92] |
| Genetic modification | Experimental model | Effect | Reference |
|---|---|---|---|
| Global HIF overexpression | UUO | Suppressed inflammation and fibrogenesis | [85] |
| Genetic HIF-2α overexpression | Adenine-induced nephropathy | At early stages, aggravated histological lesions without impairment of renal function; at late stages, improved renal function and fibrosis | [88] |
| Genetic ablation of endothelial HIF-1α | Angiotensin-II-induced renal injury | Decreased microalbuminuria and fibrosis | [91] |
| Genetic ablation of epithelial HIF-1α | UUO | Decreased tubulointerstitial fibrosis | [94] |
| Genetic PHD2 knockout | Baseline | Increased renal fibrosis, dysfunction and cysts | [83] |
| Baseline | Elevation of serum creatinine, increased interstitial fibrosis | [82] | |
| HIF-1α shRNA | Angiotensin-II-induced renal injury | Decreased albuminuria and glomerular damage | [89] |
| Unilateral clamping of the left renal artery for 3 weeks | Attenuated glomerular injury index and fibrotic markers | [90] | |
| Inducing stable expression of HIF-1/2 via Vhl knock-out in podocytes | Baseline | Induces rapidly progressing glomerulonephritis | [81] |
| Inducing stable expression of HIF-1/2 via Vhl knock-out | UUO | Increased fibrosis | [95] |
| 5/6 nephrectomy | Increased fibrosis | [95] | |
| Tubular HIF-1α overexpression | Baseline | Accelerated renal damage in ageing mice | [84] |
| Systemic HIF-1α knock-down | Streptozotocin-induced diabetic nephropathy in mice | Increased glomerular and podocytes damage | [87] |
| Systemic HIF-1α knock-out | Streptozotocin-induced diabetic nephropathy in mice | Increased histological damage, fibrosis and albuminuria | [86] |
| Mouse podocyte ablation model | Decreased glomerulosclerosis and col-1α accumulation | [93] | |
| Polycystic disease induced by Pkd deletion | In the severe phenotype, attenuated cyst growth; no effect in the mild phenotype | [96] | |
| Systemic Hif1α + Hif2α deletion | UUO | Aggravated fibrosis and inflammatory cell infiltration | [67] |
| None | MRL/lpr lupus nephritis mouse model | Decreased proteinuria and tubulointerstitial injury | [92] |
The studies showing a protective role of HIF are highlighted in green and those in favour of a detrimental role are highlighted in orange. Yellow indicates a dual role.
Genetic manipulation of the HIF pathway in murine models of renal disease yielded more contrasting results. Some studies observed a protective role of HIF on CKD progression. Indeed, systemic genetic inactivation of both HIF-1α and HIF-2α was deleterious in unilateral urinary tract obstruction (UUO) [85]. In streptozotocin-induced diabetic nephropathy, systemic deficiency in HIF-1α increased tissue damage and fibrosis in the kidneys [86] or induced more podocyte damage [87]. In contrast, HIF was detrimental for CKD progression in other studies. In the adenine model of CKD, a dual role of HIF-2α was observed, with HIF-2α tubular-specific knock-out causing aggravation of histological lesions at early stages and protection against fibrosis and renal function decline in late stages [33, 88]. In angiotensin-II-induced injury, HIF-1α short hairpin ribonucleic acid (shRNA) transfected into the kidney decreased albuminuria and glomerular damage [89] and decreased fibrotic markers in a chronic ischaemia model [90]. Endothelial-specific HIF-1α genetic ablation also decreased microalbuminuria and fibrosis [91]. Similarly, in a model of lupus nephritis, HIF-1α shRNA protected against proteinuria and tubulointerstitial injury [92]. In the NEP 25 podocyte injury mouse model, systemic HIF-1α genetic inactivation was protective against kidney lesions [93]. In the UUO model of fibrosis, HIF-1α tubular deletion was protective against fibrosis progression [94]. In the same model and also in subtotal nephrectomy, stabilization of HIF-1α and HIF-2α via tubular VHL knock-out increased fibrosis [95]. Finally, in polycystic kidney disease, an attenuation of the cystic phenotype was observed in Hif-1α-Pkd systemic double knock-out mice [96]. Altogether, genetic modulation of the HIF pathway in mouse models of CKD yields variable results according to the compartment of expression, the timing and the disease model used.
