





Abstract
Increasing evidence indicates that epidermal growth factor receptor (EGFR) has a pathogenic role in renal fibrosis. Currently no effective treatment can completely halt the progression of chronic kidney disease (CKD). This study was undertaken to investigate the renoprotective effects of erlotinib, a tyrosine kinase inhibitor that can block EGFR activity in the progression of CKD and the mechanisms involved.
Sprague Dawley rats with 5/6 nephrectomy were administered either erlotinib or vehicle from 2 weeks after surgery and for a period of 8 weeks. Blood pressure, proteinuria and serum creatinine were measured periodically. Renal morphological investigations were performed at sacrifice. In vitro, we used normal human mesangial cells (NHMCs) and human proximal tubular cells to investigate the inhibitory effects of erlotinib on renal fibrosis–associated signaling pathways by western blotting.
Erlotinib treatment significantly blunted the progression of CKD as evidenced by reduced levels of serum creatinine, proteinuria and renal cortical profibrogenic genes and scores of glomerulosclerosis and tubulointerstitial damage. Tubulointerstitial macrophage infiltration and multiple pro-inflammatory cytokine gene expression levels were also attenuated by erlotinib treatment. In vitro, heparin-binding epidermal growth factor–like growth factor-induced Akt and extracellular-regulated kinase (ERK) 1/2 activation in normal human mesangial cells and human proximal tubular cells was inhibited by pretreatment with erlotinib.
EGFR blocking by erlotinib protected against renal fibrosis in 5/6 nephrectomized rats via inhibition of Akt and ERK 1/2 signaling pathways, which are associated with renal fibrosis. Erlotinib also has anti-inflammatory properties, which may contribute to its renoprotective effects. Erlotinib represents a potential novel therapeutic strategy for the treatment of CKD.
INTRODUCTION
Epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein with an extracellular epidermal growth factor (EGF) binding domain and an intracellular domain that possesses intrinsic tyrosine kinase activity. In general, EGFR exists on the cell surface and is activated by binding of its specific ligands, including EGF, heparin-binding EGF-like growth factor (HB-EGF), transforming growth factor-α (TGF-α), amphiregulin, betacellulin, epiregulin and the neuregulins [1]. Upon activation by its growth factor ligands, the autophosphorylation of several tyrosine residues in EGFR occurs. This autophosphorylation elicits downstream activation and signaling. As a result, the downstream signaling initiates several signal transduction cascades, principally the Ras/Raf/mitogen-activated protein kinase (MAPK) and phosphatidyl inositol 3′ kinase (PI3K)/Akt pathways, which act to regulate cellular processes such as proliferation, angiogenesis, and inhibition of apoptosis, depending on the cell type [2].
A number of experimental studies support a role for EGFR in mediating renal fibrogenesis. Researchers have demonstrated that mice overexpressing a dominant negative isoform of EGFR were protected from tubulointerstitial injury during chronic angiotensin II (AT-II) infusion [3], subtotal nephrectomy [4, 5] and renal ischemia [4]. A recent report from Tang et al. [6] showed that ischemia/reperfusion renal injury was protected in Waved-2 (Wa-2) mice with a point mutation in EGFR. In addition, several reports have suggested that EGFR is associated with TGF-β-dependent tubulointerstitial fibrogenesis [7, 8] and epithelial-to-mesenchymal transition (EMT) [9].
Erlotinib (Tarceva) and gefitinib (Iressa) are small molecule tyrosine kinase inhibitors (TKIs) that compete with the binding of adenosine triphosphate to the intracellular tyrosine kinase domain of EGFR, thereby inhibiting receptor autophosphorylation and blocking downstream signal transduction [10]. They are widely used in the treatment of tumors with activated EGFR, including non-small cell lung cancer. These two TKIs of EGFR have been applied in intervention studies of tubulointerstitial fibrosis in mice established by chronic AT-II infusion and unilateral ureteral obstruction (UUO) [7, 8]. In experimental glomerular fibrosis, EGFR inhibition preserved renal function and renal histological change as well as reduced activation of MAPK and Akt [11–13]. In contrast, Helle et al. [14] showed that gefetinib improved renal blood flow but not renal function or inflammation using the same rat model. In addition, Ulu et al. [15] reported that the suppression of EGFR by PKI-166 did not have any beneficial effects on renal function and renal fibrosis in 5/6 nephrectomized rats. Accordingly, the therapeutic effects of pharmacological EGFR inhibition in renal fibrosis have not been clearly elucidated. In the present study, we investigated the effects of erlotinib on renal fibrosis induced by 5/6 renal nephrectomy in rats in order to clarify the potential of erlotinib as a therapeutic agent in chronic kidney disease (CKD).
MATERIALS AND METHODS
Experimental protocol
All animals were given humane care in compliance with institutional guidelines and following protocols approved by the Animal Care Committee of Showa University, Tokyo. Ten-week-old male Sprague Dawley (SD) rats weighing 270–320 g were purchased from Sankyo Labo Service (Tokyo, Japan) and used in all of the experiments. The animals were housed in the animal care facility of Showa University (25°C, 50% humidity, 12-h dark/light cycle) with free access to food and water. A total of 34 rats were subjected to 5/6 nephrectomy (Nx; right nephrectomy with surgical resection of the lower and upper thirds of the left kidney) [16]. Sham-operated rats (Sham, n = 9) underwent the same procedure but without surgical reduction of the kidney. Rats with 5/6 nephrectomy were then administered either erlotinib (20 mg/kg; Chugai Pharmaceutical/F. Hoffmann-La Roche, Basel, Switzerland) [erlotinib-treated 5/6 nephrectomized rats (5/6Nx-Erlotinib), n = 18] or vehicle-treated 5/6 nephrectomized rats (5/6Nx-Vehicle, n = 16) via daily oral gavage from 2 weeks after surgery for a period of 8 weeks. The 5/6Nx-vehicle rats and Sham rats received an equal volume of saline. At the end of the study the rats were anesthetized with pentobarbital (100 mg/kg), their blood was collected by cardiac puncture and their remnant kidneys were collected. The renal tissue was divided; a portion of the tissue was snap frozen in liquid nitrogen for biochemical and gene analyses and the remainder was fixed in 2% paraformaldehyde/phosphate-buffered saline (PBS) for histological analysis.
Light microscopic study
Tissues fixed in 2% paraformaldehyde/PBS were embedded in paraffin using routine protocols. Paraffin-embedded materials were sectioned at 2 μm for routine staining with periodic acid–Schiff and Masson trichrome and 1-μm-thick sections were used for the periodic acid–methenamine silver stain (silver). Glomerulosclerosis was assessed in 50 glomeruli on silver-stained sections under 400× magnification using a semi-quantitative score from 0 to 4 (0, no sclerosis; 1, sclerosis up to 25% of glomeruli; 2, sclerosis from 25 to 50% of glomeruli; 3, sclerosis from 50 to 75% of glomeruli; 4, sclerosis >75% of glomeruli) and the results were averaged. For evaluating tubulointerstitial damage, 15 fields for each section (Masson trichrome stain) were evaluated at 200× magnification using WinROOF image processing software (Mitani, Tokyo, Japan). The extent of tubulointerstitial damage was evaluated by counting the percentage of areas with tubular dilatation, interstitial infiltration and fibrosis per field of cortex. Scores from 0 to 5 were used (0, normal interstitium; 1, <10% of areas injured; 2, 11–25% of areas injured; 3, 26–50% of areas injured; 4, 51–75% of areas injured; 5, > 75% of areas injured) and the results were averaged.
Immunohistochemistry
The mouse monoclonal anti-rat ED1 antibody (BMA Biomedicals, Augst, Switzerland) was used in this study as a macrophage marker. Biotinylated rabbit anti-mouse immunoglobulin G (IgG) and horseradish peroxidase (HRP)-conjugated streptavidin (LSAB2 kit/HRP) were purchased from Dako (Glostrup, Denmark). Immunohistochemistry was performed according to our previous protocols [17].
Real-time reverse transcriptase polymerase chain reaction (RT-PCR)
Gene expressions of rat TGF-β, fibronectin, interleukin 6 (IL-6), IL-1β, monocyte chemoattractant protein-1 (MCP-1), collagen type I and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were analyzed using real-time RT-PCR. The procedure for real-time RT-PCR has been described previously [18].
Homogenization of kidney tissues
Kidney tissues (cortex) were homogenized with T-PER Mammalian Protein Extraction Reagent (20 mL/g renal tissues; Pierce Biotechnology, Rockford, IL, USA) containing 1% (v/v) protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA) using a TissueLyser (QIAGEN, Hilden, Germany). Harvested lysates were then centrifuged for 10 min at 4°C to remove the cellular debris. The supernatants were collected and stored at −80°C. Protein concentration was measured using the BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA).
Measurement of TGF-β1 protein levels in kidney tissue homogenate
Total TGF-β1 protein levels were measured in kidney tissue homogenates from each sample using the TGF-β1 enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Abingdon, Oxfordshire, UK) following the manufacturer’s instructions. To control for the difference between samples, the concentration was corrected based on the amount of total tissue protein.
Measurement of urinary total collagen levels
Urinary total collagen levels were determined by analysis of urinary hydroxyproline content as described by Kivirikko et al. [19]. Hydroxyproline values were converted to collagen content by multiplying by a factor of 6.94 (since hydroxyproline represents ∼14.4% of the amino acid composition of collagen) [20] and expressed further as a proportion of the urinary creatinine (Cr) levels (μmol of collagen/mg of urinary Cr).
Bio-plex suspension array system
The procedure for the Bio-Plex Suspension Array System (Bio-Rad Laboratories, Hercules, CA, USA) has been described previously [21].
Cell culture studies
Cell culture for normal human mesangial cells (NHMCs) and human proximal tubular cells (HK-2 cells) was carried out as described previously [22]. To evaluate the inhibitory effects of erlotinib on Akt and phosphorylated extracellular-regulated kinase (ERK) 1/2, phosphorylation induced by HB-EGF in cultured NHMCs and HK-2 cells was performed as follows: cells were initially cultured for 48 h in media containing 10% foetal bovine serum (FBS) and then cultured overnight in serum-free media. The cells were pre-incubated with erlotinib (5 μmol/L) or dimethylsulfoxide (DMSO) vehicle control for 1 h, followed by stimulation with HB-EGF (20 ng/mL) or medium alone for 5 min. Cell lysates were separated by gel electrophoresis and blotted with antibodies against phospho-Akt, total-Akt, phospho-ERK1/2 and total-ERK1/2 (1:1000; Cell Signaling Technology, Danvers, MA, USA) and GAPDH. The secondary antibody was HRP-conjugated anti-rabbit IgG antibody (1:3000; Cell Signaling Technology). Western blot analysis for NHMCs and HK-2 cells was carried out according to our previous protocols [23].
Statistical analysis
Data were recorded as mean ± SEM. The Mann–Whitney test was performed and P-values <0.05 were considered significant.
RESULTS
Effects of erlotinib on biochemical parameters in 5/6Nx rats
In comparison with the Sham rats, the 5/6Nx-vehicle and 5/6Nx-erlotinib rats had higher systolic blood pressure (BP) readings and lower body weights (BWs). Erlotinib treatment did not affect either BP or BW in the 5/6Nx rats (Figure 1A and B). Urinary total protein and serum Cr levels were significantly higher in the 5/6Nx-vehicle rats than in the Sham rats from 2 weeks after treatment and throughout the study period (Figure 1C and D). The increased urinary total protein excretion in the 5/6Nx-vehicle rats was significantly reduced by erlotinib treatment throughout the study period (Figure 1C). Erlotinib treatment significantly reduced urinary total protein excretion and serum Cr levels in the 5/6Nx rats compared with the vehicle rats throughout the study period (Figure 1C and D).
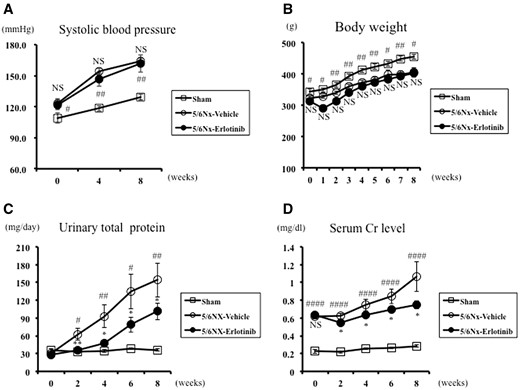
Systolic BP, body weight, urinary total protein excretion and serum Cr in the study groups. Changes in (A) systolic BP, (B) body weight, (C) urinary total protein excretion and (D) serum Cr of Sham and 5/6 nephrectomized rats treated with either vehicle (5/6Nx-vehicle) or erlotinib (5/6Nx-erlotinib) are shown. Data are expressed as mean ± SEM. Mann–Whitney test: NS, *P < 0.05, **P < 0.01: 5/6Nx-vehicle versus 5/6Nx-Erlotinib; #P < 0.05, ##P < 0.01, ####P < 0.0001: 5/6Nx-vehicle versus Sham. NS, not significant.
Effects of erlotinib on renal histology in 5/6Nx rats
Subtotal nephrectomy led to significant hypertrophy, glomerulosclerosis and tubulointerstitial fibrosis compared with the Sham rats. The glomerulosclerosis score and tubulointerstitial damage score was greater in the 5/6Nx-vehicle rats compared with the Sham rats (Figure 2 and Table 1). Erlotinib treatment significantly improved both glomerulosclerosis and tubulointerstitial damage in the 5/6Nx rats (P < 0.05) (Figure 2 and Table 1).
Morphologic evaluation of glomerular sclerosis and tubulointerstitial damage at the end of the study
| 5/6Nx-vehicle | 5/6Nx-erlotinib | Sham | |
|---|---|---|---|
| Glomerular sclerosis score (0–4) | 2.18 ± 0.20*** | 1.63 ± 0.09** | 0.38 ± 0.04 |
| Tubulointerstitial damage score (0–5) | 3.28 ± 0.30 | 2.71 ± 0.16* | 0 |
| 5/6Nx-vehicle | 5/6Nx-erlotinib | Sham | |
|---|---|---|---|
| Glomerular sclerosis score (0–4) | 2.18 ± 0.20*** | 1.63 ± 0.09** | 0.38 ± 0.04 |
| Tubulointerstitial damage score (0–5) | 3.28 ± 0.30 | 2.71 ± 0.16* | 0 |
P < 0.0001, Sham versus 5/6Nx-vehicle; *P < 0.05, **P < 0.01, 5/6Nx-vehicle versus 5/6Nx-erlotinib.
Morphologic evaluation of glomerular sclerosis and tubulointerstitial damage at the end of the study
| 5/6Nx-vehicle | 5/6Nx-erlotinib | Sham | |
|---|---|---|---|
| Glomerular sclerosis score (0–4) | 2.18 ± 0.20*** | 1.63 ± 0.09** | 0.38 ± 0.04 |
| Tubulointerstitial damage score (0–5) | 3.28 ± 0.30 | 2.71 ± 0.16* | 0 |
| 5/6Nx-vehicle | 5/6Nx-erlotinib | Sham | |
|---|---|---|---|
| Glomerular sclerosis score (0–4) | 2.18 ± 0.20*** | 1.63 ± 0.09** | 0.38 ± 0.04 |
| Tubulointerstitial damage score (0–5) | 3.28 ± 0.30 | 2.71 ± 0.16* | 0 |
P < 0.0001, Sham versus 5/6Nx-vehicle; *P < 0.05, **P < 0.01, 5/6Nx-vehicle versus 5/6Nx-erlotinib.
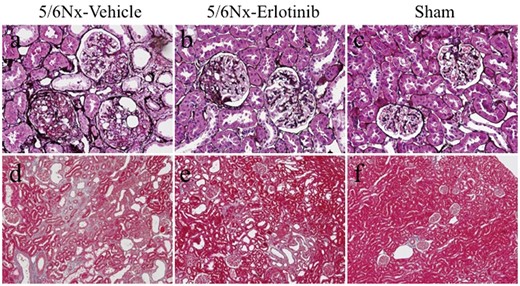
Light microscopic findings in the study groups. Representative images of kidney sections stained with (A–C) silver and (D–F) Masson trichrome in a 5/6Nx-vehicle rat (A, D), a 5/6Nx-erlotinib rat (B, E), and a Sham rat (C, F). Original magnifications, 200× (A–C) and 100× (D–F).
Effects of erlotinib on renal fibrosis in 5/6Nx rats
TGF-β and fibronectin gene expressions in the 5/6Nx-vehicle rats were significantly higher compared with the Sham rats. These expression levels were significantly reduced by erlotinib treatment (Figure 3). When compared with the Sham rats, the 5/6Nx-vehicle rats showed a significantly higher level of TGF-β1 protein (P < 0.05), which was measured in the kidney tissue homogenate. Erlotinib treatment significantly reduced TGF-β1 protein in the 5/6Nx rats (P < 0.05) (Figure 4A). There was a 50% (P < 0.05) reduction of TGF-β1 in the 5/6Nx-erlotinib rats compared with the 5/6Nx-vehicle rats (Figure 4A). In addition, a 25% reduction in the urinary collagen level, which was determined by analysis of urinary hydroxyproline content, was observed in the 5/6Nx-erlotinib rats compared with the 5/6Nx-vehicle rats (P < 0.05) (Figure 4B).

Results of real-time RT-PCR for fibrogenetic and pro-inflammatory cytokine genes in the study groups. Data are expressed as mean ± SEM. Mann–Whitney test: *P < 0.05, **P < 0.01: 5/6Nx-vehicle versus 5/6Nx-erlotinib. The values were normalized to the GAPDH values and then expressed as relative quantification to the Sham rats. The horizontal dotted line shows the expression levels in the Sham rats.
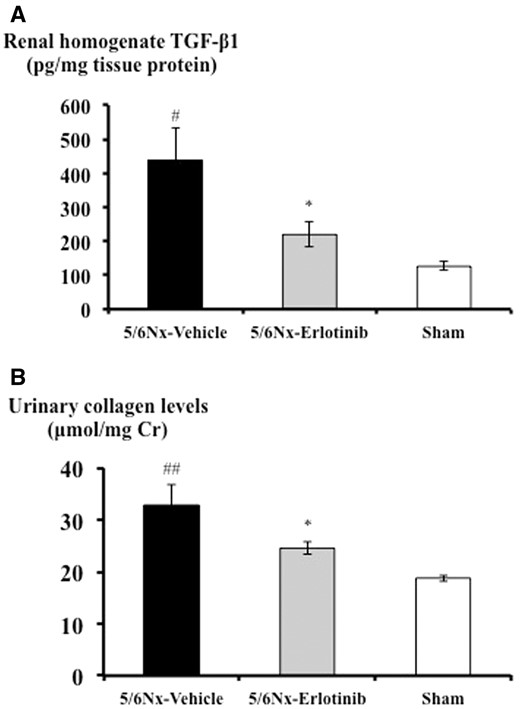
Effects of erlotinib on TGF-β1 protein synthesis and urinary collagen levels in the study groups. (A) Renal cortical homogenate TGF-β1 protein levels of each group measured by ELISA. (B) Urinary collagen levels of each group measured as described in ‘Materials and Methods’. Data are expressed as mean ± SEM. Mann–Whitney test: *P < 0.05: 5/6Nx-vehicle versus 5/6Nx-erlotinib; #P < 0.05, ##P < 0.01: 5/6Nx-vehicle versus Sham.
Effects of erlotinib on tubulointerstitial macrophage infiltration and pro-inflammatory genes in 5/6Nx rats
There was a 33% reduction of ED1-positive macrophages in the tubulointerstitium in the 5/6Nx-erlotinib rats compared with the 5/6Nx-vehicle rats (Figure 5 and Table 2). The gene expression levels of macrophage-derived pro-inflammatory cytokines such as IL-6, IL-1β and MCP-1, which are known as macrophage-associated pro-inflammatory cytokines, were much higher in the 5/6Nx-vehicle rats than in the Sham rats. Consistent with the reduction of macrophage infiltration, erlotinib treatment significantly decreased gene expression levels of IL-6 (P < 0.01), IL-1β (P < 0.05) and MCP-1 (P < 0.05) in 5/6Nx rats (Figure 3).
Semiquantitative evaluation of IHC for ED-1 in tubulointerstitium at the end of the study
| 5/6Nx- vehicle | 5/6Nx- erlotinib | Sham | |
|---|---|---|---|
| ED-1-positive macrophages score | 2.06 ± 0.28 | 1.39 ± 0.16* | 0 |
| 5/6Nx- vehicle | 5/6Nx- erlotinib | Sham | |
|---|---|---|---|
| ED-1-positive macrophages score | 2.06 ± 0.28 | 1.39 ± 0.16* | 0 |
IHC, immunohistochemistry. *P < 0.05, 5/6Nx-vehicle versus 5/6Nx-erlotinib.
Semiquantitative evaluation of IHC for ED-1 in tubulointerstitium at the end of the study
| 5/6Nx- vehicle | 5/6Nx- erlotinib | Sham | |
|---|---|---|---|
| ED-1-positive macrophages score | 2.06 ± 0.28 | 1.39 ± 0.16* | 0 |
| 5/6Nx- vehicle | 5/6Nx- erlotinib | Sham | |
|---|---|---|---|
| ED-1-positive macrophages score | 2.06 ± 0.28 | 1.39 ± 0.16* | 0 |
IHC, immunohistochemistry. *P < 0.05, 5/6Nx-vehicle versus 5/6Nx-erlotinib.
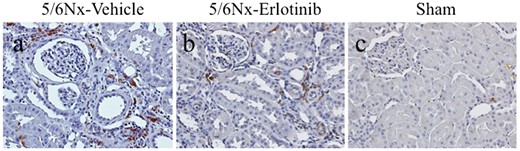
Immunohistochemical analysis of ED-1 expression in the study groups. Representative images of immunohistochemical staining for ED-1 of (A) a 5/6Nx-vehicle rat, (B) a 5/6Nx-erlotinib rat and (C) a Sham rat. Original magnification, 200×.
Effects of erlotinib on Akt and MAPK signaling pathways in 5/6Nx rats
To determine the effects of erlotinib on Akt and MAPK signaling pathways in the renal cortex, Bio-Plex suspension arrays were performed using antibodies against phospho-Akt, total Akt, phospho-MEK1 and total MEK1. As shown in Figure 6A, the level of phosphorylated Akt in the 5/6Nx-vehicle rats was higher than in the Sham rats; however, this trend did not reach statistical significance. Erlotinib treatment significantly reduced the level of phosphorylated Akt in 5/6Nx rats (P < 0.01). On the other hand, the levels of phosphorylated MEK1 were not significantly different between the study groups (Figure 6B). Neither levels of total Akt nor total MEK1 were significantly different between the study groups (data not shown).
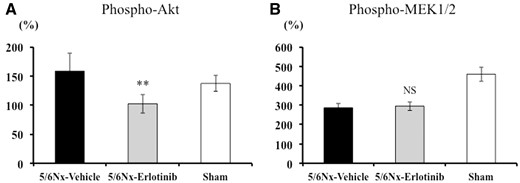
Inhibitory effects of erlotinib on the Akt and MAPK signaling pathways in the study groups. (A) Phosphorylated Akt and (B) phosphorylated MEK1/2 in the renal cortex were quantified using the Bio-Plex Suspension Array System. Data are expressed as mean ± SEM. Mann–Whitney test: NS, **P < 0.01: 5/6Nx-vehicle versus 5/6Nx-erlotinib. NS, not significant.
Effects of erlotinib on Akt and ERK1/2 phosphorylation induced by HB-EGF in cultured NHMCs and renal tubular cells (HK-2 cells) in vitro
The Akt and ERK1/2 phosphorylation induced by HB-EGF in mesangial cells and renal tubular cells and whether erlotinib could inhibit phosphorylation were investigated. Stimulation with HB-EGF for 5 min increased Akt and ERK1/2 phosphorylation, which was significantly inhibited by pretreatment with erlotinib in cultured mesangial cells (Figure 7A and B) and renal tubular cells (Figure 7C and D).
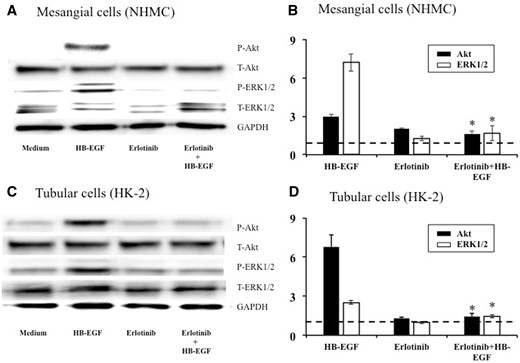
Representative immunoblots (A, C) and the results of densitometry analysis (B, D) showing inhibitory effects of erlotinib on Akt and ERK1/2 phosphorylation induced by HB-EGF in cultured mesangial cells (A, B) and renal tubular cells (C, D). Cells were initially cultured for 48 h in media containing 10% FBS and then cultured overnight in serum-free media. The cells were pre-incubated with erlotinib (5 μmol/L) or DMSO vehicle control for 1 h, followed by stimulation with HB-EGF (20 ng/mL) or medium alone for 5 min. Cell lysates were separated by gel electrophoresis and blotted with antibodies against phospho-Akt, total-Akt, phospho-ERK1/2, total-ERK1/2 and GAPDH. Densitometry values of phospho-Akt and phospho-ERK1/2 were normalized to corresponding total-Akt and total-ERK1/2 and to control (medium alone). The horizontal dotted lines show the results of the controls (medium alone). Data are expressed as mean ± SEM (n = 3). Mann–Whitney test: *P < 0.05: HB-EGF versus erlotinib + HB-EGF.
DISCUSSION
Renal fibrosis is the principal process underlying the progression of CKD to end-stage renal disease. Although a number of researchers have focused on the mechanisms underlying renal fibrosis, effective anti-fibrotic therapies that can reverse or stop the progression of CKD have yet to be developed in the clinical setting. Hence, new studies are needed to explore the cellular and molecular mechanisms of renal fibrosis and to develop novel therapeutic strategies for the treatment of CKD. Here, we found that erlotinib, a TKI that can block EGFR activity, provided functional and histologic benefits in rats with subtotal nephrectomy, as evaluated by a significant reduction in the urinary protein level, serum Cr level and the degree of glomerulosclerosis and tubulointerstitial damage. There was no toxicity in this model throughout the study period. Notably, the renoprotective effects of erlotinib in this rat CKD model were independent of lowering BP. Erlotinib treatment resulted in the reduction of fibrotic genes, including TGF-β and fibronectin, and suppression of macrophage recruitment and concomitant downregulation of pro-inflammatory cytokines. We also showed that HB-EGF-induced ERK1/2 and Akt activation in mesangial cells and renal tubular cells was significantly blunted by erlotinib in vitro.
The expression of EGFR in many cell types and the multiple signaling pathways involved in this receptor explain its importance in many cellular processes such as survival, proliferation, apoptosis and migration in the development of many organs [24]. MAPK and PI3K-Akt are the major signaling cascades downstream of EGFR activation pathways [25]. The role of ERK1/2 in the transduction of AT-II-induced renal fibrosis [7–9], aldosterone-induced renal fibrosis [26, 27] and EMT in tubular cells [28, 29] via EGFR activation has been described. In the present study, MEK1/2 signaling cascades were not activated in the remnant kidney compared with controls, and the erlotinib treatment did not modify MEK1/2 phosphorylation. This is consistent with a previous report in which ERK phosphorylation was not significantly different between sham-operated and 5/6Nx rats [30]. Since the results of signaling pathways in vivo were elusive, we therefore verified whether erlotinib could inhibit ERK1/2 signaling cascades induced by HB-EGF in renal intrinsic cells in vitro. ERK1/2 activation induced by HB-EGF in mesangial cells and renal tubular cells was shown to require EGFR kinase activity, as pretreatment with erlotinib blocked the activity. It has been shown that AT-II-dependent EGFR signaling activated ERK1/2 phosphorylation, which increased TGF-β expression and thereby mediated subsequent tubulointerstitial fibrosis, which was significantly reduced by erlotinib in mice [7]. In cultured renal interstitial fibroblasts (NRK-49 F), inhibition of EGFR by gefitinib significantly abrogated TGF-β- or serum-induced phosphorylation of EGFR, STAT3, ERK1/2, and Smad3 as well as the expression of α-smooth muscle actin (α-SMA) and extracellular matrix proteins [8]. Taken together, EGFR blockade by erlotinib induces downstream ERK1/2 inhibition, thereby exhibiting anti-fibrotic effects in renal intrinsic cells, which is one possible mechanism for its renoprotective effect.
On the other hand, the PI3K-Akt signaling pathway has been reported to be involved in the pathogenesis of renal fibrosis [31]. In mesangial cells, TGF-β-mediated expression of fibrotic proteins including fibronectin and collagen is regulated by PI3K-Akt signal transduction [32, 33]. In tubulointerstitial fibrosis following UUO, Akt plays an essential role in regulating cell proliferation, fibroblast activation and matrix production [34, 35]. In addition, Akt activation has been reported to be involved in TGF-β-induced renal EMT in vivo and in vitro [36]. In the present study, a significant increase of Akt phosphorylation in the 5/6Nx-vehicle rats compared with the Sham rats was not observed at sacrifice (8 weeks after operation), while erlotinib significantly inhibited Akt phosphorylation compared with vehicle in 5/6Nx rats. This observation was further supported by our in vitro data demonstrating erlotinib-suppressed Akt phosphorylation induced by HB-EGF. Therefore, an alternate possible mechanism of the renoprotection by erlotinib inhibition of EGFR is linked to subsequent Akt signaling pathway inhibition, which may lead to blocking of TGF-β-induced renal fibrosis.
Recently, the involvement of the EGFR pathway in inflammation [37, 38] and the effects of erlotinib have been reported [39, 40]. However, the anti-inflammatory effects of EGFR inhibition in renal diseases are rarely reported [41, 42]. In this study, erlotinib could attenuate the tubulointerstitial infiltration of macrophages in rats with remnant kidney, in which monocyte/macrophage infiltration is involved in disease progression [43, 44]. Such attenuation could be due in part to the inhibitory effects of erlotinib on the differentiation and proliferation of monocyte [45] and chemokine production, including granulocyte-macrophage colony-stimulating factor (GM-CSF) [46]. The reduction of macrophage-derived pro-inflammatory cytokines such as IL-6, IL-1β and MCP-1 was also found. IL-1β and MCP-1 were reported to be colocalized with ED1-positive macrophages in the remnant kidney model, as indicated by double immunostaining [17]. Although the association of IL-6 in renal fibrosis is controversial, IL-1β and MCP-1 are known to have a fibrogenic property in kidney [47, 48] and are expressed in the remnant kidney model [49]. Blockade of IL-1 and MCP-1 has also been reported to have therapeutic effects on experimental renal fibrosis [47, 50, 51]. Taken together, EGFR is critically involved in the production of pro-inflammatory cytokines in renal fibrosis. Therefore, another possible mechanism of renal fibrosis attenuation by erlotinib may involve the suppression of macrophage recruitment and concomitant downregulation of these pro-inflammatory cytokines in remnant kidney.
A limitation of our study is the relatively unimpressive in vivo data for the ERK1/2 and Akt signaling pathways. These signaling pathways could be influenced by the difference in protocols, such as timing of sacrifice, rat species (SD or Wistar) and operative procedures (selective ligation of renal artery or surgical resection of the kidney); thus interpretation of the results is difficult. One possible explanation is that activation of these signaling pathways might have already ceased at the time of sacrifice, therefore we were unable to observe a significant difference in ERK1/2 and Akt phosphorylation between the study groups. Kurdian et al. [30] performed a similar protocol as in the present study, in which rats were sacrificed 10 weeks after surgery, and reported that neither Akt mRNA nor ERK1/2 protein were upregulated in the remnant kidney. On the other hand, several other previous studies have reported that ERK1/2 and Akt signaling were significantly activated in remnant kidney [52–54]. This discrepancy could have been explained if we performed a time point study and used varying doses of erlotinib to show a serial change in these signals and a clear dose-dependent response to the treatment. In addition, we found that erlotinib had anti-inflammatory effects mediated via inhibition of macrophage infiltration/activation. However, nephron mass reduction also results in a general inflammatory effect and other mechanisms of inflammation, such as the activation of nuclear factor κB [55] and NLRP3 inflammasome [56]. Thus, examining these molecules would have substantially strengthened the evidence of anti-inflammatory effects by erlotinib. Nevertheless, data from the present study showed that macrophage inhibition was one of the critical anti-inflammatory mechanisms underlying erlotinib treatment that obviously resulted in reducing pro-inflammatory cytokines, which also contributed to the attenuation of renal fibrosis, as inflammation and fibrosis promote one another in the progression of CKD.
In conclusion, these in vivo and in vitro data demonstrated that EGFR blocking by erlotinib protected against renal fibrosis in 5/6Nx rats. This renoprotection by erlotinib appears to be mediated through inhibition of the ERK1/2 and Akt signaling pathways, which are closely associated with renal fibrosis. In addition, anti-inflammatory effects may contribute in part to the renoprotection effects of erlotinib. Although erlotinib is currently a promising anticancer drug, this study indicates that erlotinib represents a novel therapeutic strategy for the treatment of CKD.
ACKNOWLEDGEMENTS
The authors would like to thank Tomoko Suzuki for her excellent technical assistance. Erlotinib was a gift from Chugai Pharmaceutical/F. Hoffmann-La Roche (Basel, Switzerland), which did not otherwise participate in the design, execution or funding of this study.
FUNDING
This work was supported in part by grants from the Showa University School of Medicine Alumni Association (to M. I.) and the Yukiko Ishibashi Foundation (to M. I.).
CONFLICT OF INTEREST STATEMENT
None declared.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments