





Abstract
Accurate identification of single-nucleotide variants (SNVs) is critical in clinical diagnostics but remains challenging due to subtle free energy variations, particularly for hard-to-detect SNVs such as wobble base pairs and those in high guanine–cytosine (GC) regions. Here we report a high-energetic-penalty SNV detection (HEPSD) platform that redesigns the hybridization regions of CRISPR RNA (crRNA) in the CRISPR/Cas12a system. This system employs a binary crRNA architecture design that enables the activation of the cleavage activity of Cas12a while amplifying the energetic penalty for single-nucleotide mismatches through nonequilibrium hybridization-driven regulation. Consequently, the entire targeting region of CRISPR/Cas exhibits a marked preference for mutations in genomic DNA, while preventing false activation induced by sequences containing a single mismatched nucleotide. Moreover, HEPSD exhibits exceptional differentiation performance for hard-to-detect SNVs including wobble mutations at extreme GC contents. As proof of principle, profiling of BRAF V600E and EGFR L858R tumor mutations down to a 0.01% variant allele frequency was achieved, enabling accurate discrimination of 132 clinical sample pairs, which showed high consistency with quantitative polymerase chain reaction-based techniques and next-generation sequencing. The proven effectiveness of this platform showcases its potential for clinical molecular diagnostics and expands the fundamental scope of hybridization-based protocols.
Introduction
Single-nucleotide variants (SNVs), a prevalent form of genetic variation, are critical genetic biomarkers associated with the onset of various serious diseases, from cancer to monogenic disorders and infectious diseases [1, 2]. Accurate profiling of disease-related SNVs has been a key focus in diagnostics and precision medicine because SNVs provide extensive diagnostic and therapeutic insights, including information on genetic predispositions, therapeutic targets, drug resistance, and prognosis assessment [3, 4]. Commonly, SNVs are identified using DNA hybridization probes [5], such as TaqMan probes [6], molecular beacons [7, 8], allele-specific polymerase chain reaction (ARMS-PCR) [9], and toehold-exchange probes [10]. These probes are designed to distinguish a single mismatch in 20–30 nucleotide sequences by utilizing hybridization reactions that are sensitive to the free energy changes (ΔG) between wild-type (WT) and mutant sequences. However, the free energy differences introduced by SNVs are relatively small (typically ranging from 1.5 to 7 kcal/mol) and can be easily overshadowed by the presence of large excesses of WT sequences, posing a significant challenge for detection [11]. In particular, the type of variation and the SNV-located sequence context have significant impacts on outcome of the detection system. Several mismatched base pairs, including rG-dT, rA-dC, rC-dA, and rU-dG can adopt wobble or Watson–Crick-like conformations, and exhibit negligible ΔG changes [12]. High GC-content sequence contexts further complicate SNV assays due to the stronger triple hydrogen bonds and the base-stacking interactions that promote the formation of secondary structures [13, 14]. Therefore, developing novel strategies capable of discriminating hard-to-detect SNVs with minimal free energy differences is of utmost urgency.
Molecular assays typically seek favorable thermodynamic parameters in hybridization reactions to enhance mismatch discrimination and improve specificity. Various strategies, such as chemically modified probes [15], secondary structure [16], or multistranded DNA designs [17], strand exchange-based probes [18, 19], and mismatch-guided assembly [20], have been developed to achieve high levels of discrimination. However, these technologies frequently necessitate target preparation, such as converting target double-stranded DNA (dsDNA) into hybridizable single-stranded DNA (ssDNA) or introducing toehold structures, which can compromise genetic integrity and reduce procedural simplicity. Additionally, most assays rely on precise temperature control and stoichiometric adjustments to achieve optimal performance.
In recent years, CRISPR-Cas systems have revolutionized nucleic acid molecular diagnostics. These systems typically consist of two core components: CRISPR-derived RNAs (crRNA) and Cas proteins. Among them, the CRISPR-Cas12a system can directly recognize protospacer adjacent motif (PAM)-containing dsDNA targets and exhibits trans-cleavage activity toward ssDNA to achieve high sensitivity [21]. For CRISPR-Cas12a, it is more sensitive to mismatches between the target DNA and crRNA than CRISPR-Cas9. However, the crRNA-mediated recognition process of Cas12a does not guarantee high specificity for discriminating arbitrary single mismatches, and it is generally believed that there is still a possibility of mismatch cleavage in PAM-distal regions [22]. Recent advancement in CRISPR-based SNV detection have further expanded its utility. For instance, Cas protein optimization [23, 24], engineered crRNA [25–27], and toehold exchange strategy [28]. Parallel developments have explored ssDNA targeting paradigms as a solvable direction for applying CRISPR-Cas12a systems to SNV detection [29, 30]. Despite these significant improvements in Cas12a performance, the underlying mechanisms governing specificity enhancement remain incompletely understood. Moreover, certain challenging mutations, particularly those involving wobble base pairs or high GC-content regions, continue to pose detection difficulties and warrant further investigation.
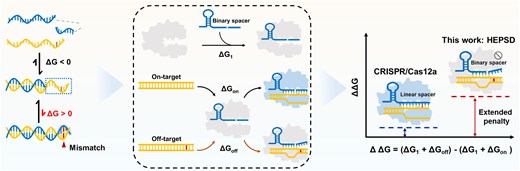
The CRISPR/Cas12a system’s activation is significantly affected by Gibbs free energy (ΔG) changes during the hybridization of target DNA with the crRNA spacer region. Specifically, a spacer length of 20–24 nucleotides is required to initiate cleavage events [31–34]. In this context, hybrids of this length are highly stable, rendering the mismatch-induced subtle thermodynamic perturbations (2–5 kcal/mol) negligible [35, 36]. Here, we demonstrate that a nonequilibrium hybridization probe can be integrated with the CRISPR/Cas12 system to establish a high–energetic–penalty-induced SNV detection platform (HEPSD), significantly enhancing selectivity for SNV recognition. In this work, a series of binary crRNA architectures were designed according to the theoretical findings of CRISPR thermodynamics. Interestingly, we found that the nonequilibrium hybridization between binary crRNA and target DNA enables effective signal output for fully matched sequences and selectively suppresses activation in the presence of mismatches. This process expands Gibbs free energy changes, resulting in markedly improved discrimination of SNVs (Scheme 1). While the use of CRISPR/Cas12a involving split crRNA is reminiscent of recent work that split crRNA into scaffold (nonhybridizing) and spacer (hybridizing) regions to enhance specificity [37], the foundational principles and molecular implementation of our study are fundamentally and mechanistically distinct. This study focuses on a binary strategy that targets the crRNA spacer region, uncovering its thermodynamic contribution to SNV detection. To date, the impact of spacer splitting on the performance of CRISPR-Cas12a remains largely unexplored. The findings reveal an extended energetic penalty originating from nonequilibrium hybridization and demonstrate exceptional capacity in profiling hard-to-detect SNVs, including wobble base pairs and those in high-GC-content sequences, which are challenging for conventional approaches. Notably, different from CRISPR-Cas12a-based detection strategies that specifically recognize SNVs in only a few specific locations, the HEPSD system exhibits specificity for point mutations across the entire targeting region, greatly broadening the application scenarios. In a study of 132 clinical samples, HEPSD successfully detected and identified BRAF V600E or EGFR L858R variants with 100% accuracy, indicating its great potential in clinical molecular diagnostics.
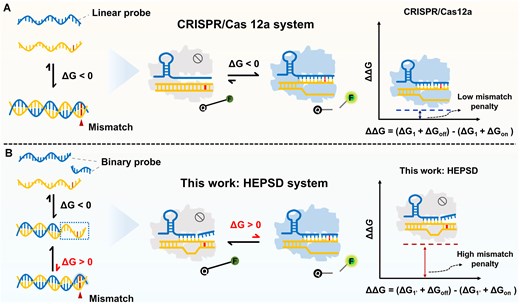
Comparison of HEPSD and conventional CRISPR/Cas12a system. (A) Low mismatch penalty drives activation by mismatched targets in linear probe (LP)-based CRISPR/Cas12a systems. (B) Schematic illustration of the HEPSD platform using binary spacer-assisted CRISPR/Cas adaptors to achieve a high mismatch penalty, allowing for precise SNV detection.
Materials and methods
Materials and reagents
All buffers were prepared using ultrapure H2O (18.2 MΩ·cm at 25°C, using a Direct-Q UV-R ultrapure water system). All chemical reagents were analytical grade. Potassium chloride (KCl), magnesium chloride hexahydrate (MgCl2·6H2O), and tetrasodium salt [ethylenediaminetetraacetic acid (EDTA)·2Na] were purchased from Titan Scientific Co., Ltd. (Shanghai, China). N, N, N’, N’-tetramethyl ethylenediamine (TEMED) was purchased from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China), Tris-(hydroxymethyl)-amino-methane (Tris), 30% acrylamide and bis-acrylamide solution (29:1), ammonium persulphate (APS), 6 × loading buffer, 10 000 × 4SGelred nucleic acid gel, 10 × Tris–borate–EDTA (TBE) buffer, diethyl pyrocarbonate (DEPC)-treated water and DNA marker were purchased from Sangon Biotech Co., Ltd. (Shanghai, China). LbCas12a protein was purchased from Bio-lifesci., Ltd. (Guangzhou, China). Genomic DNA was extracted from the tumor tissues of thyroid and lung cancer patients using the TIANamp Genomic DNA Kit purchased from Tiangen Biotech Co., Ltd. (Beijing, China); 2× Taq PCR Master Mix with dye was purchased from Tiangen Biotech Co., Ltd. The 2 × quantitative PCR (qPCR) Premix Ex TaqTM was purchased from Takara Biomedical Technology (Beijing) Co., Ltd. Sequences with 2-aminopurine (2-AP) modification were synthesized and purified by Sangon Biotech Co., Ltd. (Shanghai, China). All other synthetic DNA and RNA sequences were synthesized and purified by Hippo Biotechnology (Huzhou, China). All detailed sequence information is listed in Supplementary Table S1. The DNA sequences were dissolved in ddH2O to make stock solutions of 100 μM, which were stored at −20°C until use. All RNA sequences were dissolved in DEPC-treated water and kept at 100 μM. All dsDNA targets were annealed by hybridization with the equivalent concentration of target strand (TS) and nontarget strand (NTS) in an annealing buffer (20 mM Tris–HCl, 150 mM KCl, 1 mM EDTA, and 50 mM MgCl2, pH 7.5). The mixture was heated to 95°C for 5 min, followed by gradual cooling down to 25°C. The annealed dsDNA products were kept at −4°C before use.
Ultraviolet melting studies
Ultraviolet (UV) melting experiments were conducted using a JASCO-1500 spectropolarimeter (JASCO, Japan) equipped with a temperature-controlled water bath accessory. Measurements were performed in quartz cuvettes with an optical path length of 1 mm. A linear RNA/DNA duplex at a concentration of 2 μM was prepared in 1 × Tris–HCl buffer (pH 7.5) containing 150 mM NaCl. Melting curves were generated by monitoring absorbance at 260 nm while incrementally increasing the temperature by 1°C at a rate of 0.5°C per min. These experiments facilitated the determination of the duplex’s melting temperature (Tm) and thermal stability, providing critical insights into the conformational dynamics and binding interactions of the RNA/DNA complex.
Circular dichroism measurements
Circular dichroism (CD) spectra were recorded using a JASCO-1500 spectropolarimeter (JASCO, Japan) with a temperature-controlled water bath. Measurements were taken from 350 to 200 nm at a scan rate of 50 nm/min, using 1 mm path-length cuvettes, and under various temperature conditions. The cuvette chamber was continuously purged with dry N₂ gas to maintain stable conditions. All experiments were performed with a total strand concentration of 2 μM in 1 × Tris–HCl buffer (pH 7.5) containing 150 mM NaCl.
Quenching analysis
To investigate the interactions between LbCas12a and RNA [crRNA or binary probe (BP)-assisted crRNA] and their subsequent binding to dsDNA, we utilized the intrinsic fluorescence properties of the protein, primarily arising from tyrosine and tryptophan residues. These native fluorophores served as sensitive probes for monitoring conformational changes during the formation of protein-nucleic acid complexes. Fluorescence measurements were performed at 25°C and 37°C to evaluate temperature-dependent quenching behavior. For these measurements, 5 μM LbCas12a was incubated with various concentrations of RNA (0–50 μM) and monitored using an Edinburgh FLS1000 fluorometer equipped with an ultra-microcuvette to ensure high-sensitivity detection. Following preincubation, the LbCas12a/RNA complex was combined with dsDNA targets (0–40 μM), including both fully matched and single-nucleotide mismatch sequences. Fluorescence excitation was set at 285 nm, with emissions recorded over a range of 295–400 nm.
CRISPR-Cas12a real-time fluorescence assay and cleavage time courses
The CRISPR/Cas12a reaction system was prepared by combining 500 nM LbCas12a and 500 nM RNA (crRNA or BP-assisted crRNA) with varying concentrations of target DNA and 2 μM of the FQ reporter substrate, all within a 20-μl reaction volume containing 1 × LbCas12a reaction buffer. The Cas12a trans-cleavage reactions were carried out at 37°C for 90 min. The fluorescence intensity was measured every minute using a Thermal Cycler Dice Real-Time System III (TaKaRa). To calculate the cleavage rates of all binary spacer-assisted CRISPR/Cas12a systems in HEPSD that were successfully activated by recognizing dsDNA and ssDNA, the fluorescence intensity data were normalized to the maximum fluorescence intensity for consistent comparative analysis. The time-course data of the cleavage reactions were fitted to a single-exponential model using the equation below, employing nonlinear regression analysis in Origin software.
where F is the fluorescence intensity at time t, a is the fluorescence intensity amplitude, and k is the rate. Rates are reported per minute. Each reaction was repeated at least three times.
Gel electrophoresis
The Cas12a cis-cleavage was verified using 12% polyacrylamide gel electrophoresis (PAGE) in 1 × TBE buffer. To prepare the gel, 4 ml of 30% acrylamide/bis-acrylamide (29:1) solution, 3.93 ml of ultrapure water, and 2 ml of 5 × TBE buffer were mixed. Then, 70 μl of 10% APS solution and 7 μl of TEMED solution were added to the gel mixture for polymerization. The cleavage efficiency of BS-14 + 6 and conventional crRNA-mediated Cas12a towards fully-matched dsDNA was then verified. Briefly, a 20 μl reaction solution containing 500 nM crRNA/BS-14 + 6, 500 nM dsDNA, and 250 nM LbCas12a in 1 × LbCas12a reaction buffer was incubated at 37°C for 10 min, and compared to 30 min at 37°C. The Cas12a protein was subsequently inactivated at 95°C for 5 min and then cooled down to 25°C. Ten microliters of the cleaved products were premixed with 2 μl of 6 × loading buffer and then loaded onto a 12% PAGE gel. Electrophoresis was performed for 60 min at a constant voltage of 100 V. A 2 μl aliquot of a 25 bp DNA ladder was used as a molecular weight reference. Gel images were visualized after staining with GelRed using the GelDoc Go imaging system (Bio-Rad Laboratories Inc., USA). To compare the activation of the HEPSD system with the conventional CRISPR/Cas12a system on mismatched targets in cis-cleavage experiments, the same concentrations and procedures were used as described above, except that LbCas12a cleavage times were set to 10, 30, and 60 min before enzyme inactivation.
Molecular dynamics simulations
The system preparation process involved the use of the OL15 force field for DNA [38], OL3 force field for RNA [39], and ff14SB force field for proteins [40]. Hydrogen atoms were incorporated into each system using the LEaP module. A truncated octahedral TIP3P solvent box was added at a distance of 10 Å from the system boundary to provide a solvated environment [41]. To neutralize the system’s charge, Na+/Cl− ions were included. The resulting topology and parameter files were generated for simulation and converted to GROMACS 2021.03 compatible top and gro files using the acpype program [42]. Molecular dynamics (MD) simulations were conducted using GROMACS 2021.03 [43]. Before the simulation, energy minimization was performed with 5000 steps of steepest descent and 5000 steps of conjugate gradient methods. The system was then heated to 297 K over 300 ps under constant volume and temperature (NVT) conditions. Subsequently, a 400 ps simulation was conducted under constant pressure and temperature (NPT) conditions with solute atoms constrained by a force of 1000 KJ/(mol·nm²). Finally, a 100 ns production run was performed under NPT conditions. Non-bonded interaction cutoff distance of 1.2 nm was applied throughout the simulations, with system pressure maintained at 1 atm, using a 2 fs timestep. Trajectories were saved every 10 ps for subsequent analysis.
Measurement of fluorescence anisotropy
The fluorescence anisotropy (FA) of the HEPSD for targeting matched dsDNA and mismatched dsDNA was conducted using an FLS 1000 spectrofluorometer (Edinburgh) in FA measurement mode. The final concentrations of matched dsDNA and mismatched dsDNA were both 1 nM, the concentration of LbCas12a protein was 50 nM, and the concentration of BS-14 + 6 was maintained at 50 nM. The measurements were conducted at a constant temperature of 37°C, with the samples incubated at 37°C for 60 min before measurement. For 6-carboxyfluorescein (FAM), the excitation wavelength (Ex) was set to 490 nm, and the emission spectrum (Em) was collected over the range of 505–650 nm. The FA values were calculated from the equation:
where I represents the fluorescence intensity, with the subscripts indicating the horizontal (H) and vertical (V) orientations of the excitation and emission polarizers. The grating factor G of the fluorescence spectrophotometer is employed to adjust for the wavelength-dependent polarization response of the emission optics and detectors.
2-AP fluorescence intensity measurements
NTS with 2-AP modifications were annealed with their respective TS strands to form matched dsDNA and mismatched dsDNA at the same concentration. The final concentrations were 50 nM for crRNA/BS-14+6, 50 nM for LbCas12a, and 1 nM for dsDNA in 1×LbCas12a reaction buffer. Samples were incubated at 37°C for 60 min before measurement using an FLS 1000 spectrofluorometer (Edinburgh). The excitation wavelength (Ex) was set to 305 nm, and the emission spectrum (Em) was collected over the range of 360–450 nm.
Validation of sensitivity of the HEPSD system using synthetic sequences
To verify assay sensitivity with post-PCR, synthetic BRAF V600E and EGFR L858R templates, along with their WT alleles, were diluted to a series of concentrations, including 1 aM, 10 aM, 100 aM, 1 fM, and 10 fM. The diluted targets were PCR amplified using the Gene Explorer thermal cycler. For BRAF V600E, the PCR amplification protocol was as follows: polymerase activation at 94°C for 3 min, followed by 30 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 20 s, with a final extension at 72°C for 5 min. For EGFR L858R, the PCR procedures were as follows: initial denaturation at 94°C for 3 min, followed by 40 cycles of 94°C for 16 s, 56.5°C for 16 s, and 72°C for 25 s. The primer concentrations were fixed at 250 nM in 1 × Taq PCR MasterMix II (with dye). After PCR amplification, the amplicons were analyzed using split crRNA-mediated CRISPR-Cas12a real-time fluorescence assay.
Validation of mutant-type variants at low abundance by the HEPSD system
To determine the detection limit of mutation abundance, mutant targets and WT sequences were mixed in various ratios. The crRNAs and binary spacers targeting the SNV/mutant types were evaluated using a CRISPR-Cas12a real-time fluorescence assay. To validate detectable variant allele frequencies (VAFs) of EGFR L858R using the HEPSD system with post-PCR, samples with varying EGFR L858R to WT ratios were prepared, including 0%, 0.001%, 0.01%, 0.1%, 1%, and 10%. The 0% EGFR L858R sample confirmed that all tested strands were WT. The 20 μl post-PCR reaction mixture contained 10 μl of 2 × Taq PCR MasterMix II (with dye), 4 μl of RNase-free water, 2 μl of 4 μM forward primer, 2 μl of 4 μM reverse primer, and 2 μl of mixed strands at varying ratios. After post-PCR processing, 2 μl of the amplicons were analyzed using the CRISPR-Cas12a real-time fluorescence assay. To compare with quantitative PCR-based techniques, TaqMan minor groove binder probes were used to analyze the EGFR L858R mutation, including VAFs at 0.01%, 0.1%, 1%, and 10%.
Validation of the HEPSD system using genomic DNA extracted from clinical samples
This study was approved by the Medical Ethics Committee of Hunan Cancer Hospital (2024-Research-26). These procedures were conducted in adherence to institutional guidelines. All clinical samples were collected in the Affiliated Cancer Hospital of Xiangya School of Medicine and Second Xiangya Hospital. Blood samples were centrifuged at 2000 × for 20 min at room temperature, and the plasma supernatant was separated within 2 h of collection. The obtained plasma samples were stored at −80°C until further use. Genomic DNAs were extracted from serum and tissue samples using the Blood/Cell/Tissue Genomic DNA Extraction Kit (DP304, Tiangen Biotech Co., Beijing, China) according to the instructions of the manufacturer. The extracted genomic DNA samples were quantified using a Nano-300 microvolume spectrophotometer (Allsheng Instruments Co., Hangzhou, China). For the detection using HEPSD assay, the genomic DNA samples were further PCR amplified according to the PCR protocols abovementioned. For the detection of plasma samples, the blocker displacement amplification (BDA) protocol followed the same PCR conditions. For BDA assays, the 20 μl reaction mixture included 10 μl 2 × Taq PCR MasterMix II (with dye), 2 μl RNase-free water, 2 μl 4 μM forward primer, 2 μl 4 μM reverse primer, 2 μl 10 μM blocker, and 2 μl of the samples. After the post-PCR or BDA procedures, 2 μl of the amplicons were analyzed using the CRISPR-Cas12a real-time fluorescence assay. The normalized signal was calculated as follows:
where F represents the fluorescence intensity of the sample, Fmax denotes the maximum fluorescence of the reporter probe, and FNTC is the background fluorescence of the reporter probe in the absence of the target.
qPCR assay
A total 25 μl reaction contained 12.5 μl 2 × qPCR Premix Ex TaqTM, 4.5 μl of RNase-free water, 2 μl of 2.5 μM forward primer, 2 μl of 2.5 μM reverse primer, 2 μl of 2 μM TaqMan probe, and 2 μl of genomic DNA. qPCR cycling conditions were as follows: 30 s at 95°C, followed by 45 cycles of 95°C for 15 s, 60°C for 30 s, with signal collection set at 60°C in the FAM channel using the Thermal Cycler Dice Real-time System III. Ct values were obtained using the software of the Thermal Cycler Dice Real-time System III.
Next-Generation Sequencing (NGS) protocol
A minimum of 50 ng of DNA was required for NGS library preparation. DNA was fragmented into 200–400 bp segments using the Covaris M220 system (Covaris, Inc., Woburn, MA). Fragmented DNA underwent end-repair, phosphorylation, and adapter ligation (Beckman Coulter, Fullerton, CA). Adapter-ligated DNA was hybridized with biotinylated capture probes targeting specific genomic regions, followed by magnetic bead-based enrichment and PCR amplification to ensure sufficient yield. The library’s quality and fragment size were evaluated using the Qubit 2.0 fluorometer with the dsDNA High-Sensitivity Assay Kit (Thermo Fisher Scientific, Inc., USA). Genomic profiling was performed using a capture-based targeted sequencing panel from Burning Rock Biotech (Guangzhou, China). Sequence data were aligned to the reference genome (hg19) using BWA 0.7.10. Variant calling was performed using GATK 3.2, MuTect, and VarScan, while structural variations were analyzed with Tophat2 and Factera 1.4.3.
Sanger sequencing
Key mutations in tissue samples were further validated using Sanger sequencing. Genomic DNA extracted from tissue specimens underwent PCR amplification using a Gene Explorer thermal cycler. The PCR reaction mixture (50 μl total volume) consisted of 2 μl genomic DNA template, 25 μl 2 × Taq PCR MasterMix II (with loading dye), and 2 μl each of 10 μM forward and reverse primers, with ddH₂O added to reach the final volume. Specific thermal cycling parameters were applied, with annealing temperatures optimized for different targets: 52°C for BRAF V600E detection and 58°C for EGFR L858R analysis. The sequencing reactions were performed using BigDye Terminator v3.1 chemistry on a 3730xl DNA Analyzer (Thermo Fisher Scientific Inc., USA).
Results and discussion
RNA/DNA BPs and thermodynamics analysis
In the Cas12a system, a spacer length of 20–24 base pairs in the crRNA hybridized with the target DNA duplex is sufficient to initiate cleavage events [31]. Even with one or two mismatches, such a long helix is still stable. A potential nucleic acid detection strategy involves splitting crRNA into scaffold and spacer regions, as reported in previous studies [37, 44, 45]. However, that splitting strategy has shown limited progress in selectivity because modification of binding strength between Cas12a protein and crRNA could be insufficient to reliably discriminate single nucleotide variants at any position of a 20 nucleotide DNA target. Binary probes (BPs) consist of two hybridization arms: a longer arm and a shorter arm specifically designed to discriminate SNVs through selective hybridization [46]. We hypothesize that the spacer region of crRNA in the Cas12a system could be structurally engineered to regulate and optimize the system’s detection performance. To this end, a hybridization model based on RNA/DNA pairing was implemented (Fig. 1A). Although the thermodynamics of unstructured DNA/DNA probes has been studied, the stability of RNA/DNA remains elusive for this design. For comparison, linear probes (LPs) were also designed based on the conventional crRNA format used in CRISPR/Cas systems. As shown in Fig. 1C, in contrast to the narrow (ΔT < 10°C) and high-temperature-dependent (>68°C) discrimination range of LPs (Fig. 1B), BPs operate over a broader (ΔT > 40°C) and lower-temperature range. Additionally, CD spectroscopy was used to analyze the hybridization states of LPs and BPs with mutations. As displayed in Fig. 1D and E, without single base mismatches, both LPs and BPs exhibit characteristic RNA/DNA duplex CD features: a strong negative band at 210 nm, a positive peak at 265 nm, and a minor negative peak near 230 nm [47, 35], indicating stable hybridization. In contrast, BPs targeting mismatched sequences exhibited significantly weaker CD signals, resembling those of the longer arm hybridized alone, indicating incomplete hybridization states at room temperature (Fig. 1E). Temperature-dependent CD analysis further confirmed that BPs targeting mismatched analytes maintained prolonged binding states across a broad temperature range (Supplementary Fig. S1). These results underscore the adaptability of RNA BPs to diverse reaction conditions within the CRISPR/Cas12a system, which is typically optimized for 37°C. Through systematic evaluation across a broad temperature range (25–50°C), the binary spacer-assisted assay maintained optimal activity at the conventional 37°C (Supplementary Figs S2–S4). This temperature stability enabled direct application of standard Cas12a reaction conditions for subsequent validation experiments.
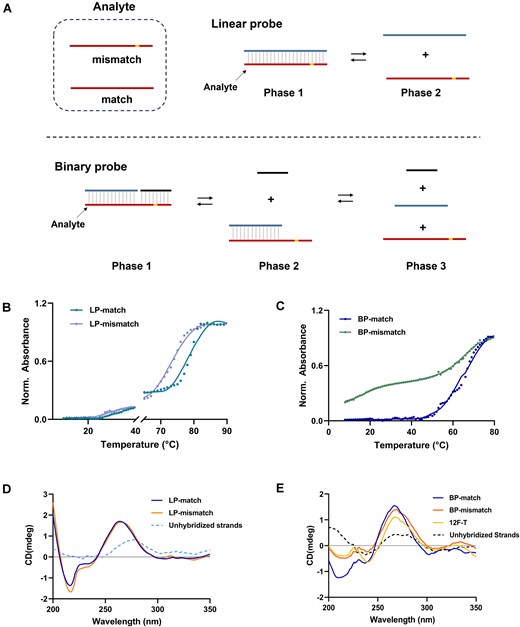
Discrimination characterization of LP and BP. (A) Schematic representation of linear and BPs across different phases. As the temperature increases, the probe-analyte duplex (phase 1) transitions to a dissociated state, with BPs potentially passing through intermediate states (phase 2) before complete dissociation (phase 3). (B, C) Comparison of the temperature window (10°C–90°C) for mismatch discrimination between LP and BP. Each RNA probe and analyte was prepared at a concentration of 2 μM. (D, E) CD plot confirming hybridization states of probes to analytes. The longer binding arm (12F) of the BP hybridizes with the analyte to form the hybridization state denoted as 12F-T. Each oligomer concentration was 2 μM, and measurements were done in an annealing buffer at 25°C.
High energy penalty SNV detection system driven by nonequilibrium hybridization
The aim of this work is to develop BPs forming two relatively short duplexes with targets in the Cas12a system could enhance specificity. However, the impact of spacer region splitting on CRISPR/Cas12a detection efficiency remains unclear. Critically, robust signal output for correct targets for an ideal discrimination probe is necessary. Therefore, we initially identified potential split sites within the spacer region of crRNA and assessed their capability to recognize both dsDNA and ssDNA targets (Supplementary Fig. S5). Orthogonal experiments with spacer-forwards and spacer-backwards demonstrated that 20 continuous base pairings between target DNA and truncated spacers are necessary for RNA-guided DNA recognition. Neither part of the split spacer alone is sufficient to activate Cas12a. Furthermore, the cleavage activity decreases significantly when Cas12a is paired with crRNA containing excess spacer bases, which might block the activity of Cas12a. Meanwhile, we observed that CRISPR/Cas12a with 14 bp spacer-forwards and 6 bp spacer-backwards (BS-14 + 6) exhibited much lower kinetics (Supplementary Figs S6 and S7). Further molecular docking simulations and trans-cleavage validations were performed (Supplementary Figs S8 and S9). The BS-14 + 6 initiated Cas12a cleavage achieved a fluorescence signal plateau within 30 min, demonstrating retained and sufficient cleavage activity.
Based on the activation performance of the BP-assisted Cas12a system, its assay efficiency for SNV discrimination and the thermodynamic landscape were investigated. We selected the rG-dT mutation, a hard-to-detect SNV tolerated by the conventional Cas12a system, as the paradigm [48, 49]. It is well known that the standard free energy change (ΔG) is critical for analyzing thermodynamic parameters. As shown in Fig. 2A, interactions between Cas12a and RNA (crRNA or BP-assisted crRNA) were denoted as ΔG1 and ΔG1’, while ΔGmatched, ΔGmatched’, ΔGmismatched, and ΔGmismatched’ were used to represent the transitions from the CRISPR/RNA complex to the ternary complexes with matched or mismatched dsDNA, respectively. Consequently, the ΔΔG was used to quantify the system’s mismatch discrimination ability. It was calculated as (ΔG1+ ΔGmismatched) − (ΔG1 + ΔGmatched) for the conventional Cas12a system and (ΔG1’ + ΔGmismatched’) − (ΔG1’ + ΔGmatched’) for the binary spacers-assisted Cas12a system, reflecting the energetic penalty of mismatched base pairs. The activation process of Cas12a occurs in two stages: crRNA interaction with Cas12a, followed by dsDNA unwinding and crRNA–DNA binding facilitated by enzymatic domains. Interestingly, we found the intrinsic fluorescence of Cas12a (attributed to tyrosine and tryptophan residues) is influenced by two hybridization processes (Supplementary Figs S10 and S11). Therefore, for the first time, we utilized a quenching mechanism [50, 51] to elucidate the binding process of the CRISPR/Cas12a system, which is mathematically described by the Stern–Volmer equation (Equation 1) [52].
where F0 and F represent the fluorescence intensity of Cas12a in the absence and presence of RNA (either crRNA or BP-assisted crRNA), respectively. To investigate the interactions between LbCas12a/RNA duplex with various concentrations of dsDNA, F0 and F are similarly defined as the fluorescence intensities of Cas12a/RNA without and with dsDNA. Ksv is the Stern–Volmer quenching constant, [Q] is the concentration of RNA or dsDNA as the quencher in each experiment. Kq is biomolecular quenching rate constant, and τ0 is the average lifetime of the fluorophore in the absence of quencher (average τ0= 10–8 s). The modified Stern–Volmer equation (Equation 2) encompasses the important issue of distinguishing the quenching is static or dynamic.
where fa is the mole fraction of accessible fluorescence. The relationship between |$\frac{{{{F}_0}}}{{{\rm{\Delta }}F}}$| and the reciprocal quencher concentration [Q]-1 is linear (Supplementary Figs S12 and S13), allowing for the determination of the binding constant K and binding sites n by plotting the fluorescence data as a double-log graph, following Equation 3 (Supplementary Figs S14 and S15):
Temperature-dependent thermodynamic parameters were calculated using the Van’t Hoff equation to clarify the binding forces between Cas12a and RNA dimers, as well as between the Cas12a/RNA complex and dsDNA. Assuming the enthalpy change (ΔH) remains relatively constant within the studied temperature range, both the enthalpy change (ΔH0) and the entropy change (ΔS0) can be evaluated based on the Equation 4:
where K represents the binding constant at a specific temperature T while R denotes the gas constant. The ΔG0 can be derived using the following equation:
From the above calculations, the thermodynamic results are presented in Fig. 2B. ΔG1 (−48.91 kJ/mol) was lower than ΔG1 (−46.31 kJ/mol). This energy difference likely originates from structural perturbations induced by spacer splitting, which correlates with the marginally reduced activity of the BS-14 + 6-mediated Cas12a system observed experimentally (Supplementary Fig. S8). Additionally, ΔGmatched, ΔGmatched’, ΔGmismatched, and ΔGmismatched’ demonstrated distinct thermodynamic profiles. In the conventional CRISPR/Cas12a system, ΔΔG (mismatched versus matched) was calculated to be 0.55 kJ/mol. By contrast, the binary spacer-assisted CRISPR/Cas12a system showed a markedly increased ΔΔG of 3.13 kJ/mol, indicating a higher energetic penalty toward single-base mismatches. This increase in ΔΔG validated the extended energy penalty SNV detection system’s capacity, showing a breakthrough in addressing the thermodynamic limitations of the conventional CRISPR/Cas12a system.
Guided by thermodynamic insights, it is hypothesized that the binary-spacers-assisted Cas12a system may exhibit incomplete hybridization, indicating a nonequilibrium hybridization process. Therefore, the hybridization dynamics of the HEPSD system in recognizing matched and mismatched targets were investigated using MD simulations. root mean square deviation (RMSD), and root mean square fluctuation analyses were performed to evaluate the overall structural stability and residue-level flexibility, respectively (Supplementary Fig. S16). In matched-target ternary complexes, RMSD values stabilized after 10 ns (Fig. 2C and D), signifying tight binding and equilibrium. In contrast, the mismatched target complexes formed by binary spacers in CRISPR/Cas12a exhibited continuously fluctuating RMSD values (Fig. 2D), reflecting dynamic instability and a lack of stable interactions. FA is an inherent property of fluorescent materials. Free fluorescent molecules rotate rapidly in solution, yielding low FA values. Upon binding, rotational motion is restricted, increasing FA values. In HEPSD, matched-target complexes showed high FA values, while mismatched targets exhibited lower values, indicating weaker interactions (Fig. 2E and F). The robust binding conformation and the continuously formed sufficient length of the R-loop between the spacer and TS serve as critical checkpoints for Cas12a activation. Therefore, the observed fluctuations in HEPSD during mismatch-guided processes provide further evidence supporting the enhanced discrimination.

Mechanism validation of BPs assisted CRISPR/Cas12a adaptor platform. (A) Schematic illustration of the ΔG workflow in conventional CRISPR/Cas12a versus the HEPSD platform. (B) Comparative analysis of ΔG and ΔΔG values between the ordinary CRISPR/Cas12a system and HEPSD for matched and mismatched targets. (C, D) RMSD analysis from MDs simulations for CRISPR/Cas12a (C) and HEPSD (D) with matched and mismatched targets. (E, F) FA analysis demonstrating the binding stability of HEPSD with matched and mismatched targets. Error bars represent the mean ± standard deviation (SD) from three independent replicates (n = 3). **P < 0.01, Student’s t-test. (G, H) Validation of nonequilibrium hybridization targeting mismatched dsDNA through 2-AP fluorescence in CRISPR/Cas12a and HEPSD platforms. Error bars represent the mean ± SD from three independent replicates (n = 3).
The underlying nonequilibrium mechanisms were explored to provide insight into the process. As previously reported [33], sequential dsDNA unwinding results in a complete R-loop, triggering allosteric activation and enabling DNA cleavage. Here, the adenine base near the PAM site in the NTS of the matched dsDNA target was replaced with 2-AP to verify dsDNA unwinding and proper R-loop formation. The (S-B)/B method was used to minimize background signals from local dsDNA concentration and quantify fluorescence enhancement induced by 2-AP. To evaluate the hybridization of HEPSD and ordinary crRNA with mismatched dsDNA, the 18th adenine at the PAM-distal end of the NTS was replaced with 2-AP. The conventional crRNA/Cas12a complex maintained crRNA-binding ability, producing a strong fluorescence signal at 377 nm (Fig. 2G). In contrast, the HEPSD system displayed minimal fluorescence enhancement relative to the DNA duplex (Fig. 2H). We therefore concluded that limited unwinding and an inactive CRISPR system caused by mismatch-induced nonequilibrium hybridization, drive the HEPSD system to effectively regulate CRISPR/Cas12a specificity.
Hard-to-detect SNV detection by HEPSD with enhanced discrimination
Having developed HEPSD and elucidated its working mechanism, its detection performance for SNV was subsequently assessed. Discriminating hard-to-detect SNVs presents a significant challenge for classic hybridization methods, primarily due to complex sequence compositions and challenging mutation types. In the CRISPR/Cas12a system, complexes formed with imperfect targets may still sufficiently promote cleavage [53, 54]. Herein, the HEPSD system was attempted to detect hard-to-detect SNVs.
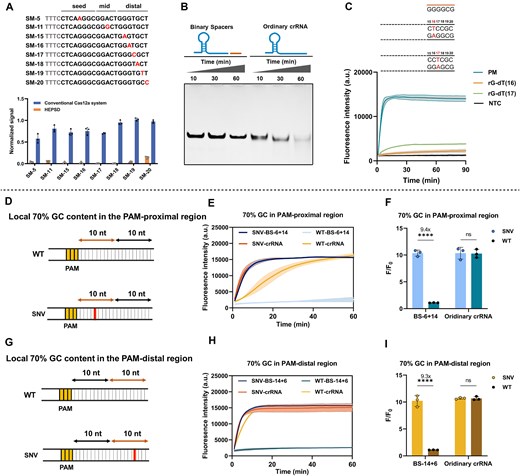
To evaluate the tolerance at each mutation site, we evaluated the single-nucleotide mismatch discrimination ability of HEPSD, conventional crRNA, and split crRNA across positions 1–20. Five types of crRNA featuring binary spacers were used for detection at each mutation site. The results shown in Fig. 3A represent the low-GC context, where the conventional crRNA-mediated CRISPR/Cas12a system exhibited significant signal reduction only for mutations at positions M2 and M17–M19. According to our experimental results and previous studies [31–34], mismatches in the seed region (5–10 nt from the PAM-proximal end) are discriminated with higher sensitivity compared to those in the PAM-distal region. However, the recognition of certain mismatched bases around position 17 from the PAM-distal may result from their impact on the cleavage domain of Cas12a. Split crRNA, which incorporates a truncated scaffold RNA as an independent component, also exhibited reduced fluorescence intensity at positions M16–M18. However, full recognition of the targeting region was not achieved in split crRNA tests, with specific nucleotide mismatches, such as M11, M12, and M1–M8, being detected. These results align with previous findings [37]. In this HEPSD system, the binary spacer-assisted Cas12a generated lower signals across a broader range of mismatch positions. Notably, BS-6 + 14 effectively distinguished mismatches across nearly all target-binding regions, while BS-14 + 6 significantly suppressed Cas12a-mediated trans-cleavage at positions M9–M20, demonstrating more distinct discrimination.
![The discrimination performance of HEPSD for mismatches at different positions. (A, B) Single mismatches (M1–M20) were introduced at different positions. PAM regions are shown as gray. Binary spacers with varying TS-binding arm lengths (varied from 6 to 14 nucleotides) were employed, including BS-6 + 14, BS-8 + 12, BS-10 + 10, BS-12 + 8, and BS-14 + 6. The trans-cleavage activity of Cas12a induced by different crRNAs after 60 min incubation at 37°C. The fluorescence intensity triggered by each mismatched target (1.0 nM) was normalized to the signal generated by the perfectly matched (PM) target (1.0 nM) for comparative analysis. The heat map represented the fluorescence intensity at time t = 30 min. Mismatch tolerance was also compared among the HEPSD system, conventional crRNA, and split crRNA (scaffold + spacer). (C–H) Kinetic rate comparison of different crRNA designs in targeting fully complementary dsDNA. The kinetic rates of conventional crRNA (C, F), split crRNA (D, G), and BS-14 + 6 (E, H) were compared at varying target concentrations and GC content. Reactions were conducted at 37°C for 30 min. [crRNA/split crRNA/BS-14 + 6] = 50 nM, [Cas12a] = 50 nM, [F-Q reporter] = 250 nM. Error bars indicate the mean ± SD (n = 3).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/53/7/10.1093_nar_gkaf287/1/m_gkaf287fig3.jpeg?Expires=1750263166&Signature=PXYOm55qyRMEwCy2R4Ch~npyVuUJ1JMWqPRNdf23AImP1L3R8lbfMFwmKZPxolXY1pK1TQs~qBb2N7-w5Ks78KVEa-WfTnvfBhb5grSOfKDuRdZjgHnezf0bPZMc6svvwzHX03Bf61~OZz4XBv4cCT13FK~Gn4flDG9ruSrEDTH7TK02V-yrbb-nPgxjeL9PnawJy80HJJ3Oh6D~LkrUZcQ~SJwK600EP-8W9SdDUOGAb7JgLVI4~SKyVYWk15PO80tFADF-CzTtEtUuW34ZXp4YIjFIZIQYSjq0FmEywrgYf4kvREkmshIt-1io~tpjxvlq8TlANX1a81GzqoUu~A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The discrimination performance of HEPSD for mismatches at different positions. (A, B) Single mismatches (M1–M20) were introduced at different positions. PAM regions are shown as gray. Binary spacers with varying TS-binding arm lengths (varied from 6 to 14 nucleotides) were employed, including BS-6 + 14, BS-8 + 12, BS-10 + 10, BS-12 + 8, and BS-14 + 6. The trans-cleavage activity of Cas12a induced by different crRNAs after 60 min incubation at 37°C. The fluorescence intensity triggered by each mismatched target (1.0 nM) was normalized to the signal generated by the perfectly matched (PM) target (1.0 nM) for comparative analysis. The heat map represented the fluorescence intensity at time t = 30 min. Mismatch tolerance was also compared among the HEPSD system, conventional crRNA, and split crRNA (scaffold + spacer). (C–H) Kinetic rate comparison of different crRNA designs in targeting fully complementary dsDNA. The kinetic rates of conventional crRNA (C, F), split crRNA (D, G), and BS-14 + 6 (E, H) were compared at varying target concentrations and GC content. Reactions were conducted at 37°C for 30 min. [crRNA/split crRNA/BS-14 + 6] = 50 nM, [Cas12a] = 50 nM, [F-Q reporter] = 250 nM. Error bars indicate the mean ± SD (n = 3).
Mismatches within high-GC-content sequences were also evaluated. As shown in Fig. 3B, single-nucleotide mismatches in high-GC dsDNA were more tolerated than those in low-GC dsDNA. Nevertheless, the binary spacers-assisted Cas12a system consistently displayed substantially reduced activity among mismatch sites. The binary spacer-assisted Cas12a system displayed varying discrimination across different mismatch regions. It was exciting to note that the combination of BS-6 + 14 and BS-14 + 6 yielded significantly reduced signals at positions M1–M20, achieving comprehensive mismatch detection in high-GC content dsDNA. Additionally, the binary spacer-assisted CRISPR/Cas12a within the HEPSD system demonstrates improved specificity in identifying single-nucleotide mismatches while retaining strong signal output for fully matched targets (Fig. 3C–H). This superior performance is likely attributed to the system’s efficient mismatch discrimination mechanism, which functions without compromising the signal output of Cas12a.
We next attempted to investigate mismatches that could form both wobble and Watson–Crick-like conformations. To have a consistent basis validation, the dsDNA with high GC content was designated. The testing covered PAM-proximal as well as mismatch-tolerant regions, including the middle and PAM-distal regions. As revealed in Fig. 4A, rG-dT wobble mismatches at various positions still almost completely suppressed Cas12a’s cleavage activity, although this mismatch has been reported to be generally tolerated even in the PAM-proximal region [49]. 12% PAGE analysis revealed only a slight reduction of wobble-mismatched dsDNA substrates after a 60-min incubation with HEPSD (Fig. 4B). Subsequently, a shorter arm with 100% GC content was designed within the HEPSD system to evaluate rG-dT tolerance (Fig. 4C). Notably, in the specified sequence context, the presence of rG-dT mismatches still resulted in only a minimal increase in fluorescence intensity at both positions over the 90-min kinetic profile.
The discrimination performances of HEPSD for wobble base pairs under different sequence settings. (A) Verification of wobble mismatch discrimination at various positions in sequences with 70% GC content. The assay was performed using 50 nM Cas12a, 50 nM binary spacers or crRNA, 200 nM F-Q reporter, and 1 nM dsDNA targets in 20-μl reaction volume (n = 3). Reactions were conducted at 37°C, and the plot represents the fluorescence signal normalized to the maximum intensity at t = 30 min. (B) Cis-cleavage characterization of Cas12a at different time points in 15% PAGE. The assay was conducted using 500 nM Cas12a, 500 nM BS-14 + 6/crRNA, and 500 nM mismatched dsDNA (rG-dT wobble mismatch) at 37°C, comparing the cleavage efficiency between the HEPSD system and the conventional CRISPR/Cas12a system. (C) Trans-cleavage response of HEPSD to the presence of PM and mismatched targets over 90 min of fluorescence measurement. A short TS-binding arm was paired with sequences containing 100% GC content to assess binding to PM targets and those forming rG-dT wobble mismatches. All experiments were repeated three times. (D–I) SNV discrimination containing wobble mismatches in the PAM-proximal (D–F) and PAM-distal (G–I) within a local 70% GC content. Kinetic profiles (E, H) and corresponding fluorescence endpoint values at t = 60 min (F, I) were measured. HEPSD performance was compared to the conventional CRISPR/Cas12a system. Error bars represent the mean ± SD from at least three independent replicates (n = 3). Statistical significance was assessed using unpaired two-tailed t-tests: ns: not significant, ****P < .0001.
As reported [55], elevated GC composition surrounding mismatches may promote robust activation. To validate the discrimination performance of HEPSD under generalized conditions, the local GC content was adjusted to match the high GC content near the wobble-mismatched nucleotide. As shown in Fig. 4D–I, BS-6 + 14 and BS-14 + 6 were designed to target SNVs within high-GC-content regions located in the PAM-proximal and PAM-distal regions, respectively. In these tests, the HEPSD targeting probe was fully complementary to the target sequence containing the SNV site, while the WT sequence harboured a single-nucleotide mismatch relative to the probe. Initial observations indicated that increasing GC content in both PAM-proximal and PAM-distal regions did not affect the recognition of the correct target by either the conventional Cas12a system or the HEPSD system. Despite these extreme conditions, the HEPSD system still exhibited brilliant discrimination ability, as evidenced by nearly no fluorescence increase on the trans-cleavage kinetics curve after 60 min for WT (Fig. 4E and H) and high discrimination factor compared with correct targets (Fig. 4F and I).
A major challenge in SNV detection is the intense background noise from the large excess of WT DNA strands. To systematically verify that the HEPSD system improves SNV discrimination in the presence of high concentrations of WT, wobble mismatched pairs in low-GC (30%) and high-GC (70%) regions within both the PAM-proximal and PAM-distal regions were examined. In a high-concentration WT background, the HEPSD system was able to detect low-abundance SNV sequences (Supplementary Figs S17 and S18), pushing the mutant abundance detection limit to 0.01% for mismatches in the PAM-proximal region. For mismatches in the PAM-distal region, with local GC content raised to 70%, HEPSD could detect mutations at frequencies as low as 0.1%. Through comprehensive validation, HEPSD exhibited outstanding detection performance, prompting our interest in evaluating its application in practical samples.
Profiling of BRAF V600E and EGFR L858R mutations by HEPSD
To evaluate the effectiveness of our optimized SNV detection method for clinically relevant cancer mutations, we selected BRAF V600E and EGFR L858R as target mutations. BRAF V600E is a highly prevalent driver mutation, accounting for ∼70%–80% of BRAF mutations [56]. This mutation is found in various cancer types, such as melanoma, papillary thyroid carcinoma, and glioma. The EGFR exon 21 L858R mutation is a key target in clinical oncology, especially in nonsmall cell lung cancer [57].
First, synthetic DNA targets were used to evaluate the sensitivity and specificity of the HEPSD system. Fluorescence intensity increased significantly with rising concentrations of mutant sequences, whereas WT sequences with single-base mismatches exhibited minimal fluorescence (Supplementary Figs S19 and S20). To better mimic clinically relevant conditions, we prepared synthetic DNA mixtures with varying VAFs to represent the coexistence of WT and mutant alleles in patient samples. The HEPSD system successfully detected BRAF V600E at VAFs as low as 0.005% and the high-GC-content EGFR L858R mutation at VAFs as low as 0.01% (Supplementary Fig. S21). Since the HEPSD system directly targets dsDNA, it is inherently compatible with nucleic acid amplification methods such as PCR. To confirm this compatibility, gradient dilutions of WT and mutant DNA templates were prepared and subjected to PCR amplification. The HEPSD system reliably detected mutant targets at template concentrations as low as 1 aM, with no detectable activation by WT amplification products (Supplementary Fig. S22). Additionally, EGFR L858R mutation and WT templates were mixed at varying VAFs and PCR amplified, demonstrating that HEPSD could robustly detect SNVs at VAFs as low as 0.01% (Supplementary Fig. S23). By combining direct dsDNA detection with strong resistance to sequence variations and base mutations, the HEPSD system achieves a detection limit comparable to existing methods while maintaining high specificity and sensitivity (Supplementary Table S2). These findings underscore its potential as a powerful tool for detecting low-abundance mutations in heterogeneous clinical samples.
We further assessed its performance using clinical samples. Genomic DNA was extracted using a commercial kit following standardized protocols, and its integrity was verified through PCR amplification and gel electrophoresis (Supplementary Fig. S26). Genomic DNA from negative controls, confirmed by NGS sequencing to be negative for both mutations, was used as a baseline reference (Supplementary Figs S24 and S25). As shown in Fig. 5, out of 104 tissue samples tested for BRAF V600E, 88 were positive and 16 were negative (Fig. 5A, Supplementary Fig. S27, and Supplementary Table S3). For EGFR L858R, 11 of the 20 tissue samples were positive for the mutation, while 9 negative samples (Fig. 5B, Supplementary Fig. S28, and Supplementary Table S4). The HEPSD system demonstrated a significantly higher signal for mutant alleles compared to WT alleles (Fig. 5C and D). In addition to tissue samples, eight plasma-derived cell-free DNA samples were analysed to assess the HEPSD system’s performance in detecting low-abundance mutations in liquid biopsies (Fig. 5E, Supplementary Fig. S29, and Supplementary Table S5). Given the inherently low total DNA quantity and VAF in plasma samples, a PCR-based SNV enrichment method, BDA [58], was employed before HEPSD analysis. Compared to NGS, the clinical sensitivity and specificity of HEPSD were both determined to be 100% for BRAF V600E and EGFR L858R (Fig. 5H and I, and Supplementary Tables S6–S8). The area under the curve (AUC) in the receiver operating characteristic (ROC) curve was 1.000 (Fig. 5J and K). To ensure the accuracy and reliability of critical mutation calls identified by NGS, targeted Sanger sequencing was performed on a subset of tissue samples harboring BRAF V600E or EGFR L858R variants (Supplementary Fig. S30). All samples were also tested using a real-time quantitative PCR-based method to analyze BRAF V600E and EGFR L858R mutations in the same set of gDNA samples (Fig. 5F and G, and Supplementary Figs S31–S33). qPCR detection of clinical samples yielded satisfactory AUC for tissue samples, with 0.999 for BRAF V600E and 1.000 for EGFR L858R, but showed limited performance in detecting mutations in plasma samples (Supplementary Fig. S34). It is worth noting that testing of the BRAF V600E samples using the HEPSD system and qPCR technique occurred several months after the initial NGS analysis. Variables such as DNA degradation, storage conditions, and elapsed time may have impacted the results. Despite these variables, the HEPSD system accurately identified mutation-positive samples, demonstrating its robustness and reliability. These results highlight the HEPSD system’s potential in clinical settings, emphasizing its value for accurate and reliable diagnostics in oncology.
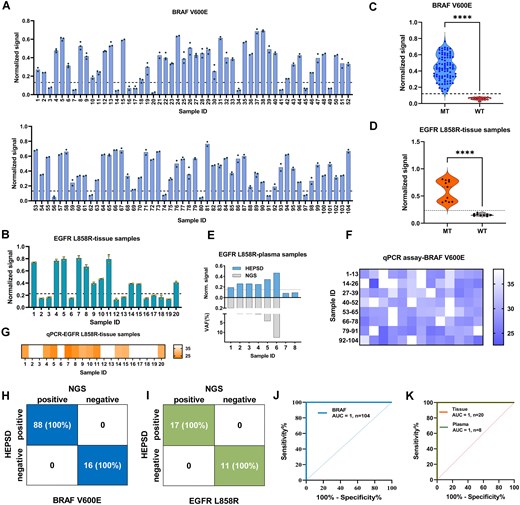
Clinical analysis of BRAF V600E and EGFR L858R samples. (A, B) Summary of test results for BRAF V600E using HEPSD system. The plot represents the fluorescence signal normalized to the maximum intensity at t = 60 min. (C, D) Normalized signal values for two clinical cohorts of BRAF (C) and EGFR (D) were analyzed using the HEPSD system. The BRAF V600E cohort included 88 positive mutant-type (MT) samples and 16 negative WT samples, while the EGFR L858R cohort comprised 11 positive MT samples and 9 negative WT samples. The normalized signal values for MT were significantly higher than those for WT (****P < .0001), Student’s t-test. (E) Test results for the analysis of L858R in eight clinical plasma samples using the HEPSD system and NGS. (F, G) Heatmap results of qPCR validation for BRAF V600E in 104 clinical tissue samples (F) and EGFR L858R in 20 clinical tissue samples (G). (H, I) Evaluation of HEPSD sensitivity and specificity for BRAF V600E (n = 104) (H) and EGFR L858R (n = 28) (I) using a confusion matrix compared with NGS. (J, K) ROC curve and AUC analysis for BRAF mutations in tissue samples (J) and for EGFR L858R in both tissue and plasma samples using the HEPSD system (K).
Conclusion
In conclusion, we developed a binary-spacer-assisted CRISPR/Cas12a platform to address key limitations of probe-hybridization and CRISPR/Cas-based methods in detecting challenging SNVs. This study provides a comprehensive thermodynamic framework for point mutation detection using an engineered Cas12a system. The modified spacer in crRNA exhibited minimal impact on Cas12a activation for correct targets. By employing mismatch-guided binary spacers, the method overcomes sequence constraints and mutation-type limitations, effectively enhancing mismatch-associated energetic penalties. The high mismatch discrimination in high-GC regions or those forming stable Watson–Crick-like mismatches was validated. Guided by these findings, SNVs in 104 BRAF V600E and 28 EGFR L858R clinical tissue and plasma samples were discriminated with 100% sensitivity and specificity. Moreover, the potential to address off-target effects in genome editing is promising, making it an attractive molecular tool for genomic DNA diagnostics and future biomedical applications.
Acknowledgements
Author contributions: Qiong Liu (Conceptualization, Data curation, Investigation, Formal analysis, Methodology, Validation, Visualization, Writing—original draft, Writing—review and editing), Zhen Zou and Ronghua Yang (Conceptualization, Funding acquisition, Resources, Formal analysis, Visualization, Supervision, Writing—original draft, Writing—review and editing), Zhou Jiang (Methodology, Resources, Validation, Data curation, Supervision, Formal analysis), Sheng Li (Methodology, Validation, Formal analysis), Yinfeng Li (Validation, Formal analysis), Yingfei Wan (Validation, Formal analysis), Zhenyu Hu (Validation, Formal analysis), and Shimeng Ma (Validation, Formal analysis).
Supplementary data
Supplementary data is available at NAR online.
Conflict of interest
the authors declare no conflict of interest.
Funding
This work was supported in part by the National Natural Science Foundation of China (22334005, 22474037), Natural Science Foundation of Hunan Province (2022JJ20038), the Scientific Research Fund of Hunan Provincial Education Department (23A0059), and the Key Project of Developmental Biology and Breeding from Hunan Normal University (2022XKQ0205).
Data availability
The data underlying this article are available in the article and in its online supplementary material.
References
Author notes
The first two authors should be regarded as Joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments