





Abstract
Often the cause of refractory lupus nephritis (RLN) remains unclear. We performed next-generation sequencing for podocyte genes in an RLN patient and identified compound heterozygosity for APOL1 risk alleles G1 and G2 and a novel homozygous c.[1049C>T]+[1049C>T] NPHS1 gene variant of unknown significance. To test for causality renal progenitor cells isolated from urine of this patient were differentiated into podocytes in vitro. Podocytes revealed aberrant nephrin trafficking, cytoskeletal structure and lysosomal leakage, and increased detachment as compared with podocytes isolated from controls. Thus, lupus podocytopathy can be confirmed as a cause of RLN by functional genetics on patient-derived podocytes.
BACKGROUND
Refractory lupus nephritis (RLN) is a term used to describe patients that undergo induction therapy for active LN and show one of the following: (i) persistent or progressive renal dysfunction, (ii) no relevant decline in proteinuria within 3–6 months or (iii) no complete response at 24 months [1]. RLN is usually addressed by escalating immunosuppressive therapy [1], although this may not always be appropriate. Here we present a case of RLN in which only genetic testing and functional analysis of patient-derived podocytes unraveled the underlying cause of ‘RLN’.
CASE REPORT
A 14-year-old Senegalese female presented with fever, perimyocarditis, 8.5 g proteinuria/day, hypocomplementaemia and antinuclear antibodies of 1:3200. Kidney biopsy confirmed LN, i.e. mesangial immune complex deposits with mild mesangial expansion, and no evidence of focal-segmental glomerulosclerosis (FSGS) in 13 glomeruli. She responded well to steroids and 1 g/day of mycophenolate mofetil (MMF) (Figure 1A). One year later, a re-biopsy displayed no immune deposits but a single focal segmental sclerotic lesion in 1/19 glomeruli (Figure 1B), so secondary FSGS was suspected. After 2 years of remission and cessation of MMF treatment the patient presented again with 10 g proteinuria/day. A third kidney biopsy was performed and showed class V and global sclerosis in 2/13 glomeruli (Figure 1B). Oral prednisolone, cyclosporine A and later MMF were started but proteinuria of 6–8 g/day persisted (Figure 1A). When 12 months later proteinuria reached 14 g/day, a fourth biopsy was performed (Figure 1A) and displayed disease class III/V (A/C), 1/8 glomeruli with global sclerosis, interstitial fibrosis and tubular atrophy of 15–20%. RLN was addressed by rituximab and methylprednisolone treatment. This ‘multitarget’ therapy controlled thrombocytopaenia and hypocomplementaemia but not nephrotic-range proteinuria (Figure 1A). Subsequently, progressive chronic kidney disease (CKD) with hyperparathyroidism and hypertension became evident. To rule out persistently active LN a fifth biopsy was performed but displayed only global sclerosis in 3/3 glomeruli and 50% interstitial fibrosis and tubular atrophy (Figure 1B), i.e. LN class VI. Six months later dialysis was required.
![Clinical course, kidney biopsy results and genotyping. (A) The graph illustrates proteinuria as assessed by 24 h urine collection (red), serum creatinine levels (blue) and the dose of oral prednisolone (green). Kidney biopsies are indicated by yellow arrows and the main result is listed above. The consecutive treatment regime is indicated below. LN, lupus nephritis; FSGS, focal segmental glomerulosclerosis; MP, intravenous methylprednisolone; MMF, oral mycophenolate mofetil; ACEI, oral angiotensin-converting enzyme inhibitor; HQL, hydroxychloroquine; ARB, angiotensin receptor blocker; CyA, cyclosporine A; RTX, rituximab; CYC, cyclophosphamide. (B) From the five kidney biopsies available the second, third and fifth are shown and the time of each biopsy is indicated. PAS and silver staining are shown at a magnification of 200×. Assessment was according to the ISN/RPS classification of lupus nephritis. Left: focal-segmental glomerulosclerosis in 1/19 glomeruli in the absence of proliferative immune complex disease. Middle: ISN/RPS class V with 15% interstitial fibrosis and tubular atrophy. Right: ISN/RPS class VI with >50% interstitial fibrosis and atrophy. (C) Schematic structure of the human APOL1 gene showing the risk alleles G1 and G2 identified in the patient affected by LN. Exons are indicated as rectangles. The family pedigree of the patient displays the recessive pattern of inheritance of the risk alleles G1 and G2 in APOL1 gene and of the variants in NPHS1 gene. The schematic structure of the human NPHS1 gene shows the variant c.[1049C>T]+[1049C>T] identified in the patient affected by LN by next-generation sequencing. Exons are indicated as rectangles.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ndt/31/9/10.1093_ndt_gfw234/1/m_gfw23401.jpeg?Expires=1748214090&Signature=acVntwN827vo3mLYWDOZZOdhuui8QZ0GZCXV3TNC51Y8RMSWzyVVePP-1RtN3-o4ePOfENt~7mc0pN8F4fiYtHYvLsMO9qCeYs~CBYrMkWaDO5kcazXkSJpt8Y-l-4XNjdqk903Mmfg9PP-0JW4Y~aVmQmS4AX4h8MNLfpNvwAUchzbR9c7CyqeGpGL9anb9FYk6YqgzuSPgJZ6b3xhpPwkcOLYyVeD8b2yPmjsbtEi-tBhkxU4Cc8vdJ9~hkNf9-UUfh-B2~M4zp-rvudSW1FDxQ16kBeSWG9R24QVI-9r2rVnEiC3aXB0doJbReHy3c0U4MjDtZTmyf1jwi7TbDg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Clinical course, kidney biopsy results and genotyping. (A) The graph illustrates proteinuria as assessed by 24 h urine collection (red), serum creatinine levels (blue) and the dose of oral prednisolone (green). Kidney biopsies are indicated by yellow arrows and the main result is listed above. The consecutive treatment regime is indicated below. LN, lupus nephritis; FSGS, focal segmental glomerulosclerosis; MP, intravenous methylprednisolone; MMF, oral mycophenolate mofetil; ACEI, oral angiotensin-converting enzyme inhibitor; HQL, hydroxychloroquine; ARB, angiotensin receptor blocker; CyA, cyclosporine A; RTX, rituximab; CYC, cyclophosphamide. (B) From the five kidney biopsies available the second, third and fifth are shown and the time of each biopsy is indicated. PAS and silver staining are shown at a magnification of 200×. Assessment was according to the ISN/RPS classification of lupus nephritis. Left: focal-segmental glomerulosclerosis in 1/19 glomeruli in the absence of proliferative immune complex disease. Middle: ISN/RPS class V with 15% interstitial fibrosis and tubular atrophy. Right: ISN/RPS class VI with >50% interstitial fibrosis and atrophy. (C) Schematic structure of the human APOL1 gene showing the risk alleles G1 and G2 identified in the patient affected by LN. Exons are indicated as rectangles. The family pedigree of the patient displays the recessive pattern of inheritance of the risk alleles G1 and G2 in APOL1 gene and of the variants in NPHS1 gene. The schematic structure of the human NPHS1 gene shows the variant c.[1049C>T]+[1049C>T] identified in the patient affected by LN by next-generation sequencing. Exons are indicated as rectangles.
The unfortunate outcome of this patient prompted us to consider previously unrecognized disease mechanisms. Patients of African origin with LN carrying APOL1 risk alleles (termed G1 and G2) are at risk for CKD progression [2], hence, we performed Sanger DNA sequencing and identified the patient to be compound heterozygous for G1 and G2, one allele each inherited from her parents (Figure 1C). However, in patients with APOL1 risk alleles proteinuria usually responds to immunosuppression [3], unlike children with steroid-resistant nephrotic syndrome, which in 30% a genetic podocytopathy can be identified by next-generation sequencing [4]. Using the same strategy, we identified the homozygous variant c.[1049C>T]+[1049C>T] in the NPHS1 gene (Figure 1C), inherited by each of the non-affected parents (Figure 1C). This previously unreported variant results in the single amino-acid substitution of serine with phenylalanine in the position 350 of the protein nephrin.
To validate the pathogenicity of these gene variants, we compared podocytes from this case with those from controls by isolating CD24+/CD133+ renal progenitor cells (RPC) from the urine, expanding them in culture, and differentiating them into podocytes as described [5]. Phalloidin staining showed no difference between patient and controls before differentiation (Figure 2A and B) but the patient's differentiated podocytes displayed an aberrant actin filament distribution in comparison with controls (Figure 2C, D, C′ and D′). They were also less viable upon differentiation (Figure 2E and F). APOL1 risk variants can also enhance podocyte death via lysosomal destabilization [6]. Consistently, the patient's podocytes displayed a significantly lower staining with Lysotracker in comparison with controls as a sign of lysosomal destabilization (Figure 2G–I). Although nephrin mRNA levels were similar in the patient and controls (data no shown), confocal microscopy for nephrin protein revealed nephrin properly localizing to the outer plasma membrane in control podocytes (Figure 2J, J′), while the patient's podocytes showed significant aberrant nephrin localization within the cytoplasm (Figure 2K, K′).
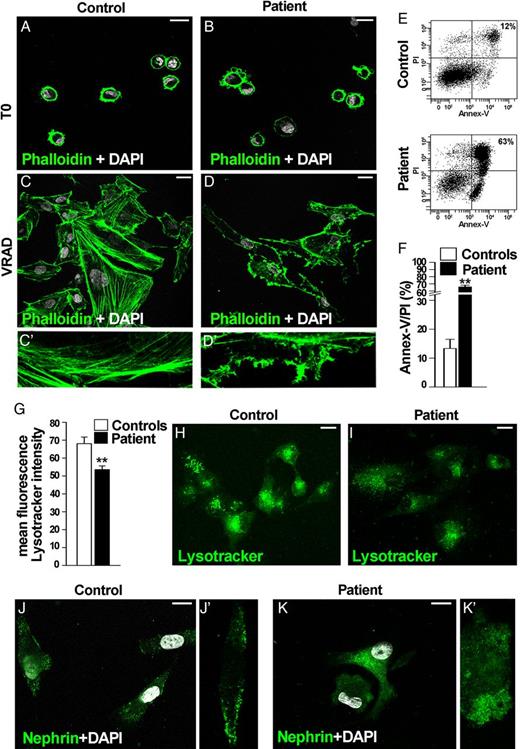
Functional characterization of urinary renal progenitor cell (RPC)-derived podocytes from the patient and controls. (A–D) Distribution of actin filaments by Phalloidin staining in RPC (A and B) and podocytes (C, C′, D, D′) is shown in green. Altered cytoskeleton architecture of podocytes obtained from urinary RPCs of the patient affected by lupus nephritis carrying the risk alleles G1/G2 in APOL1 gene and the genetic variants in NPHS1 gene (D, D′) in comparison with podocytes obtained from a control (C, C′). DAPI counterstains nuclei (white). Bars = 20 µm. Data from one out of five controls are shown. One representative out of four independent experiments is shown. (E) Percentage of dead cells following differentiation into podocytes of u-RPCs obtained from controls (above) and from the patient carrying the risk alleles G1/G2 in APOL1 gene and the genetic variants in NPHS1 gene (below), as assessed by spontaneous uptake of propidium iodide (PI) and annexin V (Annex-V) by flow cytometry analysis. Data from one out of five controls are shown. One representative out of four independent experiments is shown. (F) Graph representing percentage of Annex-V/PI-positive cells in controls (white column) and in the patient (black column), obtained in at least four separate experiments. Data are shown as mean ± SEM. **P < 0.001 by Mann–Whitney Test. (G) Graph representing mean ± SEM of fluorescence intensity of Lysotracker dye in controls (white column) and in the patient (black column). The mean fluorescence intensity was measured per region of interest. One representative out of four independent experiments is shown. **P < 0.001 by Mann–Whitney test. (H and I) Assessment of lysosomal structural integrity by Lysotracker dye (green) in the control (H) and the patient (I). DAPI counterstains nuclei (white). Bars = 20 µm. Data from one out of five controls are shown. One representative out of four independent experiments is shown. (J, J′, K, K′) Expression of nephrin (green) after differentiation into podocytes of RPC obtained from control (J, J′) and the patient carrying genetic variants in NPHS1 gene (K, K′). DAPI counterstains nuclei (white). Bars = 20 µm. Data from one out of five controls are shown. One representative out of four independent experiments is shown.
DISCUSSION
We had speculated that the entity of RLN may also include patients with genetic lupus podocytopathies [7]. Indeed, our patient was a carrier of a homozygous variant in the NPHS1 gene, one allele each inherited from each of her non-proteinuric heterozygous parents, in line with the autosomal-recessive transmission of nephrotic syndrome of the Finnish type. A point mutation in the same position causing substitution of serine with proline was reported in a compound heterozygous status in a case with congenital nephrotic syndrome and is listed in Human Gene Mutation Database [8, 9]. This amino acid is highly conserved and its replacement by phenylalanine is predicted as potentially damaging by PolyPhen and PMut prediction tools. This disease most commonly presents as congenital nephrotic syndrome, but cases with a juvenile onset have been reported, depending on the genomic impact on protein amount, shape and location [10]. A careful structural and functional characterization of podocytes derived from urinary RPC is a powerful diagnostic tool for genetic podocytopathies [5]. In our patient, RPC-derived podocytes displayed disturbed nephrin trafficking to the cell surface, an abnormal cytoskeletal structure, and reduced cell viability similar to what has been previously reported in patients with nephrotic syndrome of the Finnish type [9]. These findings validate the pathogenicity of the patient's NPSH1 gene variant and provide a possible explanation for: (i) accelerated podocyte loss and glomerulosclerosis, (ii) the delayed onset of nephrotic syndrome and (iii) the multidrug resistance of the patient. In addition, the genetic status of two APOL1 risk alleles (termed G1 and G2) put out patient at high risk for an accelerated progression of CKD to end-stage renal disease [2], probably because of accelerated podocyte loss via lysosomal leakage-driven podocyte detachment and death [6].
Together, our case underlines the need for genetic screening in RLN. Early diagnosis of genetic disease is important for prognosis prediction, to avoid unnecessary immunosuppressive therapy and for genetic counselling of family members. Our observations raise the question on the prevalence of genetic kidney disease among patients with RLN.
CONFLICT OF INTEREST STATEMENT
The results presented in this paper have not been published previously in whole or part, except in abstract format.
ACKNOWLEDGEMENTS
This study was supported by the European Research Council under the Consolidator Grant RENOIR to P.R. (ERC-2014-CoG, grant 648274). H.-J.A. was supported by European Union's Horizon 2020 research and innovation programme under grant agreement No. 668036 (project RELENT).
REFERENCES
Author notes
These authors contributed equally to this work.


{kind=link}
{kind=link}
Comments