





Abstract
Pseudohypoaldosteronism type II (PHAII) is a hereditary disease characterized by salt-sensitive hypertension, hyperkalemia and thiazide sensitivity. Mutations in with-no-lysine kinase 1 (WNK1) and WNK4 genes are reported to cause PHAII. Rigorous studies have demonstrated that WNK kinases constitute a signaling cascade with oxidative stress-responsive gene 1 (OSR1), Ste20-related proline-alanine-rich kinase (SPAK) and the solute carrier family 12a (SLC12a) transporter, including thiazide-sensitive NaCl cotransporter. The WNK–OSR1/SPAK-SLC12a signaling cascade is present in the kidneys and vascular smooth muscle cells (VSMCs) and regulates salt sensitivity physiologically, i.e. urinary sodium excretion and arterial tone by various hormonal and dietary factors. However, although it was clear that the abnormal activation of this signaling cascade is the molecular basis of PHAII, the molecular mechanisms responsible for the physiological regulation of WNK signaling and the effect of WNK4 mutations on PHAII pathogenesis are poorly understood. Two additional genes responsible for PHAII, Kelch-like 3 (KLHL3) and Cullin 3 (CUL3), were identified in 2012. WNK1 and WNK4 have been shown to be substrates of KLHL3–CUL3 E3 ubiquitin ligase both in vitro and in vivo. In PHAII, the loss of interaction between KLHL3 and WNK4 induces increased levels of WNK kinases due to impaired ubiquitination. These results indicate that WNK signaling is physiologically regulated by KLHL3/CUL3-mediated ubiquitination. Here, we review recent studies investigating the pathophysiological roles of the WNK signaling cascade in the kidneys and VSMCs and recently discovered mechanisms underlying the regulation of WNK signaling by KLHL3 and CUL3.
INTRODUCTION
The kidneys play an essential role in linking salt intake with blood pressure. This function is regulated by sodium reabsorption through several ion transporters expressed throughout the nephron. The thiazide-sensitive NaCl cotransporter (NCC) is essential for sodium reabsorption in the distal convoluted tubules (DCTs) of the kidneys [1]. A loss-of-function NCC mutation causes Gitelman's syndrome, an inherited disease that exhibits salt-losing phenotypes [2]. Pseudohypoaldosteronism type II (PHAII) is an autosomal dominant hereditary disease characterized by salt-sensitive hypertension, hyperkalemia and metabolic acidosis [3]. All disorders of PHAII can be managed with chronic low-salt diet, indicating that PHAII is a ‘salt-sensitive’ disease. Another important characteristic of PHAII is thiazide sensitivity. The fact that thiazide, a pharmacological inhibitor of NCC, can successfully treat all manifestations of PHAII indicates that abnormally increased NCC expression could be the main cause of PHAII [4]. However, NCC mutations have not been found in PHAII patients. Instead, mutations in with-no-lysine kinase 1 (WNK1) and WNK4 genes have been reported to cause PHAII [5].
PHAII-CAUSING WNK1 AND WNK4 MUTATIONS
WNK kinase is a unique kinase as the lysine (K) residue in subdomain II present in the majority of kinases is not conserved [6]. In mammals, four members of the WNK kinase family (WNK1–4) exist [7]. In all WNK kinases, a kinase domain exists at the N-terminus followed by an autoinhibitory domain and a coiled-coil domain [8]. Interestingly, among six missense WNK4 mutations identified in PHAII patients, five mutations were found in a region termed the ‘acidic domain,’ which is located immediately after the first coiled-coil domain [5, 9, 10]. As shown in Figure 1, this acidic domain is well conserved in all WNK kinase isoforms. In contrast, all WNK1 mutations involved large deletions in intron 1. Identified WNK1 mutations increased their transcription in leukocytes of patients with PHAII [5], indicating the activation of WNK1-mediated signaling that resulted in PHAII. Subsequently, the existence of two WNK1 isoforms, a full-length WNK1 and a kidney-specific WNK1 lacking a kinase domain, was reported [11, 12]. Although a WNK1+/− mouse model did not exhibit decreased NCC phosphorylation [13], a PHAII-causing WNK1 deletion mutant mouse model (WNK1+/FHHt) exhibited increased renal transcription of full-length WNK1, resulting in increased phosphorylation of NCC [14]. This study further demonstrated that phosphorylation of NCC was increased in WNK1+/FHHt mice in a WNK4-independent and SPAK-dependent manner [15].
![Structures of WNK signaling cascade components. PHAII-causing mutations of WNK4 are predominantly clustered in the acidic domain. The acidic domain is located immediately after the first coiled-coil domain and conserved among all WNK kinases. WNK kinases activate OSR1 and SPAK by phosphorylating threonine residues in the kinase domain (T185 and T233). CCT domains in OSR1 and SPAK bind to RFx[V/I] motifs of the WNK and SLC12a transporter family. Phosphorylation of NCC/NKCC by OSR1/SPAK leads to activation and plasma membrane localization.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ndt/31/9/10.1093_ndt_gfv259/3/m_gfv25901.jpeg?Expires=1750304348&Signature=2L9DODYsGoulwR1BL2NNFyySIK6ld8tlBvU0bcIldU9O5XHxO2GxKiQsSfE-10r5JXBtV-ErDmQjBBW7li0toJrX7BgryCef37F19oO8L1mscLSl7qv6fx~XHQWNRewT55QhuBhRZnwKb9X-rnInzL1XCD1j~CQBH0Ule~4RnlpaqltW9PVK8bXcUz1EO07eWWMXKJW-UYpEfiv9Md-y4DvpJNvMD~MBy35pAU0faFWZluSbK8EtT4Dy2i0nxMp~C9QcrNhts-Ltk98H~c8M5BQLBZkGYepVnJ6OYsFz18d7efR0antsJwmHTYXxBZapt0XQtjdYZeq721ilaLnq6g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Structures of WNK signaling cascade components. PHAII-causing mutations of WNK4 are predominantly clustered in the acidic domain. The acidic domain is located immediately after the first coiled-coil domain and conserved among all WNK kinases. WNK kinases activate OSR1 and SPAK by phosphorylating threonine residues in the kinase domain (T185 and T233). CCT domains in OSR1 and SPAK bind to RFx[V/I] motifs of the WNK and SLC12a transporter family. Phosphorylation of NCC/NKCC by OSR1/SPAK leads to activation and plasma membrane localization.
SPAK AND OSR1 SUBSTRATES OF WNK KINASES
There have been many studies investigating the relationship between WNK kinases and NCC. However, because NCC is not a direct substrate of WNK kinases, the detailed pathophysiological effects of WNK4 mutations in PHAII are yet to be fully elucidated.
In 2005, two groups reported that oxidative stress-responsive gene 1 (OSR1) and Ste20-related proline-alanine-rich kinase (SPAK) were substrates of WNK1 and WNK4 [16, 17]. As shown in Figure 1, all WNK kinases in mammals phosphorylate SPAK and OSR1 at two conserved residues: threonine in a T-loop (Thr233 in SPAK and Thr185 in OSR1) and serine in an S-motif (Ser373 in SPAK and Ser325 in OSR1). T-loop phosphorylation activates SPAK and OSR1 [17, 18]. OSR1 and SPAK are related serine–threonine kinases that possess a conserved C-terminal (CCT) domain. The CCT domains of OSR1 and SPAK have been shown to interact with the RFv[V/I] motif in WNK kinases and solute carrier family 12 (SLC12) transporters. It was demonstrated that SPAK and OSR1 kinases phosphorylate three conserved residues present at the N-terminal region of SLC12a2 (NKCC1) [19–23]. In addition, two other SLC12a family transporters, SLC12a3 (NCC) and SLC12a1 (NKCC2), were found to be phosphorylated by OSR1 and SPAK at three residues, homologous to residues of NKCC1, in vitro and in vivo [16, 24, 25]. Pacheco-Alvarez et al. [26] demonstrated that phosphorylation of NCC increases its transporter activity in the Xenopus laevis oocyte expression system. In addition, a phosphorylation-specific NCC antibody used to demonstrate phosphorylated NCC was exclusively present on the apical plasma membranes of DCT in vivo [24], suggesting that phosphorylation regulates plasma membrane expression of NCC in addition to its transporter activity. Moreover, it was reported that phosphorylation of NCC decreased its ubiquitination at the plasma membrane [27], suggesting that decreased endocytosis and/or degradation of phosphorylated NCC may lead to increased accumulation of phosphorylated NCC at the apical plasma membrane.
Therefore, WNK kinases have finally been linked with NCC through SPAK and OSR1. Phosphorylation of SPAK and OSR1 was also reported to be increased in Wnk4D561A/+ knockin mice carrying a human PHAII-causing mutation [24], suggesting that WNK–OSR1/SPAK–NCC signaling is present in the kidneys and that over-activation of NCC by PHAII-causing WNK4 mutations leads to abnormally increased sodium reabsorption. Subsequently, by crossing Wnk4D561A/+ mice with SPAK and OSR1 knockin mice in which the T-loop Thr residues in SPAK (Thr243) and OSR1 (Thr185) were mutated to Ala to prevent activation by WNK kinases [28, 29], it was demonstrated that NCC phosphorylation and PHAII phenotypes in Wnk4D561A/+ mice are dependent on WNK–OSR1/SPAK signaling. This confirms that WNK phosphorylates and activates OSR1/SPAK, which then phosphorylates and activates NCC, constituting WNK–OSR1/SPAK–NCC signaling cascade in the kidneys.
PHYSIOLOGICAL REGULATION OF WNK SIGNALING
The discovery of WNK–OSR1/SPAK–NCC phosphorylation cascade has implications not only for rare inherited disease but also for NaCl homeostasis and blood pressure regulation under normal physiological conditions. Therefore, physiological regulators of WNK signaling have been rigorously studied (Figure 2). Salt intake regulates this cascade; NCC phosphorylation is increased by a low-salt diet and decreased by a high-salt diet through the actions of aldosterone [30], a strong regulator of NCC phosphorylation. Disruption of this mechanism is essential for increased salt sensitivity in PHAII and ‘salt-sensitive hypertension’ in the general population. In fact, a high-salt diet could not downregulate WNK–OSR1/SPAK–NCC signaling in a PHAII mouse model because of the constant active sodium reabsorption via NCC, resulting in the failure of proper urinary sodium excretion, leading to salt-sensitive hypertension [30]. In addition, angiotensin II [31–34] and vasopressin [35–38] have been reported as activators of WNK signaling.
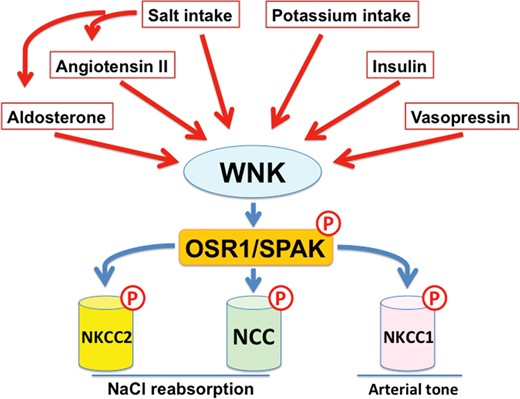
Regulators of the WNK–OSR1/SPAK-SLC12a signaling cascade. Aldosterone, angiotensin II, vasopressin and insulin are major regulators of WNK signaling in the kidney. In addition, dietary salt and potassium intake also regulates WNK signaling in the kidney. The WNK1/WNK3–SPAK–NKCC1 phosphorylation cascade also regulates arterial tone. Angiotensin II increases arterial tone through the activation of WNK3 signaling.
Insulin also stimulates WNK4 signaling through the phosphatidylinositol 3-kinase/Akt pathway [39–43]. This effect of insulin on WNK signaling may explain the increased salt sensitivity seen in patients with hyperinsulinemia, including those with metabolic syndrome [44]. Potassium intake also regulates this signaling cascade, and high potassium intake decreases NCC phosphorylation despite increased plasma aldosterone levels. [45–51]. This phenomenon has been termed the ‘aldosterone paradox.’ In contrast, low potassium intake increases WNK signaling [45, 46, 49, 50, 52]. Naito et al. [53] reported that extracellular potassium levels directly regulate WNK1 activity in cultured cells. Recently, the Ellison group reported that plasma potassium modulates DCT cell membrane voltage and intracellular chloride [49]. A WNK1 crystal structure demonstrated that WNK1 binds Cl− near the catalytic lysine in subdomain 1, inhibiting its autophosphorylation and activity [54]. Therefore, low intracellular chloride because of low plasma potassium may stimulate the activation of NCC by WNK kinases, although the involvement of WNK4 in this mechanism remains unknown.
WNK–OSR1/SPAK KINASE SIGNALING REGULATES ARTERIAL TONE
NKCC1 is involved in the regulation of vascular smooth muscle cell (VSMC) contractions via intracellular Cl− accumulation, membrane depolarization and activation of voltage-gated Ca channels, leading to increased peripheral arterial tone [55]. Indeed, NKCC1 knockout mice demonstrated lower blood pressure due to decreased arterial tone only in mice fed a low-salt diet compared with those fed a normal diet [56], indicating that NKCC1 plays an important role in the regulation of arterial myogenic tone in response to dietary salt intake. Because WNK–OSR1/SPAK signaling has been shown to regulate the phosphorylation of NKCC1 in addition to NCC, the roles of WNK1 and WNK3, WNK kinases expressed in VSMCs, in the regulation of arterial tone have been investigated. Initially, SPAK knockout mice were reported to exhibit decreased aortic phosphorylation of NKCC1, resulting in a decreased arterial response to phenylephrine [25]. Similarly, WNK1+/− mice demonstrated decreased aortic NKCC1 phosphorylation and decreased myogenic tone in mesenteric arteries, indicating that WNK–SPAK–NKCC1 signaling plays a substantial role in the maintenance of arterial tone [13, 57]. Bergaya et al. [57] also reported that WNK1 plays a role in vasoconstriction by adrenergic stimulation, but not by angiotensin II stimulation, in vivo. Later, Zeniya et al. [58, 59] demonstrated that angiotensin II regulates WNK3–SPAK–NKCC1 signaling in VSMCs in vivo and in vitro. Taken together, WNK1 and WNK3 appear to play different roles in VSMCs. WNK1 may be important for the maintenance of basal NKCC1 phosphorylation and responses to adrenergic stimulation. In contrast, WNK3 may have a key role in the response to stimulation by angiotensin II.
TWO NOVEL PHAII-CAUSING GENES: KLHL3 AND CUL3
In 2012, Boyden et al. [60] identified mutations in KLHL3 and Cullin 3 (CUL3), and Louis-Dit-Picard et al. [61] identified mutations in KLHL3, in families with PHAII using whole-exome sequencing. The identification of these two novel genes elucidated a detailed mechanism underlying the regulation of WNK signaling. KLHL3 is a member of the BTB–BACK–Kelch family, known to be substrate adapters of CUL3-based E3 ubiquitin ligase complexes [62]. The kelch domain of KLHL3 forms one blade of a β-propeller structure allowing substrate binding. Initially, it was reported that KLHL3 was able to interact with NCC and regulate its intracellular localization in vitro [61]. However, Ohta et al. [63] and Wakabayashi et al. [64] reported that both WNK1 and WNK4 were substrates of KLHL3–CUL3 E3 ubiquitin ligase. It has been demonstrated that the loss of KLHL3 and WNK4 interactions induces impaired WNK4 ubiquitination and increased protein levels of WNK4. Two further reports [65, 66] corroborated these findings, demonstrating that WNK4 is a target of the KLHL3–CUL3 E3 ligase. Finally, to clarify the role of KLHL3 mutations in PHAII pathogenesis in vivo, Susa et al. [67] generated KLHL3R528H/+ knockin mice carrying the same mutation as found in PHAII patients. These mice revealed increased protein levels of WNK1 and WNK4 kinases because impaired binding of mutant KLHL3 R528H with WNK kinases caused PHAII in vivo, indicating that both WNK1 and WNK4 kinases are physiologically regulated by KLHL3–CUL3-mediated ubiquitination. Boyden et al. [60] reported more severe symptoms in PHAII patients with KLHL3 mutations than with WNK1 or WNK4 mutations. Increased protein levels of both WNK1 and WNK4 kinases resulting from KLHL3 mutations may result in further increased phosphorylation of downstream components of WNK signaling as opposed to an increased protein level of a single WNK kinase.
Furthermore, PHAII-causing mutations in WNK4, KLHL3 and CUL3 have revealed the interaction between each of these protein products. As mentioned earlier, PHAII-causing WNK4 mutations involve the acidic domain, which is highly conserved in all WNK kinases [5]. Fluorescent correlation spectroscopy has demonstrated that the acidic domain of WNK kinases directly binds to KLHL3 and that WNK4 mutations within the acidic motif disrupt the binding of KLHL3 and WNK4 kinases [64]. Impaired binding of WNK4 to KLHL3 results in decreased ubiquitination and increased protein expression of the mutant WNK4. Increased levels of WNK4 protein have been demonstrated in the kidneys of Wnk4D561A/+ mice [67].
In contrast with the clustering of WNK4 mutations within the acidic domain, PHAII-causing mutations in KLHL3 are not restricted to kelch repeat domains, but are present in BTB and BACK domains [60, 61]. KLHL3 mutations at the BTB domain (E85A) and BACK domain (C164F) decrease the binding affinity of KLHL3 for CUL3 [68], whereas mutations in the Kelch domains affect the ability of KLHL3 to bind WNK kinases [63–66]. The S410L mutation of the Kelch domain significantly decreases intracellular stability [68]. In addition, a PHAII-causing mutation at S433 has provided further insight into the physiological regulation of KLHL3 binding to WNKs. Recently, Shibata et al. [69] reported that phosphorylation of KLHL3 at S433 decreased the binding affinity for WNK kinases, leading to impaired WNK degradation. Moreover, angiotensin II has been shown to phosphorylate KLHL3 at S433 via protein kinase C, clarifying one of the mechanisms by which angiotensin II regulates WNK signaling.
In the case of CUL3, PHAII-causing mutations are found around the splice donor and acceptor sites of exon 9 [60]. These mutations have been shown to result in skipping of exon 9 in cultured cells and leukocytes from PHAII patients [60]. Wakabayasi et al. [64] reported that this CUL3 mutation (CUL3 Δ403–459), involving the loss of a portion of exon 9, did not reduce the binding affinity of CUL3 for KLHL3; however, the E3 ligase activity toward WNK4 significantly decreased. Recently, the Ellison group reported that PHAII-causing mutant CUL3 is more heavily neddylated and activated than wild-type CUL3 [70]. In cultured cells, activated CUL3 Δ403–459 strongly ubiquitinates KLHL3, leading to depletion of KLHL3 and the prevention of WNK degradation. A PHAII mouse model carrying a CUL3 gene lacking a portion of exon 9 would provide more detailed information regarding the pathophysiology of CUL3 mutations.
As a result of recent studies, PHAII is now considered a disease caused by the abnormal activation of the WNK–OSR1/SPAK–NCC phosphorylation cascade due to increased levels of WNK kinases because of dysregulation of either the transcription or ubiquitination of WNK kinases (Figure 3).
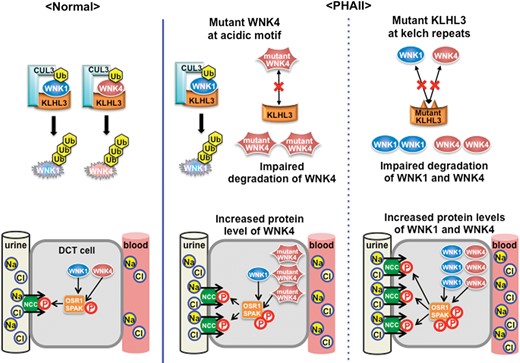
Regulation of WNK signaling by KLHL3–CUL3 complex and molecular pathogenesis of PHAII. Under normal conditions, protein levels of WNK1 and WNK4 in DCT are maintained by degradation through ubiquitination by the KLHL3–CUL3 E3 ligase complex. However, PHAII-causing mutations in the acidic domain of WNK4, and in the Kelch domains of KLHL3, interrupt binding of the KLHL3–CUL3 E3 ligase complex, resulting in impaired ubiquitination and degradation of WNK kinases. PHAII-causing CUL3 mutations lack a portion of exon 9 shown to have decreased ubiquitination and degradation of WNK kinases. Thus, PHAII-causing mutations in three different genes share a common mechanism: decreased ubiquitination and increased WNK1/WNK4 protein levels in DCT cells. Both WNK1 and WNK4 are increased in the kidneys with PHAII caused by KLHL3 and CUL3 mutations. This could lead to more severe phenotypes compared with PHAII caused by single WNK1 or WNK4 mutations. Ub, ubiquitin; DCT, distal convoluted tubule.
This newly identified pathophysiology of PHAII supports the notion that WNK4 is an activator of NCC. The effect of WNK4 on NCC has been debated for a long time [71]. After WNK4 was identified as causative gene of PHAII, many studies investigated the relationship between WNK kinases and NCC, with the majority of studies demonstrating an inhibitory effect of WNK4 on electrolyte transporters in the X. laevis oocyte expression system and in cultured cells [72–75]. However, Castaneda-Bueno et al. [31] and Takahashi et al. [43] reported that WNK4 knockout mice demonstrate markedly decreased phosphorylation of NCC, indicating that WNK4 is a positive regulator of NCC in vivo. There is little evidence that WNK4 is a negative regulator of NCC in vivo, except that WNK4 BAC transgenic mice carrying a wild-type WNK4 transgene exhibit a Gitelman syndrome-like phenotype [76]. However, Wakabayashi et al. [64] generated several lines of WNK4 BAC transgenic mice and demonstrated that phosphorylation of OSR1, SPAK and NCC robustly increases as renal WNK4 protein levels increase. As increased WNK4 protein levels resulting from KLHL3 mutation have been conclusively shown to increase OSR1/SPAK–NCC phosphorylation in vivo, it is natural to assume that WNK4 positively regulates the downstream OSR1/SPAK–NCC phosphorylation cascade.
KLHL2 REGULATES WNK SIGNALING IN VSMCs
To explore the interaction of WNKs with other Kelch-like proteins, Takahashi et al. [77] focussed on KLHL2 (Mayven), a human homolog of the Drosophila Kelch protein that has the highest homology with KLHL3. Similar to KLHL3, KLHL2 directly interacts with WNK kinases, leading to their ubiquitination and degradation [78]. This suggested that KLHL2–CUL3 also functions as an E3 ligase for WNK isoforms. KLHL2, but not KLHL3, has been shown to be present in murine aortic and VSMCs [79]. As mentioned earlier, angiotensin II activates the WNK3–OSR1/SPAK–NKCC1 phosphorylation cascade in VSMCs [58], leading to increased arterial tone. Zeniya et al. [79] recently reported that ubiquitination by KLHL2 is the major regulator of WNK3 protein levels in response to angiotensin II stimulation. As shown in Figure 4, angiotensin II decreases KLHL2 and increases WNK3 levels in VSMCs within minutes. Furthermore, angiotensin II induces p62-mediated selective autophagic degradation of KLHL2. Reduction in KLHL2 leads to increased WNK3 protein levels which activate downstream SPAK–NKCC1 phosphorylation signaling.

Mechanism underlying the effect of WNK3 signaling on angiotensin II-induced vasoconstriction through KLHL2. In vascular smooth muscle cells, WNK3 is degraded by KLHL2–CUL3-mediated ubiquitination. Angiotensin II stimulation degrades KLHL2 via selective p62-mediated autophagy, leading to the activation of the WNK3–SPAK–NKCC1 phosphorylation cascade and vasoconstriction. Ub, ubiquitin; AngII, angiotensin II; VSMC, vascular smooth muscle cell.
SUMMARY AND FUTURE DIRECTIONS
Studies of the WNK signaling pathway and the discovery of two novel PHAII-causing genes encoding the E3 ligase complex have clarified the molecular pathogenesis of PHAII. Investigation of the pathophysiology of PHAII is extremely important in elucidating the physiological mechanisms of renal electrolyte homeostasis in addition to increasing knowledge of rare inherited disease. WNK kinases, regulated by ubiquitination and degradation induced by KLHL2 and/or KLHL3, are the major regulators of the WNK–OSR1/SPAK–SLC12A signaling cascade. The discovery of this novel mechanism underlying salt-sensitive hypertension highlighted the role of KLHL2 and KLHL3 in the physiological regulation of WNK signaling. In addition to the transcriptional regulation of KLHL2 and KLHL3, investigation of protein product modifications such as phosphorylation of KLHL3 at S433 [69] and autophagic degradation [79] would be important for further understanding of the physiological regulation of the WNK signaling pathway. As WNK signaling regulates arterial tone in addition to urinary sodium excretion, pharmacological inhibitors of this signaling cascade may represent a novel class of antihypertensive drugs. Recently, Mori et al. [80] discovered two novel compounds that disrupt the binding of WNK to SPAK and demonstrated dose-dependent inhibition of WNK signaling in cultured cell lines. Kikuchi et al. [81] developed a new ELISA-based screening system to identify novel SPAK inhibitors and discovered two agents that inhibit SPAK-regulated phosphorylation and activation of NCC and NKCC1 in vitro and in vivo. These compounds may have great potential as novel antihypertensive drugs. Furthermore, KLHL2 and KLHL3 may represent future drug discovery targets aimed at regulating WNK kinase activity.
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest associated with this manuscript.
ACKNOWLEDGEMENTS
This work was supported by Grants-in-Aid for Scientific Research (S) (25221306-00) from the Japanese Society for the Promotion of Science and a Health Labor Science Research Grant from the Ministry of Health Labor and Welfare, Challenging Exploratory Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Salt Science Research Foundation (1422), the Takeda Science Foundation, Banyu Foundation Research Grant and the Vehicle Racing Commemorative Foundation.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments