





Abstract
Accumulating evidence indicates that the pathological changes of the endothelium may contribute to the development of cardiovascular complications in chronic kidney disease (CKD). Non-traditional risk factors related to CKD are associated with the incidence of cardiovascular disease, but their role in uraemic endothelial dysfunction has often been disregarded. In this context, soluble α-Klotho and vitamin D are of importance to maintain endothelial integrity, but their concentrations decline in CKD, thereby contributing to the dysfunction of the endothelial lining. These hormonal disturbances are accompanied by an increment of circulating fibroblast growth factor-23 and phosphate, both exacerbating endothelial toxicities. Furthermore, impaired renal function leads to an increment of inflammatory mediators, reactive oxygen species and uraemic toxins that further aggravate the endothelial abnormalities and in turn also inhibit the regeneration of disrupted endothelial lining. Here, we highlight the distinct endothelial alterations mediated by the abovementioned non-traditional risk factors as demonstrated in experimental studies and connect these to pathological changes in CKD patients, which are driven by endothelial disturbances, other than atherosclerosis. In addition, we describe therapeutic strategies that may promote restoration of endothelial abnormalities by modulating imbalanced mineral homoeostasis and attenuate the impact of uraemic retention molecules, inflammatory mediators and reactive oxygen species. A clinical perspective on endothelial dysfunction in CKD may translate into reduced structural and functional abnormalities of the vessel wall in CKD, and ultimately improved cardiovascular disease.
INTRODUCTION
Cardiovascular complications are more frequent and severe in patients with chronic kidney disease (CKD) as compared with the general population [1]. This complex association cannot be fully explained by the presence of traditional risk factors such as hypertension, hyperlipidaemia and diabetes. Alternatively, non-traditional risk factors related to a reduced kidney function provide some insights into the mechanisms of increased risk of cardiovascular events in CKD [1, 2]. These CKD-specific factors, besides proteinuria, include disturbed mineral metabolism and bone disease, inflammation, oxidative stress and the accumulation of uraemic toxins. Most of these factors are associated with reduced heart function, vascular stiffness and calcification, typically and most prominently of the medial layer. When compared with the role of the medial layer, attention to disturbed endothelial structure and function in CKD lags behind. The vascular endothelium constitutes a monolayer of endothelial cells, forming the inner lining of the entire circulatory system. The preservation of endothelial barrier function is crucial for the normal functioning of the vascular system and requires tightly regulated intercellular junctions and endothelial cell adhesion to the basement membrane. From this perspective, endothelial cell dysfunction (ECD) can be viewed as a compromised regulation of these vital properties and comprises structural changes in the actin cytoskeleton, reduced proliferative and migratory capacities, breakdown of endothelial cell–cell contacts and impairment of the barrier function. This progressive structural remodelling dampens the proper communication between endothelial cells and vascular smooth muscle cells (VSMCs), fundamental for vascular function, resulting in the earliest detectable changes of atherosclerosis [3]. As mentioned, CKD can also drive VSMC dysfunction or vessel structural alterations without disturbing the endothelial function [4]. However, non-atherosclerotic endothelial disturbances most likely exist as well in CKD, and are the focus of this review.
Despite strong suggestions that ECD may critically impact cardiovascular health [5], in the clinical setting of CKD, there is only limited information indicating whether ECD provides important prognostic information, or actually causes future cardiovascular complications [6]. However, the data that are available strongly suggest that, also in patients with CKD, ECD contributes to cardiovascular morbidity. In patients with CKD, impaired endothelial function has been associated with arterial thickness [6, 7], abnormal left ventricular structure and function [8], and importantly, excess of cardiovascular mortality in CKD [9]. However, while these few studies highlight the importance of vascular dysfunction as a marker for cardiovascular risk, the potential impact of ECD in the progression of cardiovascular complications remains to be elucidated. As a result of these limitations, there is insufficient knowledge supporting the concept of whether targeting vascular dysfunction and in particular ECD in CKD may beneficially impact cardiovascular disease and clinical outcome.
In view of these considerations, we aim to review available information on the morphological and functional abnormalities in the endothelial lining during CKD, and to evaluate how CKD-related, non-traditional risk factors critically impact endothelial integrity. Finally, we discuss some plausible therapeutic strategies aimed at targeting these CKD-associated disturbances, to possibly prevent progression of endothelial injury and thereby attenuate cardiovascular disease.
ENDOTHELIAL DYSFUNCTION DURING CKD
Dysfunctional endothelium in patients with CKD has been demonstrated in both large and small arteries [10, 11]. Patients with impaired renal function frequently display some common adverse endothelial characteristics that provide a better understanding of the impact of CKD on this cell type (Figure 1). In particular, impaired flow-mediated dilation (FMD), reflecting abnormal endothelium-dependent vasodilatory function, has been frequently reported in CKD patients, and its impairment is associated with the severity of renal damage [12, 13]. This non-invasive approach to assess endothelial function measures the ability of the artery to respond, by the release of the endothelium-derived relaxing factor nitric oxide (NO), to the 5-min occlusion of the branchial artery with a blood pressure cuff (reactive hyperaemia). Reduced NO bioavailability [14], however, is a critical feature and characteristic for patients with CKD [5, 15]. This abnormality is accompanied by a decreased expression or limited activation of the endothelial NO synthase due to the presence of renal disease-related toxins contributing to a reduced vasodilatory capacity [16]. Importantly, FMD provides crucial information about the vasodilatory status of the endothelium, but it is not a direct assessment of the production of vasoactive molecules. A more invasive approach can overcome this limitation through the infusion of acetylcholine, which dilates normal coronary arteries in the presence of intact endothelium by stimulating NO production. In the presence of ECD, however, acetylcholine may even induce vasoconstriction through a direct effect on the underlying VSMC. In this regard, the measurement of endothelial-dependent relaxation after acetylcholine stimulation in CKD animal models reflects a valuable approach to assess vascular function and test therapeutic strategies [17, 18].
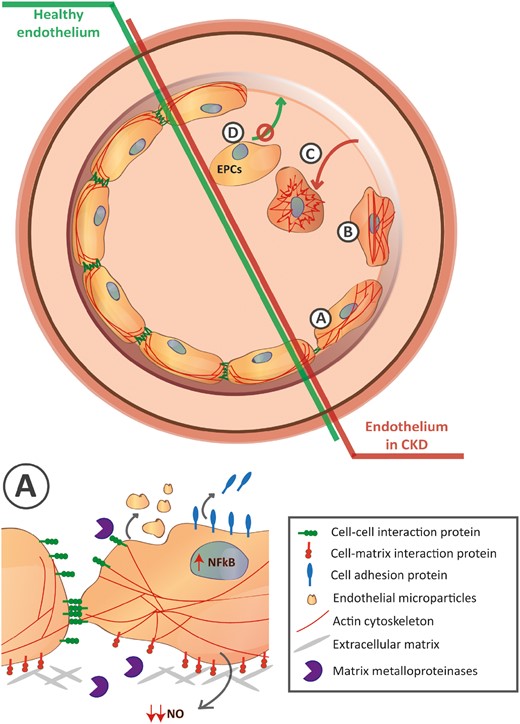
CKD disrupts the endothelial cell lining through different mechanisms. Representative different pathological mechanisms are highlighted from A to D. (A) Enlargement in the bottom. Activation of the NF-κB results in elevated soluble cell adhesion molecules characteristic of endothelial cell activation accompanied by the increase of matrix metalloproteinases that disrupt the endothelial cell–cell and cell–matrix interactions. Rearrangement of F-actin cytoskeleton towards the formation of stress fibers results in changes in endothelial cell morphology. Uraemia also enhances the release of endothelial cell microparticles and reduces the NO bioavailability. (B) Those mechanisms result in the contraction of the endothelial cell and disassociation of adjacent cells and base membrane. (C) As a result, endothelial cells detach reflecting the loss of the endothelial cell lining. (D) In addition, the limited amount of circulating EPCs accompanied by an impaired normal progenitor function and limited adhesion capacity results in an abnormal recovery of the endothelium against the uraemia-mediated damage.
Given the difficulties of assessing the structural changes of the vascular endothelium, the analysis of soluble factors is sometimes used as a non-invasive approach to explore the CKD-induced pathological consequences. During CKD, the endothelium loses its quiescent phenotype and becomes activated [5], which is exemplified by elevated levels of circulating cell adhesion molecules such as soluble Intercellular Adhesion Molecule 1 (sICAM-1), Vascular Cell Adhesion Molecule 1 (sVCAM-1), sE-selectin and platelet adhesion molecule von Willebrand factor (vWf) (as a first step of thrombus formation) in serum from patients with CKD [19, 20]. Interestingly, the presence of these ECD biomarkers has been associated with a defective FMD in CKD, which suggests that these endothelial structural changes may co-exist with an impaired endothelial function [20]. In addition, the analysis of circulating endothelial microparticles (EMPs), released into the extracellular space after endothelial injury, provides further clinical information of the endothelial damage upon CKD [21]. Similarly, as the endothelial activation markers, EMPs levels are associated with loss of FMD and increased pulse wave velocity in patients with end-stage renal failure, reinforcing the hypothesis that endothelial damage results in both morphological and functional alterations [21]. Alternatively, patients with different degrees of impaired renal function also display increased levels of circulating endothelial cells (CECs) themselves [22]. This subpopulation of cells—originating from the blood vessel wall—is detached due to endothelial damage and detachment, and reflects ongoing injury. Finally, CKD reduces the number of circulating endothelial progenitor cells (EPCs), a bone marrow-derived CEC population that can be recruited to sites of endothelial injury and then mature, playing a major role in vascular repair to restore endothelial function [23, 24]. In this regard, CKD not only dampens the availability of circulating EPCs but also impacts on the normal functioning of EPCs, resulting in abnormal colony formation together with impaired adhesion and incorporation, further worsening the repair capacity of the vascular system [23, 25].
Mechanistically, the enhanced transcription of the abovementioned adhesion molecules, vWf or EMPs is preceded by activation of the nuclear factor-κB (NF-κB) signalling pathway [5]. In experimental studies, the most prominent changes observed in endothelial cells exposed to human uraemic serum are suggested to be mediated by NF-κB signalling, substantiating its key role in the development of ECD during CKD [26, 27]. However, the harmful effects of uraemic media are not limited to activation of the NF-κB pathway but extend to NF-κB-independent structural alterations such as lower expression of Vimentin and Annexin A2, which are both involved in cell–cell and cell–matrix interactions [28]. In line with this, it was shown that uraemia modulates the expression of matrix metalloproteinases in endothelial cells leading to a remodelling of the extracellular matrix, thereby promoting endothelial detachment from the basement membrane and its subsequent loss [29]. The importance of the loss of endothelial cell–cell interactions in CKD was also recently highlighted by our group where we confirmed that uraemic plasma from pre-dialysis CKD patients was impairing the stability of the endothelial barrier function by reducing the vascular endothelial (VE)–cadherin adherens junctions on the cell surface [30]. Here, exposure to uraemic media also resulted in the re-organization of the F-actin cytoskeleton towards increased stress fibers formation [30]. Similarly, Maciel et al. [31] recently confirmed that human renal arteries from CKD patients displayed reduced VE–cadherin and Zona occludens-1 (ZO-1) protein expression and that a uraemic environment downregulated VE–cadherin and Vinculin in vitro [31]. These structural alterations make the endothelial barrier more susceptible to disruption upon electric wound or following exposure to the pro-permeability factor thrombin [30]. Finally, prolonged exposure to a uraemic environment could affect the integrity of the vascular endothelium leading to enhanced permeability and endothelial cell detachment, as confirmed in a 3/4 nephrectomized rodent model [32].
Taken together, CKD-induced disturbances of the vascular endothelium are complex and involve a large number of mechanisms including impaired cell–cell and cell–matrix interaction, which contributes to detachment from the vessel wall, increased endothelial cell activation, lost vasodilating properties and limited repair capacity of damaged endothelial surfaces, all leading to loss of endothelial barrier function.
THE INFLUENCE OF SPECIFIC CKD-RELATED FACTORS ON ENDOTHELIAL HEALTH
Recently, many CKD-specific factors such as disturbed mineral metabolism, accumulation of uraemic retention molecules, inflammation and oxidative stress have been identified as possibly being involved in ECD. Indeed, the vascular pathological features observed following exposure to these non-traditional risk factors in experimental uraemic animal models or cell cultures resemble many clinical manifestations described in CKD patients, thus reinforcing that these specific factors may actually contribute to the pathogenesis of human ECD.
Disturbances in mineral metabolism
Compelling evidence suggests that the unavoidable progressive derangement in mineral homoeostasis due to progressive kidney failure may trigger or accelerate cardiovascular disease, at the level of both the medial layer and the intimal layer. Already in early CKD, the plasma concentrations of the kidney-derived protein α-Klotho decrease, while fibroblast growth factor-23 (FGF23) levels increase. The latter is probably responsible for decreased plasma 25 hydroxyvitamin D [25(OH)D] and 1,25-dihydroxyvitamin D [1,25(OH)2D] concentrations and all these factors, along with phosphate exposure, contribute to secondary hyperparathyroidism. The imbalance of each component worsens with advancing CKD and numerous studies established associations of these disturbances with cardiovascular calcification and heart disease. Experimental evidence, described below, also demonstrates that a disturbed mineral homoeostasis contributes to the development of a dysfunctional endothelium; however, this association is not well established in CKD patients, possibly due to a lack of clinically available tools to assess the endothelial function or structure.
α-Klotho
Originally identified as an anti-ageing protein, α-Klotho is now also recognized as a major player in mineral homoeostasis. Interestingly, clinical CKD shares many biochemical and histological features with the phenotype of α-Klotho-deficient mice, including its cardiovascular manifestations [33, 34]. The vascular abnormalities in α-Klotho mutant mice, such as impaired angiogenesis, insufficient endothelium-derived NO formation and reduced levels of circulatory EPCs, may contribute to the development of ECD [34]. Membrane-bound α-Klotho is predominantly expressed in the distal tubule of the nephron. The mechanisms responsible for α-Klotho deficiency in CKD are not fully understood but are likely to be multifactorial [35]. Following tubular production and insertion in the plasma membrane, the ectodomain of membrane α-Klotho is cleaved from the cell surface by membrane-anchored proteases and released into the circulation, where it is suggested to be continuously required to maintain vascular health [36]. In this regard, one of the first vasculo-protective activities described for α-Klotho was its role in the maintenance of endothelial homoeostasis [33, 37]. Exposure of human umbilical vein endothelial cells to α-Klotho increased NO production and induced eNOS phosphorylation and inducible NOS expression [38]. Along the same line, α-Klotho has been shown to suppress the expression of the adhesion molecules ICAM and VCAM by the attenuation of NF-κB signalling pathway upon tumour necrosis factor-α (TNF-α) stimulation [39]. Another mechanism by which α-Klotho protects the endothelium was demonstrated by Kusaba et al., who showed that α-Klotho mediated the internalization of the transient receptor potential canonical 1 and vascular endothelial growth factor receptor 2 (VEGFR2) complex, thereby preventing hyperpermeability and endothelial apoptosis through an increase of calcium influx in endothelial cells incubated with VEGF [40]. Although extensive research has already provided much information on the beneficial effects of α-Klotho on endothelial damage, the relationship between α-Klotho and vascular dysfunction in patients with CKD remains poorly established. In CKD patients, lower α-Klotho levels were found to be an independent biomarker of arterial stiffness and defective FMD [41], and correlated with circulating von Willebrand factor levels [42]. However, while a deficiency of serum α-Klotho has been linked to cardiovascular complications in some studies [41, 43], this issue is still debated as Seiler et al. [2] found no relationship between soluble α-Klotho and cardiovascular outcomes in a cohort of CKD Stages 2–4 patients. Taken together, while experimental studies strongly suggest that α-Klotho preserves endothelial integrity in many different ways, there currently is no strong clinical evidence for a role of α-Klotho deficiency in CKD-mediated endothelial injury.
Vitamin D
Vitamin D deficiency, defined as serum 25(OH)D concentrations <20 ng/ml (50 nmol/L), is associated with both an increased prevalence and incidence of cardiovascular morbidity and mortality in CKD [44, 45]. In the kidney, 25(OH)D is converted by 1α-hydroxylase to its active form 1, 25(OH)2D to exert its effects on distant target tissue [46]. By binding the vitamin D receptor, 1,25(OH)2D activates both genomic and non-genomic pathways related to cellular proliferation and differentiation, and also on the endocrine and immune system [46]. As a consequence of CKD, there generally is a deficiency of 25(OH)D and a reduced production of active vitamin D, both contributing to reduced vitamin D actions on target tissues, including the vascular endothelium [45, 46]. Vitamin D deficiency is associated with decreased FMD in patients with CKD of different stages [47, 48]. In experimental models of CKD, active vitamin D analogues restored abnormal expression of aortic genes and improved endothelial function in a 5/6 nephrectomy rat model [18, 49]. Similarly, active vitamin D also protected against vascular leakage and endothelial cell detachment in in vivo models of CKD [32]. As a novel and potential protective mechanism, active vitamin D was shown to improve cell–cell interaction, disrupted after exposure to human uraemic plasma, leading to preservation of the endothelial barrier function [30]. In patients with CKD Stages 3 and 4, improvement of FMD by active vitamin D under low 25(OH)D circumstances has been reported [50]. Similar results were also observed in dialysis patients with vitamin D deficiency, where active vitamin D improved FMD of the brachial arteries [51–53]. In contrast, however, no effect of active vitamin D on brachial artery FMD or biomarkers of inflammation and oxidative stress was found in patients with advanced CKD and type 2 diabetes, and in the majority of clinical trials among diverse populations vitamin D administration has failed to show an improvement of endothelial function [54–57]. In addition, other clinical studies showed no significant effect of oral active vitamin D on left ventricular mass index in CKD patients (the PRIMO and OPERA trials) [58, 59] and no reduction of cardiovascular events in haemodialysis patients without secondary hyperparathyroidism (J-DAVID) [60]. These contradicting findings urge the search for better positioning the potential role of vitamin D administration in patients with CKD, especially in relation to disturbances in endothelial function and structure.
Phosphate and FGF23
CKD impairs phosphate balance, ultimately resulting in hyperphosphataemia [61]. In clinical studies, hyperphosphataemia and even high–normal serum phosphate concentrations represent one component of the increased risk of cardiovascular complications and mortality in both the general and CKD population [62, 63]. Recently, a number of studies suggested that phosphate may exert direct toxic effects on endothelial cells [64]. Specifically, in vitro experiments with endothelial cells demonstrated that high-phosphate concentration increases oxidative stress and decreases NO synthesis via inhibiting phosphorylation of eNOS [65]. This finding is in line with a clinical study in healthy subjects, which demonstrated that high dietary phosphate loading impaired flow-mediated vasodilation, indicating acute endothelial dysfunction [65]. In addition, exposure of endothelial cells to high-phosphate concentration also promoted the formation of EMPs with impaired capacity of angiogenesis [66, 67] and downregulated VE–cadherin and reduced ZO-1 protein levels, which are similar effects as found in endothelial cells exposed to uraemic media [31]. Importantly, in both healthy and CKD mice, it was reported that a high-phosphate diet promoted endothelial inflammation and dysfunction, and increased endothelial cell detachment [68].
To compensate for the decreased glomerular filtration of phosphate in the setting of CKD, FGF23, synthesized by osteocytes/osteoblasts, inhibits tubular reabsorption of phosphate, thereby restoring its net excretion. Besides phosphate exposure, other factors such as hyperparathyroidism and exogenous 1,25(OH)2D, calcium loading and inflammation also contribute to the elevation of plasma FGF23 concentration in CKD [69]. Although FGF23 may contribute to cardiovascular disease by the disturbance of mineral metabolism, FGF23 itself is independently associated with cardiovascular complications in different stages of CKD [70], and also with impaired vasoreactivity and increased arterial stiffness in patients with impaired renal function [70]. Furthermore, experimental ex vivo data suggest that FGF23 can directly impair endothelium-dependent relaxation upon acetylcholine stimulation [71]. This effect appeared to be mediated by the reduction of NO bioavailability due to an accumulation of either asymmetrical dimethyl arginine (ADMA) [71] or superoxide levels [72]. Remarkably, the presence of a receptor for FGF23 is not firmly established on endothelial cells, and therefore the molecular mechanisms that underlie have remained obscure so far. Overall, further clinical studies are warranted to delineate the pathological mechanisms linking phosphate and FGF23 with endothelial cell abnormalities in CKD patients.
Uraemic toxins
Progression of CKD leads to the accumulation in blood and tissues of uraemic retention solutes [73]. As a result, the cardiovascular system is constantly exposed to the potentially toxic effects of a range of uraemic retention solutes inducing, among other complications, endothelial damage [74]. One well-characterized uraemic toxin is ADMA, which is known to exert a negative impact on endothelial cell stability in both in vivo and in vitro experimental models [74]. Indeed, ADMA is considered as a circulating endogenous inhibitor of eNOS [75], and its accumulation has been associated with ECD in patients with CKD [75, 76]. In CKD mice, increased serum concentration of ADMA caused attenuated endothelium-dependent vasodilation of aortic rings by inhibiting eNOS phosphorylation, by its property of being a competitor of L-arginine (the precursor of NO) as substrate for eNOS [77]. Furthermore, ADMA induces stress fibers and focal adhesion formation in a RhoA and Rho kinase-dependent pathway leading to a limited endothelial repair [78]. Importantly, ADMA also impairs the regeneration of injured endothelium by reducing the differentiation, mobilization and function of EPCs [79].
Formed by complex pathways, the covalently protein-bound toxins advanced glycation end products (AGEs) are the result of non-enzymatic glycation and oxidation of proteins, lipids and nuclear acids, and they accumulate in CKD [80]. In various cell types, AGEs exert diverse cellular responses via the multiligand cell-surface receptor for AGEs (RAGEs) [81]. The activation of RAGE in endothelial cells in vitro induced expression of adhesion molecules, increased endothelial permeability, impaired NO production and increased reactive oxygen species (ROS) formation [82]. Moreover, in patients with CKD, decreased endothelial reactivity has been correlated with increased circulating levels of AGEs [83]. Using an in vitro approach, this study demonstrated that AGEs isolated from serum of patients with CKD induced suppression of eNOS, and this effect was attenuated after RAGE blockade [83].
Recently, several studies demonstrated that other (non-covalently) protein-bound uraemic toxins such as p-cresyl sulphate (PCS) and indoxyl sulphate (IS) exert critical toxic effects on endothelial cells in CKD. In patients with CKD, PCS is the main circulating form of p-cresol and is independently associated with cardiovascular complications [84]. In addition, markers of endothelial damage such as EMPs are directly associated with free-serum p-cresol concentrations in haemodialysis patients [85]. The same study demonstrated in vitro that PCS induced a dose-dependent increase of shedding EMP, whereas this effect was prevented by inhibition of Rho kinase [85]. An in vitro study confirmed the role of the Rho-kinase pathway in PCS-mediated toxicity. Upon exposure to p-cresol, an increased endothelial permeability and barrier disruption were induced by alterations of VE–cadherin membrane distribution [86].
IS is another critical player in the development of vascular disease and is also associated independently with elevated mortality rate in patients with CKD [87]. IS is associated with worsened FMD and arterial stiffness in CKD patients [88]. This study also demonstrated that IS impaired the chemotactic motility and colony-forming ability of EPCs, suggesting that IS contributes to the pathogenesis of ECD by limiting the vascular repair capacity [88]. In addition, several in vitro studies showed that IS can directly disrupt the stability of the endothelial cells through other molecular pathways. Specifically, IS increased EMPs release and impaired endothelial wound healing capacity [89, 90]. Moreover, it promotes endothelial activation by ROS-induced activation of NF-κB. Similar to p-cresol, cell culture exposure to IS resulted in endothelial gap formation by VE–cadherin disassembly and stress fiber formation [91].
Overall, uraemic toxins may impact the vasculature by disrupting the integrity of the endothelial cell barrier, promoting endothelial activation and weakening its recovery capacity by impairing the EPCs function. Interestingly, as highlighted in Figure 2, the deleterious effects induced by uraemic toxins in experimental research share many characteristics with the endothelial abnormalities present in CKD patients or cell-based assays with endothelial cells exposed to uraemic media, suggesting that they are important mediators in the development of CKD-induced ECD in patients.
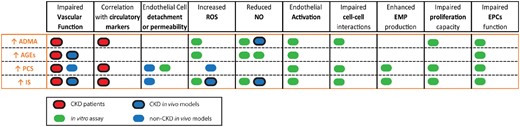
Summary of the impact of different uraemic toxins in high concentrations. Effects of the uraemic toxins ADMA, AGEs, PCS and IS in endothelial function, circulatory markers, structural changes in the vascular endothelium and in the endothelial repair capacity are highlighted as follows: patients (red), in vivo animal models (blue) and cell-based assays (green). Dark circles indicate that the study was performed in a CKD setting while no circle shows studies performed by the addition of exogenous uraemic toxin.
Oxidative stress and inflammation
Numerous studies have demonstrated that CKD is associated with increased oxidative stress and inflammation [92, 93]. Oxidative stress can be considered as accumulation of ROS in parallel with impaired or overwhelmed endogenous antioxidant mechanisms [94]. ROS are classically defined as partially reduced metabolites of oxygen that possess strong oxidizing capabilities [94]. The high production of ROS in CKD may contribute directly or indirectly to the pathogenesis of the cardiovascular disease by inducing endothelial injury [95]. Findings in animal models of chronic renal failure confirmed that enhanced generation of ROS leads to decreased NO bioavailability and impairment of the normal function of the endothelium [96]. Furthermore, increased levels of oxidative stress markers are associated with impaired endothelial function in CKD patients [97]. Moreover, chronic or prolonged ROS production is tightly connected to inflammatory processes [98], by activating transcription factors such as NF-κB, triggering a pro-inflammatory, pro-adhesion (of leucocytes) and pro-oxidant phenotype [98]. In addition, the activation of NF-κB pathway in endothelial cells is also triggered by inflammatory cytokines such as interleukin-6 and TNFα [99]. These pro-inflammatory molecules are known to be elevated in patients with CKD and cause ECD [8, 100]. Taken together, the development of a pro-inflammatory and pro-oxidative state during renal dysfunction is associated with oxidative stress, vascular NF-κB activation and inflammation, thus forming a vicious cycle amplifying ECD.
THERAPEUTIC STRATEGIES TO PROTECT THE ENDOTHELIUM IN CKD
Detailed knowledge of factors in CKD that induce ECD can pave the way to endothelial-protective therapeutic strategies, aiming to ameliorate cardiovascular disease in CKD. Based on the above, several options emerge and their clinical and experimental evidence are summarized in Table 1. The overarching approach might be the restoration of mineral metabolism network by correcting hormonal disturbances and counteracting the potential deleterious influence of uraemic toxins, inflammatory mediators and ROS. Recently, exogenous α-Klotho therapy has been shown to be effective in attenuating high-phosphate diet-induced renal and cardiac fibrosis and accelerated renal recovery after acute kidney injury in mice [101, 102]. Although the protective effects of exogenous α-Klotho administration in uraemia-mediated ECD in animal models remain to be investigated, in vitro data suggest that the endothelial-protective properties of α-Klotho are worthy of being tested in vivo. In this context, α-Klotho protein exerts protective effects by reducing the NF-κB translocation in cultured endothelial cells upon exposure to serum of Stage 5 CKD patients [103]. Moreover, exogenous α-Klotho attenuates in vitro the endothelial damage induced by the uraemic toxin IS and modulates the FGF23-mediated impaired NO synthesis and increased oxidative stress [104, 105].
Reported effective treatments against endothelial dysfunction in patients with CKD, in vivo CKD models and cell-based experiments
| Treatment | CKD patients | In vivo CKD | Cell-based assays exposed with | |||||
|---|---|---|---|---|---|---|---|---|
| Vascular function | Vascular function | Structural changes | Uraemic media | FGF23 | Phosphate | AGEs | IS | |
| α-Klotho | 103 | 105 | 104 | |||||
| Active vitamin D | 50–53 | 18, 49 | 32 | 30 | ||||
| Sevelamer | 107 | 68 | 108 | 68 | 108 | |||
| Anti-FGF23 | 71 | |||||||
| AST-120 | 113 | 112 | 112 | |||||
| Treatment | CKD patients | In vivo CKD | Cell-based assays exposed with | |||||
|---|---|---|---|---|---|---|---|---|
| Vascular function | Vascular function | Structural changes | Uraemic media | FGF23 | Phosphate | AGEs | IS | |
| α-Klotho | 103 | 105 | 104 | |||||
| Active vitamin D | 50–53 | 18, 49 | 32 | 30 | ||||
| Sevelamer | 107 | 68 | 108 | 68 | 108 | |||
| Anti-FGF23 | 71 | |||||||
| AST-120 | 113 | 112 | 112 | |||||
Positive effects from the different treatments in the different conditions are highlighted in green.
Reported effective treatments against endothelial dysfunction in patients with CKD, in vivo CKD models and cell-based experiments
| Treatment | CKD patients | In vivo CKD | Cell-based assays exposed with | |||||
|---|---|---|---|---|---|---|---|---|
| Vascular function | Vascular function | Structural changes | Uraemic media | FGF23 | Phosphate | AGEs | IS | |
| α-Klotho | 103 | 105 | 104 | |||||
| Active vitamin D | 50–53 | 18, 49 | 32 | 30 | ||||
| Sevelamer | 107 | 68 | 108 | 68 | 108 | |||
| Anti-FGF23 | 71 | |||||||
| AST-120 | 113 | 112 | 112 | |||||
| Treatment | CKD patients | In vivo CKD | Cell-based assays exposed with | |||||
|---|---|---|---|---|---|---|---|---|
| Vascular function | Vascular function | Structural changes | Uraemic media | FGF23 | Phosphate | AGEs | IS | |
| α-Klotho | 103 | 105 | 104 | |||||
| Active vitamin D | 50–53 | 18, 49 | 32 | 30 | ||||
| Sevelamer | 107 | 68 | 108 | 68 | 108 | |||
| Anti-FGF23 | 71 | |||||||
| AST-120 | 113 | 112 | 112 | |||||
Positive effects from the different treatments in the different conditions are highlighted in green.
As a potential option to restore impaired mineral balance and protect the endothelium, vitamin D replacement has raised great expectations to treat cardiovascular complications in CKD patients. However, as mentioned previously, data regarding the beneficial effects of vitamin D supplementation on cardiovascular disease including endothelial function are conflicting. In CKD animal models, active vitamin D treatment mitigates the impact of uraemia not only in endothelial function but also in structural alterations [18, 32, 49]. In randomized trials, several active vitamin D analogues lead to favourable changes on the vascular function in CKD patients of Stages 3–4 undergoing haemodialysis with or without vitamin D deficiency [50–53]; mean while, other studies reported no improvement in FMD with patients of advanced CKD [57, 106]. Overall, active vitamin D may potentially play different roles in protecting the vascular endothelium during CKD, but further studies are needed in this area.
Given its potential role in ECD, direct neutralization of the effects of phosphate and FGF23 may be another therapeutic option to protect the development of ECD. Options to accomplish the reduction of serum phosphate concentrations include treatment with phosphate binders. As an example, the phosphate-binder sevelamer hydrochloride was shown to ameliorate the phosphate-induced ECD in uraemic mice [68]. Furthermore, in hyperphosphataemic patients with Stage 4 CKD, sevelamer improved FMD, possibly mediated by parallel declines in FGF23 levels [107]. In vitro, sevelamer was effective also in protecting against endothelial activation upon uraemic media and AGEs exposure [108]. Thus, declining serum phosphorus concentrations might lead to better endothelial function and cardiovascular health in CKD patients. Alternatively, strategies to counteract high serum FGF23 concentrations such as the application of monoclonal antibodies has already been tested and shown to be effective for improving ex vivo vasodilator responses to acetylcholine in uraemic mice [71]. However, as demonstrated by Shalhoub et al. [109], the beneficial effects achieved by the neutralization of FGF23 signalling can be outbalanced by incrementing serum phosphate. Thus, inhibiting pathological FGF23-mediated pathways and lowering phosphate serum concentrations simultaneously may be a potential therapeutic strategy to reduce endothelial damage in CKD.
Because of their harmful effects on endothelial cells, reducing concentrations of uraemic toxins, ROS and inflammatory cytokines in CKD patients by dialysis may promote endothelial cell health [110, 111]. Indeed, ECD induced in vitro by serum from CKD patients led to remodelling of the extracellular matrix and this effect was mitigated in cells treated with serum from the same patients after haemodialysis therapy [29]. Unfortunately, most protein-bound uraemic retention molecules cannot be removed by dialysis. To overcome this limitation, as an absorbent of the uraemic toxin IS, AST-120 has shown to be effective to improve vascular relaxation in uraemic mice [112] and ameliorating the microvascular dysfunction in haemodialysis patients [113]. Other therapeutic approaches require components that may counterbalance the deleterious effects of oxidative stress, inflammation or toxicity, possibly by using anti-oxidants or inflammatory mediators [74]. Finally, patients with dialysis-dependent CKD following renal transplantation have improved endothelial function [114, 115]. These benefits also include the normalization of the functions of the EPCs, contributing to a better repair [114]. Overall, future studies should focus on the effective removal of these retention solutes in uraemic patients in order to attenuate ECD and promote endothelial repair.
CONCLUSIONS
In patients with CKD, ongoing endothelial damage in the vascular system exists and is frequently overlooked. However, endothelial damage is thought to be a central driver of progressive cardiovascular complications. The pathogenesis of ECD in patients with renal dysfunction results from an imbalance between increased endothelial damage and impaired regeneration. In addition, limited vasoreactivity, in particular vasodilatory properties, exists. These processes may result from the progressive loss of the vasculoprotective factors vitamin D and α-Klotho together with an increment of ECD mediators such as FGF23, uraemic toxins, ROS and inflammatory cytokines. Therapeutic strategies aiming at a better endothelial health should be based on correcting the derangements of the mineral homoeostasis, removing the retention solutes and limiting oxidative stress.
CONFLICT OF INTEREST STATEMENT
None declared.


{kind=link}
{kind=link}
Comments