Besides genetic manipulations of the HIF system, pharmacologic HIs can be used to induce the HIF pathway [97]. Each different inhibitor possesses specific properties and they may not activate the HIF pathway to the same degree. In addition, at least some HIs may have off-target effects [97]. In contrast to the genetic activation of the HIF pathway, which repeatedly appeared to be detrimental for the kidney, rescue of a potentially suboptimal activation of HIF in CKD by pharmacological means appears rather beneficial (Table 2). L-Mimosine was able to improve renal function in the model of rat subtotal nephrectomy [98, 99, 100], even though early long-term treatment actually increased fibrosis [100]. Dimethyloxalylglycine (DMOG) improved histological damage and proteinuria [101], mitochondrial function [102] and cell survival [103] in the same model. DMOG also improved glomerulosclerosis, fibrosis, renal function and proteinuria in two models of hypertensive rats [104, 105]. Cobalt chloride (CoCl2) improved glomerular filtration rate, albuminuria and tubulointerstitial damage in a rat model of streptozotocin-induced diabetic nephropathy [106]. CoCl2 was also protective in the Thy1 nephritis rat model [107] in the subtotal nephrectomy rat model [108, 109] and in Type 2 diabetic nephropathy [110]. PHD inhibition with isoquinoline derivative 2-(1-chloro-4-hydroxyisoquinoline-3-carboxamido) acetate (also called FG-2216, YC-1: 3-(5′-Hydroxymethyl-2′-furyl)-1-benzyl indazole (BIQ or IOX3) and roxadustat prevented renal function decline and histological damage in two models of nephropathies: adenine- and crystal-induced nephropathies [34]. Enoradustat also reduced albuminuria and improved morphological damage in the ob/ob model of diabetic nephropathy [111]. In the rat subtotal nephrectomy model, enoradustat was also protective, with less fibrosis and inflammation [112]. Deferoxamine led to less tubulointerstitial fibrosis in UUO [113]. In contrast to these beneficial effects, pharmacological HIF activation was detrimental in renal cystic disease [96]. Regarding direct HIF inhibition, the YC-1 HIF-1α inhibitor reduced extracellular matrix accumulation, oxidative stress and albuminuria in OVE26 Type 1 diabetic mice [114]. Zinc, another proposed inhibitor of HIF, was protective in a model of streptozoticin-induced diabetic mice [115]. However, the selectivity of YC-1 and zinc for HIF is unclear and hence they may have had effects in addition to the inhibition of HIF.
Experimental studies of the effect of HIF in rodent models of CKD: pharmacological modulation of the HIF pathway
| Treatment | Experimental model | Effect | Reference |
|---|---|---|---|
| CoCl2a | Hypertensive, Type 2 diabetic rats with nephropathy (SHR/NDmcr-cp) | Reduced proteinuria and histological kidney injury | [110] |
| Subtotal nephrectomy in rats | Attenuated renal tubulointerstitial injury | [108] | |
| Subtotal nephrectomy in rats | Increased GFR | [109] | |
| Streptozotocin-induced diabetic nephropathy in rats | Improved renal function (GFR), reduced proteinuria and tubulointerstitial damage | [106] | |
| Uninephrectomized Thy1 nephritis rat model | Attenuated renal tubulointerstitial injury | [107] | |
| Deferoxaminea | UUO | Prevented tubulointerstitial fibrosis | [113] |
| DMOGa | Dahl salt-sensitive rats | Attenuated glomerulosclerosis and interstitial fibrosis | [104] |
| DOCA/salt-hypertensive rats | Improved renal function, proteinuria and fibrosis | [105] | |
| Subtotal nephrectomy in rats | Attenuated of glomerulosclerosis, tubulointerstitial injury and proteinuria | [101] | |
| Subtotal nephrectomy in rats | Globally inhibited apoptosis in STN kidneys, improves cell survival | [103] | |
| Subtotal nephrectomy in rats | Improved mitochondrial function | [102] | |
| Subtotal nephrectomy in rats | Increased GFR | [109] | |
| Enarodustata | BTBR ob/ob mice | Reduced albuminuria and improved glomerular and endothelial damage | [111] |
| Subtotal nephrectomy in rats | Attenuated fibrosis and reduced inflammation | [112] | |
| ICAa | Adenine-induced nephropathy, cristal-induced nephropathy | Attenuation of renal dysfunction and tubulointerstitial damage | [34] |
| Polycystic disease induced by Pkd deletion | Severe aggravation of polycystic disease with loss of renal function | [96] | |
| L-Mimosinea | Subtotal nephrectomy in rats | Advanced medium-term treatment attenuated but early long-term treatment increased fibrosis and renal dysfunction | [100] |
| Subtotal nephrectomy in rats | Improved renal function and fibrosis | [99] | |
| Subtotal nephrectomy in rats | Attenuated renal tubulointerstitial injury and improves renal function | [98] | |
| Roxadustata | Adenine-induced nephropathy, cristal-induced nephropathy | Attenuation of renal dysfunction and tubulointerstitial damage | [34] |
| YC-1b | Type 1 diabetic mouse model OVE26 | Reduced ECM accumulation, albuminuria and oxidative stress | [114] |
| Zincb | Streptozotocin-induced diabetic nephropathy in rats | Attenuation of tubular EMT and attenuated renal tubulointerstitial fibrosis | [115] |
| Treatment | Experimental model | Effect | Reference |
|---|---|---|---|
| CoCl2a | Hypertensive, Type 2 diabetic rats with nephropathy (SHR/NDmcr-cp) | Reduced proteinuria and histological kidney injury | [110] |
| Subtotal nephrectomy in rats | Attenuated renal tubulointerstitial injury | [108] | |
| Subtotal nephrectomy in rats | Increased GFR | [109] | |
| Streptozotocin-induced diabetic nephropathy in rats | Improved renal function (GFR), reduced proteinuria and tubulointerstitial damage | [106] | |
| Uninephrectomized Thy1 nephritis rat model | Attenuated renal tubulointerstitial injury | [107] | |
| Deferoxaminea | UUO | Prevented tubulointerstitial fibrosis | [113] |
| DMOGa | Dahl salt-sensitive rats | Attenuated glomerulosclerosis and interstitial fibrosis | [104] |
| DOCA/salt-hypertensive rats | Improved renal function, proteinuria and fibrosis | [105] | |
| Subtotal nephrectomy in rats | Attenuated of glomerulosclerosis, tubulointerstitial injury and proteinuria | [101] | |
| Subtotal nephrectomy in rats | Globally inhibited apoptosis in STN kidneys, improves cell survival | [103] | |
| Subtotal nephrectomy in rats | Improved mitochondrial function | [102] | |
| Subtotal nephrectomy in rats | Increased GFR | [109] | |
| Enarodustata | BTBR ob/ob mice | Reduced albuminuria and improved glomerular and endothelial damage | [111] |
| Subtotal nephrectomy in rats | Attenuated fibrosis and reduced inflammation | [112] | |
| ICAa | Adenine-induced nephropathy, cristal-induced nephropathy | Attenuation of renal dysfunction and tubulointerstitial damage | [34] |
| Polycystic disease induced by Pkd deletion | Severe aggravation of polycystic disease with loss of renal function | [96] | |
| L-Mimosinea | Subtotal nephrectomy in rats | Advanced medium-term treatment attenuated but early long-term treatment increased fibrosis and renal dysfunction | [100] |
| Subtotal nephrectomy in rats | Improved renal function and fibrosis | [99] | |
| Subtotal nephrectomy in rats | Attenuated renal tubulointerstitial injury and improves renal function | [98] | |
| Roxadustata | Adenine-induced nephropathy, cristal-induced nephropathy | Attenuation of renal dysfunction and tubulointerstitial damage | [34] |
| YC-1b | Type 1 diabetic mouse model OVE26 | Reduced ECM accumulation, albuminuria and oxidative stress | [114] |
| Zincb | Streptozotocin-induced diabetic nephropathy in rats | Attenuation of tubular EMT and attenuated renal tubulointerstitial fibrosis | [115] |
HIF HI, bHIF(-1α) inhibition.
BTBR ob/ob: BTBR T+ Itpr3tf/J mouse strain with ob/ob mutation; DOCA, deoxycorticosterone acetate; ICA, Isoquinoline derivative 2-(1-chloro-4-hydroxyisoquinoline-3-carboxamido) acetate; ECM, extra-cellular matrix; OVE26: FVB(Cg)-Tg(Cryaa-Tag, Ins2-CALM1)260ve/PneJ strain; STN, subtotal nephrectomy.
The studies indicating a protective role of HIF induction/activity are highlighted in green and those in favour of a detrimental role are highlighted in orange. Yellow indicates a dual role. It should be taken into account that HIs may lead to effects in addition to HIF induction (Sulser et al. FASEB J 2019).
Experimental studies of the effect of HIF in rodent models of CKD: pharmacological modulation of the HIF pathway
| Treatment | Experimental model | Effect | Reference |
|---|---|---|---|
| CoCl2a | Hypertensive, Type 2 diabetic rats with nephropathy (SHR/NDmcr-cp) | Reduced proteinuria and histological kidney injury | [110] |
| Subtotal nephrectomy in rats | Attenuated renal tubulointerstitial injury | [108] | |
| Subtotal nephrectomy in rats | Increased GFR | [109] | |
| Streptozotocin-induced diabetic nephropathy in rats | Improved renal function (GFR), reduced proteinuria and tubulointerstitial damage | [106] | |
| Uninephrectomized Thy1 nephritis rat model | Attenuated renal tubulointerstitial injury | [107] | |
| Deferoxaminea | UUO | Prevented tubulointerstitial fibrosis | [113] |
| DMOGa | Dahl salt-sensitive rats | Attenuated glomerulosclerosis and interstitial fibrosis | [104] |
| DOCA/salt-hypertensive rats | Improved renal function, proteinuria and fibrosis | [105] | |
| Subtotal nephrectomy in rats | Attenuated of glomerulosclerosis, tubulointerstitial injury and proteinuria | [101] | |
| Subtotal nephrectomy in rats | Globally inhibited apoptosis in STN kidneys, improves cell survival | [103] | |
| Subtotal nephrectomy in rats | Improved mitochondrial function | [102] | |
| Subtotal nephrectomy in rats | Increased GFR | [109] | |
| Enarodustata | BTBR ob/ob mice | Reduced albuminuria and improved glomerular and endothelial damage | [111] |
| Subtotal nephrectomy in rats | Attenuated fibrosis and reduced inflammation | [112] | |
| ICAa | Adenine-induced nephropathy, cristal-induced nephropathy | Attenuation of renal dysfunction and tubulointerstitial damage | [34] |
| Polycystic disease induced by Pkd deletion | Severe aggravation of polycystic disease with loss of renal function | [96] | |
| L-Mimosinea | Subtotal nephrectomy in rats | Advanced medium-term treatment attenuated but early long-term treatment increased fibrosis and renal dysfunction | [100] |
| Subtotal nephrectomy in rats | Improved renal function and fibrosis | [99] | |
| Subtotal nephrectomy in rats | Attenuated renal tubulointerstitial injury and improves renal function | [98] | |
| Roxadustata | Adenine-induced nephropathy, cristal-induced nephropathy | Attenuation of renal dysfunction and tubulointerstitial damage | [34] |
| YC-1b | Type 1 diabetic mouse model OVE26 | Reduced ECM accumulation, albuminuria and oxidative stress | [114] |
| Zincb | Streptozotocin-induced diabetic nephropathy in rats | Attenuation of tubular EMT and attenuated renal tubulointerstitial fibrosis | [115] |
| Treatment | Experimental model | Effect | Reference |
|---|---|---|---|
| CoCl2a | Hypertensive, Type 2 diabetic rats with nephropathy (SHR/NDmcr-cp) | Reduced proteinuria and histological kidney injury | [110] |
| Subtotal nephrectomy in rats | Attenuated renal tubulointerstitial injury | [108] | |
| Subtotal nephrectomy in rats | Increased GFR | [109] | |
| Streptozotocin-induced diabetic nephropathy in rats | Improved renal function (GFR), reduced proteinuria and tubulointerstitial damage | [106] | |
| Uninephrectomized Thy1 nephritis rat model | Attenuated renal tubulointerstitial injury | [107] | |
| Deferoxaminea | UUO | Prevented tubulointerstitial fibrosis | [113] |
| DMOGa | Dahl salt-sensitive rats | Attenuated glomerulosclerosis and interstitial fibrosis | [104] |
| DOCA/salt-hypertensive rats | Improved renal function, proteinuria and fibrosis | [105] | |
| Subtotal nephrectomy in rats | Attenuated of glomerulosclerosis, tubulointerstitial injury and proteinuria | [101] | |
| Subtotal nephrectomy in rats | Globally inhibited apoptosis in STN kidneys, improves cell survival | [103] | |
| Subtotal nephrectomy in rats | Improved mitochondrial function | [102] | |
| Subtotal nephrectomy in rats | Increased GFR | [109] | |
| Enarodustata | BTBR ob/ob mice | Reduced albuminuria and improved glomerular and endothelial damage | [111] |
| Subtotal nephrectomy in rats | Attenuated fibrosis and reduced inflammation | [112] | |
| ICAa | Adenine-induced nephropathy, cristal-induced nephropathy | Attenuation of renal dysfunction and tubulointerstitial damage | [34] |
| Polycystic disease induced by Pkd deletion | Severe aggravation of polycystic disease with loss of renal function | [96] | |
| L-Mimosinea | Subtotal nephrectomy in rats | Advanced medium-term treatment attenuated but early long-term treatment increased fibrosis and renal dysfunction | [100] |
| Subtotal nephrectomy in rats | Improved renal function and fibrosis | [99] | |
| Subtotal nephrectomy in rats | Attenuated renal tubulointerstitial injury and improves renal function | [98] | |
| Roxadustata | Adenine-induced nephropathy, cristal-induced nephropathy | Attenuation of renal dysfunction and tubulointerstitial damage | [34] |
| YC-1b | Type 1 diabetic mouse model OVE26 | Reduced ECM accumulation, albuminuria and oxidative stress | [114] |
| Zincb | Streptozotocin-induced diabetic nephropathy in rats | Attenuation of tubular EMT and attenuated renal tubulointerstitial fibrosis | [115] |
HIF HI, bHIF(-1α) inhibition.
BTBR ob/ob: BTBR T+ Itpr3tf/J mouse strain with ob/ob mutation; DOCA, deoxycorticosterone acetate; ICA, Isoquinoline derivative 2-(1-chloro-4-hydroxyisoquinoline-3-carboxamido) acetate; ECM, extra-cellular matrix; OVE26: FVB(Cg)-Tg(Cryaa-Tag, Ins2-CALM1)260ve/PneJ strain; STN, subtotal nephrectomy.
The studies indicating a protective role of HIF induction/activity are highlighted in green and those in favour of a detrimental role are highlighted in orange. Yellow indicates a dual role. It should be taken into account that HIs may lead to effects in addition to HIF induction (Sulser et al. FASEB J 2019).
In humans, HIF prolyl HIs emerged as a new therapeutic drug for the treatment of renal anaemia [116]. The first large studies with roxadustat (FG-4592) were non-inferior to parenteral epoetin-α in dialysed patients [117] and superior to placebo in non-dialysed patients [118]. A Phase 2 study with vadadustat [119] showed a similar ability to maintain hemoglobin (Hb) levels as EPO in dialysis settings and other trials with vadadustat are ongoing [120]. Daprodustat (GSK1278863) had also a promising profile in several clinical studies [121–124] and the Anaemia Studies in CKD: Erythropoiesis via a Novel PHI Daprodustat (ASCEND) trials are currently further testing daprodustat in dialysis and non-dialysis settings with various schemes [125]. DS1093a is also being tested in CKD patients [126]. Molidustat is a further prolyl HI in Phase 3 clinical trials [127]. Although the trial is too recent to conclude about an effect on CKD progression, no detrimental effect by HIs on renal function was reported so far.
Altogether, most pre-clinical studies using HIs showed a protective effect against CKD progression in different rodent models, except in polycystic kidney disease. This may be mediated through HIF-α induction, but also potentially through HIF-independent effects of these drugs [128–130]. In humans, further studies of the current clinical trials will be needed to ensure the absence of detrimental effect of HIs on CKD progression and/or to reliably analyse potential beneficial effects.
CONCLUSIONS
From the available literature, the role of hypoxia in CKD progression is still unresolved. Further studies with multiple and complementary chronic models and renal function assessment are needed to definitely answer this question. The impact of hypoxia on cells and tissues should be differentiated from the activation of HIF, which is primarily an adaptive mechanism. The HIF pathway may have time-, isoform- and compartment-specific action in CKD progression. Pharmacological HIF activation by HIs appears mostly protective in experimental CKD progression in various models, either through HIF stimulation or through modulation of other pathways.
FUNDING
Funding was provided by the NCCR.kidney.ch. Sophie de Seigneux is supported by a SNF grant PP00P3 157454. Anna Faivre is supported by SNF grant 323530_191224.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
Search of: vadadustat - List Results - ClinicalTrials.gov [Internet]. [cited 2020 Mar 18]. Available from: https://clinicaltrials.gov/ct2/results?cond=&term=vadadustat&cntry=&state=&city=&dist=
Meadowcroft AM, Cizman B, Holdstock L et al. Daprodustat for anemia: a 24-week, open-label, randomized controlled trial in participants on hemodialysis. Clin Kidney J 2019; 12: 139–48
Holdstock L, Cizman B, Meadowcroft AM et al. Daprodustat for anemia: a 24-week, open-label, randomized controlled trial in participants with chronic kidney disease. Clin Kidney J 2019; 12: 129–38
Tsubakihara Y, Akizawa T, Nangaku M et al. A 24-week anemia correction study of daprodustat in Japanese dialysis patients. Ther Apher Dial Off Peer-Rev J Int Soc Apher Jpn Soc Apher Jpn Soc Dial Ther. 2020; 24: 108–14
Bailey CK, Caltabiano S, Cobitz AR et al. A randomized, 29-day, dose-ranging, efficacy and safety study of daprodustat, administered three times weekly in patients with anemia on hemodialysis. BMC Nephrol 2019; 20: 372
Search of: daprodustat - List Results - ClinicalTrials.gov [Internet]. [cited 2020 Mar 18]. Available from: https://clinicaltrials.gov/ct2/results?cond=&term=daprodustat&cntry=&state=&city=&dist=
Search of: DS1093a - List Results - ClinicalTrials.gov [Internet]. [cited 2020 Mar 18]. Available from: https://clinicaltrials.gov/ct2/results?cond=&term=DS1093a&cntry=&state=&city=&dist=
Yamamoto H, Taguchi M, Matsuda Y et al. Molidustat for the treatment of renal anaemia in patients with non-dialysis-dependent chronic kidney disease: design and rationale of two phase III studies. BMJ Open 2019; 9: e026704


{kind=link}
{kind=link}
Comments