





Abstract
Sister chromatid cohesion is mediated by a conserved multiprotein complex called cohesin. The loading of cohesin onto chromosomes involves the RSC (remodels the structure of chromatin) chromatin remodeling complex. Here, we demonstrate that the fission yeast Phi1, a palmitoyl hydrolase inactive protein 1, serves to bridge the interaction between cohesin and the RSC complex. Phi1 interacts with Rad21 in cohesin and Snf21, the RSC complex ATPase, to promote chromosome loading of cohesin. The identified characteristic features of Phi1 are conserved in the human homologues Apt1 and Apt2, which interact with Rad21 and Brg1, the human homologue of Snf21, in an enzymatic-independent manner. Intriguingly, the cohesin–Apt1–Brg1 complex is upregulated in C4-2B prostate cancer cells, and co-depletion of Apt1 and Apt2 by small interfering RNA triggers mitotic catastrophe in these cells. In addition, Apt1 nuclear localization is associated with poor clinical outcomes in prostate cancer. These results suggest a pro-survival function against mitotic stress for the complex.

Introduction
The faithful passage of genetic information into daughter cells relies on accurate chromosome duplication and the subsequent equal segregation of sister chromatids at mitosis. During DNA replication in eukaryotes, the newly replicated sister chromatids become stably associated by cohesion from the time of their synthesis in the S phase until they are separated in anaphase. Cohesion is a prerequisite for the bipolar attachment of chromatid pairs to the spindle apparatus in mitosis [1]. Dissolution of cohesion initiates the poleward movement of chromosomes that enables the equal segregation of the duplicated genome to daughter cells long after DNA replication has occurred.
Sister chromatid cohesion is mediated by a conserved multiprotein complex called cohesin, which consists of three core components: two SMCs (structural maintenance of chromosome) and a kleisin subunit [2]. The two SMC subunits, Psm1Smc1 and Psm3Smc3 (in the protein nomenclature of the fission yeast Schizosaccharomyces pombe), form a stable heterodimer via hinge domains situated at one end of the long antiparallel coiled-coil domains. At the other ends of the SMC coiled-coil domains lie the ATPase heads. A kleisin subunit, Rad21Scc1, bridges the two ATPase heads to complete the cohesin ring. Additional HEAT-repeat subunits Psc3Scc3 and Pds5 contact the kleisin subunit to regulate the SMC–kleisin ring. At the metaphase-to-anaphase transition, cleavage of the Rad21Scc1 subunit of cohesin initiates the dissolution of cohesion, allowing sister chromatid separation [3]. In most organisms, cohesin protein complexes are enriched at centromeres. The presence of a specialized heterochromatin structure at the centromere mediated by the RNA interference (RNAi) pathway is vital for tight physical cohesion between sister centromeres to ensure accurate chromosome segregation [4].
Cohesin loading onto chromosomes requires a specialized cohesin loader, a second HEAT-repeat heterodimeric complex comprised of the Mis4Scc2 and Ssl3Scc4 subunits. In vitro, Mis4Scc2 alone is sufficient to promote cohesin loading onto DNA [5], whereas Ssl3Scc4 serves as an in vivo chromatin adaptor through interaction with the RSC (remodels the structure of chromatin) chromatin remodeling complex [6]. The function of the cohesin loader is not limited to facilitating cohesin loading. The RSC acts as a docking platform that recruits the cohesin loader via a direct interaction; the cohesin loader in turn feeds back onto the nucleosome landscape, suggesting a two-way functional crosstalk between primary chromatin structure and cohesin function [7]. In addition to the best-known function in chromatin structure, cohesins also play vital roles not only in regulating gene expression by connecting promoters with their distal enhancers but also in stabilizing replication forks, promoting DNA repair, and recruitment of checkpoint proteins [2].
Protein S-palmitoylation is a post-translational modification that involves a thioester linkage between palmitic acid and the cysteine residue of a protein [8]. S-palmitate is added by a family of transmembrane zinc finger DHHC (Asp–His–His–Cys) containing protein acyl transferases and is removed by palmitoyl hydrolase/fatty acyl protein thioesterases [9–11]. The reversible nature of S-palmitoylation enables fine-tuned regulation of protein functions. More importantly, the proper function of many membrane proteins, such as surface receptors and signaling proteins, requires palmitoylation-mediated partitioning into lipid rafts [12]. The functions of S-palmitoylation in telomere anchoring [13] and compartmentalized DNA repair [14] to the nuclear membrane have also been reported. In this study, we described a noncanonical function of palmitoyl hydrolase in sister chromatid cohesion. An enzymatic-independent function of palmitoyl hydrolase in loading cohesin onto chromosomes is demonstrated both in the fission yeast and in humans. Furthermore, we demonstrate that this function is required to suppress aberrant mitosis in prostate cancer cells and can be a potential prognostic indicator for poor clinical outcome.
Materials and methods
Fission yeast strains and methods
We constructed all strains according to standard procedures. Information of oligonucleotides for gene disruption or modification can be obtained upon request. The original snf21ts strain was a gift from Frank Uhlmann. A complete list of the strains used in this study is given in Table 1. Conditions for growth, maintenance, and genetic manipulation of fission yeast were as described previously [15]. Except otherwise stated, strains were grown at 30°C in yeast extract (YE) or Edinburgh Minimal Medium (EMM2) with appropriate supplements. Where necessary, gene expression from the nmt1 promoter was repressed by the addition of 15 μM thiamine to the culture medium. To inactive Snf21, the thermosensitive snf21-36 mutant was grown at the restrictive temperature of 36°C for 3 h.
Schizosaccharomycespombe strains used in this study
| Genotype | Source |
|---|---|
| h− 972 | Lab stock |
| h−pah1::hyh | This study |
| h−phi1::hyh | This study |
| h−akr1::hyh | Lab stock |
| h−erf2::hyh | Lab stock |
| h−erf4::hyh | Lab stock |
| h−pfa3::hyh | Lab stock |
| h−pfa5::hyh | Lab stock |
| h−cid12::ura4 leu1 ura4 | Lab stock |
| h−his2 ade6-M210 Ch16 (ade6-M216) | Lab stock |
| h−phi1::hyh ade6-M210 Ch16 (ade6-M216) | This study |
| h−hht2-GFP::ura4 ura4 | Lab stock |
| h−hht2-GFP::ura4 phi1::hyh ura4 | This study |
| h−mad2::ura4 ade6 leu1 ura4 | Lab stock |
| h−mad2::ura4 phi1::hyh ura4-D18 | This study |
| h−bub1::LEU2 ade6 leu1 ura4 his1 | Lab stock |
| h−bub1::LEU2 phi1::hyh leu1 | This study |
| h−rad21-GFP::LEU2 leu1 | Lab stock |
| h−rad21-GFP::LEU2 phi1::hyh leu1 | This study |
| h−nda3-KM311 leu1 ura4 cut3<<GFP (cut3::lacOp his7::lacI-GFP-NLS) | Lab stock |
| h−nda3-KM311 leu1 ura4 cut3<<GFP (cut3::lacOp his7::lacI-GFP-NLS) phi1::hyh | This study |
| h−otr1R (Sph1)::ade6 ade6 leu1 ura4 | Lab stock |
| h−otr1R (Sph1)::ade6 ade6 leu1 ura4 cid12::ura4 | Lab stock |
| h−otr1R (Sph1)::ade6 ade6 leu1 ura4 phi1::hyh | This study |
| h−GFP-swi6 (p81-GFP-swi6::LEU2) ade6 leu1 ura4 | Lab stock |
| h−GFP-swi6 (p81-GFP-swi6::LEU2) phi1::hyh ade6 leu1 ura4 | This study |
| h−GFP-phi1 (phi1<<kanr-nmt1-GFP-phi1) pREP1-GST leu1 | This study |
| h−GFP-phi1 pREP1-phi1-GST leu1 | This study |
| h−GFP-phi1 pREP1-phi1-N-GST leu1 | This study |
| h−GFP-phi1 pREP1-phi1-C-GST leu1 | This study |
| h−pREP1-phi1-6xHis leu1 | This study |
| h−GFP-phi1 Snf21-Flag::kanr | This study |
| h−pREP1-phi1-GST leu1 | This study |
| h−Snf21-GFP::kanr | This study |
| h−Snf21-GFP::kanr phi1::hyh | This study |
| h−GFP-phi1 | This study |
| h−GFP-phi1 snf21ts | This study |
| h−Snf21-Flag::kanr pREP1-GFP leu1 | This study |
| h−Snf21-Flag::kanr rad21-GFP::LEU2 leu1 | This study |
| h−Snf21-Flag::kanr rad21-GFP::LEU2 phi1::hyh leu1 | This study |
| h−mis4-HA::LEU2 leu1 | Lab stock |
| h−mis4-HA::LEU2 phi1::hyh leu1 | This study |
| h−ssl3-GFP::kanr | This study |
| h−ssl3-GFP::kanr phi1::hyh | This study |
| h−pREP1-Apt1-his leu1 | This study |
| h−pREP1-Apt2-his leu1 | This study |
| h−pREP1-Apt1S119A-his leu1 | This study |
| h−pREP1-Apt2S122A-his leu1 | This study |
| h−pREP1-Rad21-GST leu1 | This study |
| h−pREP2-Apt1-Flag ura4 | This study |
| h−pREP2-Apt2-Flag ura4 | This study |
| Genotype | Source |
|---|---|
| h− 972 | Lab stock |
| h−pah1::hyh | This study |
| h−phi1::hyh | This study |
| h−akr1::hyh | Lab stock |
| h−erf2::hyh | Lab stock |
| h−erf4::hyh | Lab stock |
| h−pfa3::hyh | Lab stock |
| h−pfa5::hyh | Lab stock |
| h−cid12::ura4 leu1 ura4 | Lab stock |
| h−his2 ade6-M210 Ch16 (ade6-M216) | Lab stock |
| h−phi1::hyh ade6-M210 Ch16 (ade6-M216) | This study |
| h−hht2-GFP::ura4 ura4 | Lab stock |
| h−hht2-GFP::ura4 phi1::hyh ura4 | This study |
| h−mad2::ura4 ade6 leu1 ura4 | Lab stock |
| h−mad2::ura4 phi1::hyh ura4-D18 | This study |
| h−bub1::LEU2 ade6 leu1 ura4 his1 | Lab stock |
| h−bub1::LEU2 phi1::hyh leu1 | This study |
| h−rad21-GFP::LEU2 leu1 | Lab stock |
| h−rad21-GFP::LEU2 phi1::hyh leu1 | This study |
| h−nda3-KM311 leu1 ura4 cut3<<GFP (cut3::lacOp his7::lacI-GFP-NLS) | Lab stock |
| h−nda3-KM311 leu1 ura4 cut3<<GFP (cut3::lacOp his7::lacI-GFP-NLS) phi1::hyh | This study |
| h−otr1R (Sph1)::ade6 ade6 leu1 ura4 | Lab stock |
| h−otr1R (Sph1)::ade6 ade6 leu1 ura4 cid12::ura4 | Lab stock |
| h−otr1R (Sph1)::ade6 ade6 leu1 ura4 phi1::hyh | This study |
| h−GFP-swi6 (p81-GFP-swi6::LEU2) ade6 leu1 ura4 | Lab stock |
| h−GFP-swi6 (p81-GFP-swi6::LEU2) phi1::hyh ade6 leu1 ura4 | This study |
| h−GFP-phi1 (phi1<<kanr-nmt1-GFP-phi1) pREP1-GST leu1 | This study |
| h−GFP-phi1 pREP1-phi1-GST leu1 | This study |
| h−GFP-phi1 pREP1-phi1-N-GST leu1 | This study |
| h−GFP-phi1 pREP1-phi1-C-GST leu1 | This study |
| h−pREP1-phi1-6xHis leu1 | This study |
| h−GFP-phi1 Snf21-Flag::kanr | This study |
| h−pREP1-phi1-GST leu1 | This study |
| h−Snf21-GFP::kanr | This study |
| h−Snf21-GFP::kanr phi1::hyh | This study |
| h−GFP-phi1 | This study |
| h−GFP-phi1 snf21ts | This study |
| h−Snf21-Flag::kanr pREP1-GFP leu1 | This study |
| h−Snf21-Flag::kanr rad21-GFP::LEU2 leu1 | This study |
| h−Snf21-Flag::kanr rad21-GFP::LEU2 phi1::hyh leu1 | This study |
| h−mis4-HA::LEU2 leu1 | Lab stock |
| h−mis4-HA::LEU2 phi1::hyh leu1 | This study |
| h−ssl3-GFP::kanr | This study |
| h−ssl3-GFP::kanr phi1::hyh | This study |
| h−pREP1-Apt1-his leu1 | This study |
| h−pREP1-Apt2-his leu1 | This study |
| h−pREP1-Apt1S119A-his leu1 | This study |
| h−pREP1-Apt2S122A-his leu1 | This study |
| h−pREP1-Rad21-GST leu1 | This study |
| h−pREP2-Apt1-Flag ura4 | This study |
| h−pREP2-Apt2-Flag ura4 | This study |
Schizosaccharomycespombe strains used in this study
| Genotype | Source |
|---|---|
| h− 972 | Lab stock |
| h−pah1::hyh | This study |
| h−phi1::hyh | This study |
| h−akr1::hyh | Lab stock |
| h−erf2::hyh | Lab stock |
| h−erf4::hyh | Lab stock |
| h−pfa3::hyh | Lab stock |
| h−pfa5::hyh | Lab stock |
| h−cid12::ura4 leu1 ura4 | Lab stock |
| h−his2 ade6-M210 Ch16 (ade6-M216) | Lab stock |
| h−phi1::hyh ade6-M210 Ch16 (ade6-M216) | This study |
| h−hht2-GFP::ura4 ura4 | Lab stock |
| h−hht2-GFP::ura4 phi1::hyh ura4 | This study |
| h−mad2::ura4 ade6 leu1 ura4 | Lab stock |
| h−mad2::ura4 phi1::hyh ura4-D18 | This study |
| h−bub1::LEU2 ade6 leu1 ura4 his1 | Lab stock |
| h−bub1::LEU2 phi1::hyh leu1 | This study |
| h−rad21-GFP::LEU2 leu1 | Lab stock |
| h−rad21-GFP::LEU2 phi1::hyh leu1 | This study |
| h−nda3-KM311 leu1 ura4 cut3<<GFP (cut3::lacOp his7::lacI-GFP-NLS) | Lab stock |
| h−nda3-KM311 leu1 ura4 cut3<<GFP (cut3::lacOp his7::lacI-GFP-NLS) phi1::hyh | This study |
| h−otr1R (Sph1)::ade6 ade6 leu1 ura4 | Lab stock |
| h−otr1R (Sph1)::ade6 ade6 leu1 ura4 cid12::ura4 | Lab stock |
| h−otr1R (Sph1)::ade6 ade6 leu1 ura4 phi1::hyh | This study |
| h−GFP-swi6 (p81-GFP-swi6::LEU2) ade6 leu1 ura4 | Lab stock |
| h−GFP-swi6 (p81-GFP-swi6::LEU2) phi1::hyh ade6 leu1 ura4 | This study |
| h−GFP-phi1 (phi1<<kanr-nmt1-GFP-phi1) pREP1-GST leu1 | This study |
| h−GFP-phi1 pREP1-phi1-GST leu1 | This study |
| h−GFP-phi1 pREP1-phi1-N-GST leu1 | This study |
| h−GFP-phi1 pREP1-phi1-C-GST leu1 | This study |
| h−pREP1-phi1-6xHis leu1 | This study |
| h−GFP-phi1 Snf21-Flag::kanr | This study |
| h−pREP1-phi1-GST leu1 | This study |
| h−Snf21-GFP::kanr | This study |
| h−Snf21-GFP::kanr phi1::hyh | This study |
| h−GFP-phi1 | This study |
| h−GFP-phi1 snf21ts | This study |
| h−Snf21-Flag::kanr pREP1-GFP leu1 | This study |
| h−Snf21-Flag::kanr rad21-GFP::LEU2 leu1 | This study |
| h−Snf21-Flag::kanr rad21-GFP::LEU2 phi1::hyh leu1 | This study |
| h−mis4-HA::LEU2 leu1 | Lab stock |
| h−mis4-HA::LEU2 phi1::hyh leu1 | This study |
| h−ssl3-GFP::kanr | This study |
| h−ssl3-GFP::kanr phi1::hyh | This study |
| h−pREP1-Apt1-his leu1 | This study |
| h−pREP1-Apt2-his leu1 | This study |
| h−pREP1-Apt1S119A-his leu1 | This study |
| h−pREP1-Apt2S122A-his leu1 | This study |
| h−pREP1-Rad21-GST leu1 | This study |
| h−pREP2-Apt1-Flag ura4 | This study |
| h−pREP2-Apt2-Flag ura4 | This study |
| Genotype | Source |
|---|---|
| h− 972 | Lab stock |
| h−pah1::hyh | This study |
| h−phi1::hyh | This study |
| h−akr1::hyh | Lab stock |
| h−erf2::hyh | Lab stock |
| h−erf4::hyh | Lab stock |
| h−pfa3::hyh | Lab stock |
| h−pfa5::hyh | Lab stock |
| h−cid12::ura4 leu1 ura4 | Lab stock |
| h−his2 ade6-M210 Ch16 (ade6-M216) | Lab stock |
| h−phi1::hyh ade6-M210 Ch16 (ade6-M216) | This study |
| h−hht2-GFP::ura4 ura4 | Lab stock |
| h−hht2-GFP::ura4 phi1::hyh ura4 | This study |
| h−mad2::ura4 ade6 leu1 ura4 | Lab stock |
| h−mad2::ura4 phi1::hyh ura4-D18 | This study |
| h−bub1::LEU2 ade6 leu1 ura4 his1 | Lab stock |
| h−bub1::LEU2 phi1::hyh leu1 | This study |
| h−rad21-GFP::LEU2 leu1 | Lab stock |
| h−rad21-GFP::LEU2 phi1::hyh leu1 | This study |
| h−nda3-KM311 leu1 ura4 cut3<<GFP (cut3::lacOp his7::lacI-GFP-NLS) | Lab stock |
| h−nda3-KM311 leu1 ura4 cut3<<GFP (cut3::lacOp his7::lacI-GFP-NLS) phi1::hyh | This study |
| h−otr1R (Sph1)::ade6 ade6 leu1 ura4 | Lab stock |
| h−otr1R (Sph1)::ade6 ade6 leu1 ura4 cid12::ura4 | Lab stock |
| h−otr1R (Sph1)::ade6 ade6 leu1 ura4 phi1::hyh | This study |
| h−GFP-swi6 (p81-GFP-swi6::LEU2) ade6 leu1 ura4 | Lab stock |
| h−GFP-swi6 (p81-GFP-swi6::LEU2) phi1::hyh ade6 leu1 ura4 | This study |
| h−GFP-phi1 (phi1<<kanr-nmt1-GFP-phi1) pREP1-GST leu1 | This study |
| h−GFP-phi1 pREP1-phi1-GST leu1 | This study |
| h−GFP-phi1 pREP1-phi1-N-GST leu1 | This study |
| h−GFP-phi1 pREP1-phi1-C-GST leu1 | This study |
| h−pREP1-phi1-6xHis leu1 | This study |
| h−GFP-phi1 Snf21-Flag::kanr | This study |
| h−pREP1-phi1-GST leu1 | This study |
| h−Snf21-GFP::kanr | This study |
| h−Snf21-GFP::kanr phi1::hyh | This study |
| h−GFP-phi1 | This study |
| h−GFP-phi1 snf21ts | This study |
| h−Snf21-Flag::kanr pREP1-GFP leu1 | This study |
| h−Snf21-Flag::kanr rad21-GFP::LEU2 leu1 | This study |
| h−Snf21-Flag::kanr rad21-GFP::LEU2 phi1::hyh leu1 | This study |
| h−mis4-HA::LEU2 leu1 | Lab stock |
| h−mis4-HA::LEU2 phi1::hyh leu1 | This study |
| h−ssl3-GFP::kanr | This study |
| h−ssl3-GFP::kanr phi1::hyh | This study |
| h−pREP1-Apt1-his leu1 | This study |
| h−pREP1-Apt2-his leu1 | This study |
| h−pREP1-Apt1S119A-his leu1 | This study |
| h−pREP1-Apt2S122A-his leu1 | This study |
| h−pREP1-Rad21-GST leu1 | This study |
| h−pREP2-Apt1-Flag ura4 | This study |
| h−pREP2-Apt2-Flag ura4 | This study |
Plasmid construction and recombinant protein
Plasmids for the expression of S. pombe Phi1 and Rad21, and human Apt1, Apt2, and Rad21 were constructed by polymerase chain reaction (PCR) amplification of the corresponding open reading frame from complementary DNA (cDNA) and subsequently ligated into plasmids derived from pREP1 or pREP2 with a green fluorescent protein (GFP), GST, His, and Flag-tag inserted at the 3′ end of the multiple cloning sites. The Apt1 (S119A) and Apt2 (S122A) mutants were created using PCR-based site-directed mutagenesis and verified by DNA sequencing. The GST-tagged fusion proteins from yeast were purified by Glutathione Sepharose™ 4B (170756-01, GE Healthcare) according to the manufacturer’s recommendations. The levels of purification and protein stability are shown in Supplementary Fig. S1.
Cell culture and small interference RNA experiments
Human RWPE-1 (ATCC-CRL-11609), PC-3 (ATCC-CRL-1435), and C4-2B (ATCC-CRL-3315) prostate cells were cultured in RPMI medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (FCS) and 100 IU/ml penicillin, 0.1 mg/ml streptomycin, and maintained at 37°C in a humidified incubator containing 5% CO2 in air. For transfection studies, the human embryonic kidney cell line HEK293T cells (ATCC-CRL-3216) (1 × 106) were plated and cultured in a p-100 dish in 10% FCS serum in Dulbecco’s modified Eagle’s medium (DMEM) at 37°C for 24 h and transfected with 4 μg of pEGFP-APT1 and pEGFP-APT2 or pEGFP empty vector using 10 μl of Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Cells were incubated at 37°C for an additional 2 days, and whole-cell extract was subjected to immunoprecipitation and western blotting.
The ON-TARGET plus SMARTpool siRNA targeting human APT1 (L-010007), APT2 (L-009256), and nontargeting pool scramble small interfering RNA (siRNA) (D-001810) were purchased from Thermo Scientific,Dharmacon (Illkirch, France). 5 × 105 cells plated and cultured in a p-60 dish in RPMI medium (10% FCS serum) at 37°C for 24 h were transfected with siRNA (200 nM final concentration) or control siRNA in RPMI using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) at 37°C for 4 h. One-third volume of RPMI (30% FCS) was added and cells were incubated at 37°C for an additional 2 days before use. Cell viability after introduction of siRNA was assessed by the XTT-1 cell proliferation kit (Biological Industries/Sartorius) according to the manufacturer’s protocol.
Antibodies and immunoprecipitation
The antibodies against GFP (AE012), APT1 (A4419), APT2 (A15792), and Rad21 (A18850) were purchased from ABclonal Technology. Anti-phospho H2A.X-Ser139 (9718S) antibody was purchased from Cell Signaling Technology. Anti-HA (GTX115044) was purchased from GeneTex, Irvine, CA, USA. Anti-Brg1 (sc-17796) was purchased from Santa Cruz Biotechnology. Anti-His (YH-8003) and anti-GST (YH-8002) were purchased from Yao-Hong Biotechnology Inc. (New Taipei City, Taiwan). The anti-Flag M2 antibody was purchased from Sigma–Aldrich (Saint Louis, MO, USA). Antibody against α-tubulin (Sigma–Aldrich, Saint Louis, MO, USA) was used as a control.
For immunoprecipitation from yeast extracts, 2 × 108 yeast cells were lysed in NP-40 lysis buffer (6 mM Na2HPO4, 4 mM NaH2PO4, 1% NP-40, 150 mM NaCl, 2 mM EDTA, 50 mM NaF, 0.1 mM Na3VO4, 1 mM phenylmethanesulfonylfluoride (PMSF), 1 mM dithiothreitol (DTT), complete protease inhibitor cocktail) by vortexing with acid-washed glass beads. The lysate was clarified by centrifugation, and GFP fusion proteins were retrieved using GFP-Trap-coupled agarose beads (Chromotek, Martinsried, Germany) following the manufacturer’s instructions. His-tagged proteins from yeast were purified using Ni-NTA agarose (Qiagen, Hilden, Germany).
Microscopy
Visualization of GFP in living cells was performed at room temperature. Images acquired from a Leica DM RA2 microscope equipped with a Leica DC 350F camera were assembled using Adobe Photoshop. Visualization of Hht2-GFP in living cells embedded in 0.6% LMP agarose was performed at room temperature as previously described [4].
GFP protein identification
The purified proteins were separated on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gels and visualized by Sypro Ruby (Invitrogen, Carlsbad, CA, USA). Protein bands were excised from gels, treated with trypsin, and subjected to analysis for protein identification using the Waters NanoAcquity UPLC-Synapt G2 HDM instrument by the Core Facilities for Proteomics at National Health Research Institutes. Data were processed for database searching using ProteinLynx Global Server (PLGS) against the Swiss-Prot database with the Mascot program.
GST pull-down assay
The human recombinant proteins Rad21-GST combined with APT1-His, APT2-His, APT1-S119A-His, and APT-S122A-His (1 μg) were incubated with 50 μl of 50% Glutathione Sepharose 4B resin (GE Healthcare) at 4°C for 24 h in a binding buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 2 mM EDTA, 1% NP-40, 10% glycerol, protease inhibitors, and 1 mM PMSF. After three washes with the binding buffer, the samples were subjected to SDS–PAGE and western blotting analysis.
Immunoprecipitation using GFP-Trap
GFP-tag proteins (GFP-Phi1, Snf21-GFP, and Rad21-GFP) were extracted from yeasts and immunoprecipitated using GFP-Trap agarose beads in NP-40 lysis buffer incubated at 4°C by rotating for 4 h. After three washes using the lysis buffer, the GFP-Trap agarose beads were incubated with 1 μg GST protein (Phil-GST, Phil-N-GST, and Phil-C-GST) in lysis buffer at 4°C by rotating overnight. The beads were washed three times with lysis buffer and subjected to SDS–PAGE and western blotting analysis.
Acyl-resin-assisted capture assay
The palmitoylation assay was conducted based on the acyl resin-assisted capture (acyl-RAC) assay described previously [15]. Cell lysate extracted from 3 × 109S. pombe cells in ice-cold lysis buffer [phosphate buffered saline (PBS), pH 7.4, 0.5 mM EDTA, protease inhibitor (PI), and 1 mM phenylmethylsulfonyl fluoride] was incubated with 1% Triton X-100 and 25 mM N-ethylmaleimide (NEM) for 1 h. The protein pellet was collected by chloroform–methanol (CM) precipitation, which may be stored at −80°C for up to 2 weeks for the following assay.
For acyl-RAC, the protein pellet was dissolved in a small volume of 4% SDS buffer (4% SDS, 50 mM Tris/HCl, pH 7.4, 5 mM EDTA) incubated at 37°C to improve dissolution before blocking free cysteines with 20 mM NEM in LB buffer (150 mM NaCl, 50 mM Tris/HCl, pH 7.4, 5 mM EDTA) overnight. Three sequential CM precipitations were processed to remove excessive NEM and the sample was resolved in LB buffer with 0.2% SDS before treatment with neutral 1 M hydroxylamine (HAM) to cleave the thioester bond between cysteine residues and a fatty acid moiety. Thirty microliters of thiopropyl resin (G-Biosciences) was added to every tube and incubated for 1–2 h at room temperature. The beads were washed five times with washing buffer (LB buffer with 0.1% Triton X-100) to remove unbound proteins, and the palmitoylated proteins were eluded in an SDS sample buffer with DTT detected by western blotting.
ChIP and RT-PCR analysis
Chromatin immunoprecipitation (ChIP) was performed as described previously [4]. For immunoprecipitation, GFP-Trap or anti-HA antibody with protein G beads (GE Healthcare) was used and then PCR amplified with primer pairs specific for the centromeric dh repeat (GAAAACACATCGTTGTCTTCAGAG and CGTCTTGTA GCTGCATGTGAA) and for fbp1 (AATGACAATTCCCCACT AGCC and ACTT CAGCTAGGATTCACCTGG).
The cDNA synthesis and reverse transcription (RT) PCR analysis was performed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems), according to the manufacturer’s instructions. Primer pairs specific for human Brg1 (GCAGCAGAC AGACGAGTAC and ATCCCTTGTAAGACAC), Rad21 (ATGTTCTACGCACAT TTTGTTC and TATAATATGGAACCTTGGTC), SMC1 (GTATGACAAGCGAAA GAAGG and CATCTCCTTCTCCAGCTCAT), SMC3 (TCCTATGATCAGCAAAC TGA and ACTGTCTGTTTTCCTGCTGA), APT1 (ATGGCCGGCAATAACATGT CAACCCC and TCACTTGTCATCGTCATCCTTGTA), APT2 (TGCGGCAATAA CATGTC and GCGACAGGAGGCAGCAGCTTC), and GAPDH (AGAAGGCTGGGG CTCATTTG and TTGGCAGGTTTTTCTAGACG) were used. The messenger RNA level of each gene was normalized to that of GAPDH in the same sample.
Quantitative real-time PCR
Chromosome DNA, together with specific primers, was mixed with KAPA SYBR® FAST qPCR Master Mix (2X) (Sigma–Aldrich) following the supplier’s protocol. The DNA copy numbers were determined by real-time quantitative PCR (qPCR) using the QuantStudio 3 Real-Time PCR (QS3) (Thermo Fisher). Primer pairs specific for cut3 (GCTTGACGTTTCTGCTTGTGAT and GGCCTCAACTTCCTTGTCCA TA), dh (TGCTGTCAGCTCACTCAAGTCCAA and AGGACTAAGCCCAAGCAC CGTATT), and rDNA (GGACGGTGGCCATGGAA and CATTCGGCCGGTGAGTTG) were used.
Metaphase chromosome spreads
C4-2B cells transfected with siAPT1 and siAPT2 or negative control siRNA using Lipofectamine 2000 were incubated for 48 h. Before harvesting, colchicine (0.02 μg/ml final concentration) was added to the medium for 16 h. The cells were harvested by trypsin and washed twice in ice-cold PBS before resuspending the pellet with pre-warmed (37°C) 75 mM KCl and incubated for 30 min at 37°C. After centrifugation for 10 min at 200 × g to remove the supernatants, cell pellets were fixed in 10 ml fixative solution (methanol:glacial acetic acid 3:1) for 1 h and washed once with fixative solution before resuspending in a small volume of fixative solution. Few drops of cell suspensions were dropped onto clean, cold slides and allowed to air dry before staining with DAPI.
Tissue microarray immunohistochemistry
Prostate cancer tissue microarray (MPR1005sur) with survival data, including TNM and clinical stage, was purchased from TissurArray.com, containing 90 cases of prostate cancer, 10 cases of normal prostate hyperplasia tissue, and single cores per case. Immunohistochemistry (IHC) was performed according to standard protocol. In brief, the slide was deparaffinized at 60°C for 1 h, rehydrated, and then antigen unmasking was performed using citrate buffer (pH 6.0) for 30 min at 95°C. The slide was incubated with 3% of hydrogen peroxide for 5 min to block endogenous peroxidase activity and was incubated with 2.5% goat serum for 1 h to block nonspecific binding. Rabbit-anti-APT1 (LYPLA1) (ABclonal A4419, 1:150) primary antibody diluted in 2.5% goat serum was incubated on the slide in a humidified chamber overnight at 4°C. The slide was washed three times with 1× PBS buffer for 5 min each and then incubated with anti-rabbit-HRP antibody (Vector Labs MP-7451, 1:200) for 1 h. After the wash steps, the signal of IHC was developed with a DAB kit (Dako) with the following counterstain, dehydration, mounting, and slide scanning. APT1 nuclear localization and intensity were defined according to 0 and 1 (weak), 2 and 3 (strong).
Results
Fission yeast Phi1, a palmitoyl hydrolase inactive protein
A role of protein S-palmitoylation in budding yeast heterochromatin formation has recently been described [13]. We set out to determine the potential roles of S-palmitoylation in chromatin organization by characterizing the fission yeast palmitoylation enzymes. In addition to the five palmitoyl acyltransferases Akr1, Erf2-Erf4, Pfa3, Pfa5, and Swf1 encoded in the S. pombe genome [15], there are five genes referred to as phi1 (palmitoyl hydrolase inactive protein 1 for the reason explained below) (SPAC9G1.08c), pah1 (palmitoyl hydrolase 1) (SPAC8E11.04c), pah2 (SPAC22H12.03), bem46, and pdf1 annotated with the term palmitoyl hydrolase activity, catalysis of a hydrolase reaction that removes a palmitoyl moiety from some substrates (Fig. 1A) (PomBase genomic data) [16]. In a genome-wide Orfeome localization study [17], Phi1 and Pah1 were found to be enriched in the nucleus, suggesting a potential nuclear function, whereas Bem46, Pdf1, and Pah2 were associated with ER and mitochondria, respectively. All proteins contained the characteristic Alpha/Beta hydrolase fold domain structure of the family. However, despite the structural similarity, the triple Ser, Asp, and His residues of the fatty acyl protein thioesterase active site as indicated in Pah1 (PomBase AlphaFold structure predictions) were not conserved in Phi1 protein (Fig. 1B), suggesting that Phi1 might be inactive for the palmitoyl hydrolase activity. In support of this idea, in contrast to the elevated level of S-palmitoylated proteins observed in the pah1 mutant as determined by acyl-RAC assay [15], no significant changes in the global level of S-palmitoylated proteins were observed when phi1 was deleted (Fig. 1C). Although we could not rule out that Phi1 might have activity toward a subset of substrates or under specific conditions, the lack of an active site suggested that Phi1 was an inactive palmitoyl hydrolase.
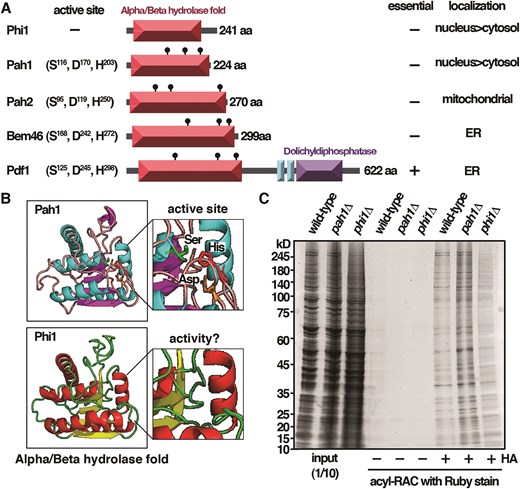
Fission yeast Phi1, a palmitoyl hydrolase inactive protein. (A) Schematic representation of the domain structure of palmitoyl hydrolases in S. pombe. (B) AlphaFold structure of Pah1 and Phi1. (C) Samples from acyl-RAC experiments were subjected to immunoblotting with Ruby stain.
Phi1 mutant is defective in chromosome segregation
The lack of enzymatic activity of Phi1 prompted us to investigate the biological function of this interesting protein. In the laboratory, we routinely used the spindle poison thiabendazole (TBZ), which interfered with the progress of mitosis, as a sensitized condition to determine nuclear function of candidate proteins in chromosome segregation, such as Cid12 [4], shown in the lower panel of Fig. 2A. Among Phi1, Pah1, and the four nonessential S-palmitoytransferases (Akr1, Erf2-Erf4, Pfa3, and Pfa5), only phi1 mutant similar to cid12 was hypersensitive to TBZ, suggesting a potential function of Phi1 in chromosome segregation. Experiments were performed to determine whether there was any chromosome segregation defect in the phi1 mutant.

Phi1 mutant is defective in chromosome segregation. (A) Ten-fold serial dilutions of strains indicated were spotted onto YES agar containing 15 μg TBZ or no drug control. Plates were photographed after 3–5 days of incubation at 30°C. (B) Rates of mini-chromosome loss were calculated for phi1, cid12, and pdc5 strains and expressed as mean chromosome loss per division (500 cells were scored, average of three independent clones). Asterisks indicated statistical significance (P < .05, t-test unpaired) versus wild type. (C) Fluorescence micrographs showing lagging chromosomes in living phi1 cells. Scale bar, 5 μm. Percentage of phi1 cells with lagging chromosomes in late anaphase was given (200 cells were scored, average of three independent clones). (D) Time-lapse imaging of Hht2-GFP was performed at 4-min intervals with the phi1 strain. Scale bar, 5 μm. (E) Ten-fold serial dilutions of strains indicated were spotted onto YES. Plates were photographed after 3 days of incubation at 30°C. Doubling time in liquid cultures and survival rates (plating efficiency: 500 cells were scored, average of three independent clones) were calculated for each strain as indicated.
We found that deletion of phi1 led to an increased chromosome segregation failure rate by the use of a standard mini-chromosome assay with strains containing the nonessential ade6-M216-marked Ch16 mini-chromosome derivative of chromosome 3 [4] (Fig. 2B). The ade6-M216 allele complemented an unlinked ade6-M210 marker in these strains such that they remained ade+ as long as the mini-chromosome was maintained. As shown in the left panel of the upper figure, red colonies, indicative of the loss of the ade6-M216-marked Ch16 mini-chromosome, were readily detectable in the phi1 mutant. Like those of the nonessential cohesin pds5 and silencing cid12 mutants, quantitative measurements indicated that the phi1 mutant lost the mini-chromosome at least 17-fold more frequently than the control strain (lower panel).
In line with the elevated rate of mini-chromosome loss, as shown in Fig. 2C using Hht2 (histone H3) GFP fusion protein as the nuclear marker, the phi1 mutant had a significantly high percentage of cells with unequal segregation or lagging chromosomes (Fig. 2D) (42% binucleated cells) that were not seen in the wild-type cells. Segregation defects at the frequency seen could account for the elevated rate of mini-chromosome loss in phi1 mutant cells (Fig. 2B).
Taken together, the results described above suggested a function of Phi1 in chromosome segregation. Nonetheless, deletion of phi1 was viable, indicating that the spindle checkpoint might be activated in these cells as in the case of cid12 [4]. To address the point, we determined the genetic interaction between phi1 and the spindle checkpoint genes mad2 and bub1. In support of the idea, we observed a synthetic growth defect in the double mutants. This was reflected in the prolonged generation time and the poor survival rates of the double mutant cells (Fig. 2E). These results suggested that the spindle checkpoint was required to prevent potential chromosome segregation defects in phi1 mutant cells for their survival. Experiments were performed to determine the underlying molecular mechanism of Phi1 in chromosome segregation.
Centromeric cohesion, but not gene silencing, is defective in phi1 mutant
One possible explanation for the mitotic defects in phi1 cells would be that, in the absence of Phi1, some aspect of centromere function was defective in a significant proportion of cells. To test whether the observed chromosome segregation defects were in part due to defects in centromeric cohesion, we performed a ChIP assay (Fig. 3A) analyzed by qPCR (Fig. 3B) with strains carrying an epitope-tagged version of the cohesin subunit Rad21 [4]. As shown in Fig. 3A, Rad21 was preferentially enriched at the centromeric dh repeats, but not on the open reading frame of the fbp1 gene as determined by a CHIP analysis. We observed a considerable reduction in its localization at the centromeres of the phi1 cells (Fig. 3B). Similar results were observed for the other region of chromosomes, including cut3 locus at the arm of chromosome II and rDNA at the end of chromosome III. These results suggested that, not only was it limited to the centromere regions, but Phil1 was also required for the general loading of cohesion onto chromosomes. In line with these results, fluorescence microscopy revealed that Rad21-GFP foci were significantly reduced in phi1 cells (24% as compared to 78% in control cells, Fig. 3C). These results suggested that phi1 mutant was defective in the recruitment of cohesin to chromosomes, which might affect chromosome segregation during mitosis.
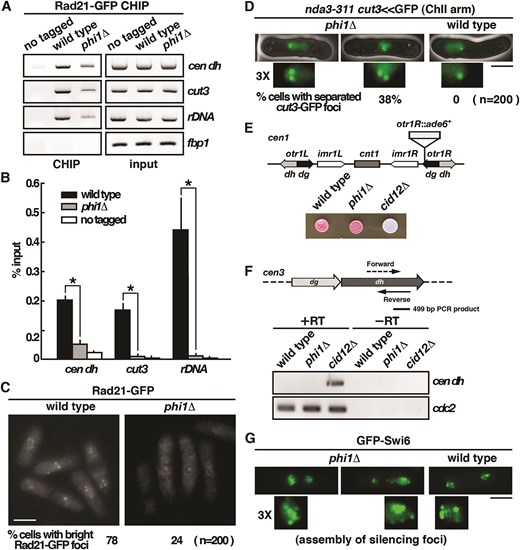
Centromeric cohesion, but not gene silencing, is defective in phi1 mutant. (A) ChIP analysis of Rad21-GFP in phi1 mutant using GFP-Trap affinity resin. As a control, wild-type cells in which Rad21 was not tagged were used. (B) The DNA recovered from the IP was amplified using qPCR and SYBR Green with primers specific to the genes indicated normalized to that of the fbp1 gene. Relative enrichment (% input) was calculated. Data were expressed as means ± standard deviation in triplicate. Asterisks indicated statistical significance (P < .05, t-test unpaired) versus wild type. (C) Fluorescence micrographs showing Rad21-GFP in living phi1 cells. Percentage of cells with bright Rad21-GFP foci was given (200 cells were scored, average of three independent clones). Scale bar, 5 μm. (D) Fluorescence micrographs showing cut3<<GFP localization at metaphase in nda3-311 phi1 cells after 8-h incubation at 20°C. Scale bar, 5 μm. Percentage of cells with separated GFP signals associated with the chromosome 2 arm at the cut3 locus was shown (200 cells were scored, average of three independent clones). (E) Strains indicated, all with an otr1R-inseted ade6+ as shown, were spotted onto a YE agar plate and photographed after 3 days of incubation. (F) Schematic representation of a portion of centromere III indicating the location of fragments amplified. Total RNA from cultures of the indicated strains was reverse transcribed using random primers and then PCR amplified with primer pairs specific for the dh repeat and cdc2 (normalizer gene). A non-reverse-transcribed negative control was included (−RT). (G) Fluorescence micrographs showing GFP-Swi6 in living phi1 cells. Scale bar, 5 μm.
Next, we further examined the cohesion function in phi1 cells by monitoring GFP associated with the chromosome arm at the cut3 locus (cut3-GFP, which was located at the middle of the left arm of chromosome 2). Cells were arrested in metaphase by inactivation of nda3 [4]. As shown in Fig. 3D, a single cut3-GFP signal was detected in the mitotically arrested nda3-KM311 cells, whereas in many phi1 nda3-KM311 cells (38%), cut3-GFP signals were clearly separated, forming two distinct spots in cells. These results suggested that Phi1 was required for sister chromatid cohesion in establishing bipolar attachments to the spindle microtubules and hence for accurate chromosome segregation.
The presence of a specialized heterochromatin structure at centromeres was vital for tight physical cohesion between sister centromeres to ensure accurate chromosome segregation. Proteins such as Cid12 in the RNAi pathway have been implicated in heterochromatin assembly and are critical for accurate chromosome segregation [4]. Cells lacking Cid12 failed to initiate heterochromatin assembly and were defective in centromeric gene silencing and chromosome segregation. To determine whether the chromosome segregation defects in phi1 cells could be due to defects in heterochromatin assembly as in the cid12 mutant, we determined the effect of phi1 deletion on centromeric gene silencing. As shown in Fig. 3E, we found that deletion of cid12 abolished silencing of the reporter gene ade6 located at the outermost centromeric repeat region otr1R and produced white colonies on plates limiting for adenine, whereas wild-type and phi1 cells efficiently silenced otr1R::ade6 expression, and the colonies turned red due to accumulation of intermediate metabolite of adenine. These results suggested that centromeric gene silencing was not affected by the deletion of phi1.
This was further explored by analysis of the centromeric transcripts with total cellular RNA extracted from the phi1 cells, followed by RT-PCR analysis. As shown in Fig. 3F, pericentromeric transcripts were quickly converted into siRNA by the RNAi machinery and were barely detectable in the wild-type cells. Conversely, these transcripts became accumulated in mutants defective in RNAi machinery, such as cid12. We found that, like the wild-type cells, the pericentromeric transcripts were barely detectable in phi1 cells, suggesting that the centromeric genes were silenced. In line with these results, despite the chromosome segregation defects, the GFP-Swi6 silencing foci could still form in the phi1 cells (Fig. 3G). Together, these results suggested that the chromosome segregation defects in the phi1 cells were not due to defects in the heterochromatin structure at the centromeres for enrichment of cohesin loading.
Phi1 dimerized and interacted with Snf21 to associate with centromeres
Next, we characterized the fission yeast Phi1 protein. Apt1, the homologue of Phi1 in high eukaryotes, dimerized in humans [18] and formed tetramers in plants [19]. In an in vitro binding assay, experiments were performed to determine whether Phi1 self-interacted. As shown in Fig. 4A, purified recombinant Phi1 specifically interacted with itself mediated through its N-terminal 1–127 amino acids, but not with the GST control in the pull-down experiment. In line with these results, recombinant Phi1 protein appeared as a dimer in native gel and moved as a monomer when separated in SDS–PAGE (Fig. 4B). Together, these results suggested that Phi1 normally functioned as a dimer.
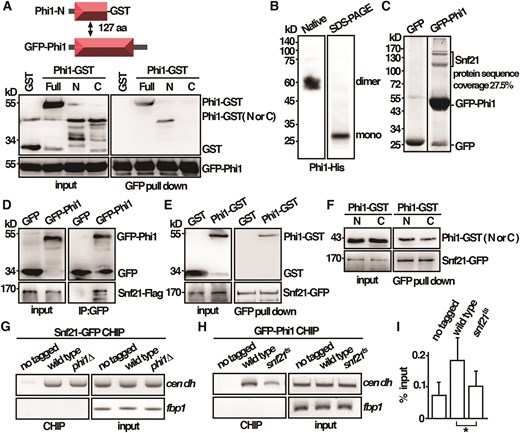
Phi1 dimerized and interacted with Snf21 to associate with centromeres. (A) Purified full-length GFP-Phi1 and GST-tagged N (1–127 amino acids) and C (128–241 amino acids) terminal half of the Phi1 proteins from yeast cells were subjected to a GFP pull-down assay followed by western immunoblotting analysis with antibodies against GST and GFP. (B) Western blot analysis of Phi1–His fusion proteins separated on native and SDS–PAGE gel detected by antibodies against His. (C) GFP-Trap pull down of GFP-Phi1 proteins resolved by SDS–PAGE was visualized by SYPRO Ruby staining. The identities of the constituent proteins identified by Matrix-assisted laser desorption/ionization tandem mass spectrometry (MALDI-MS/MS) analysis were indicated. (D) Coimmunoprecipitation was performed with extracts prepared from Snf21-Flag tagged strains expressing GFP-Phi1 or GFP control proteins. GFP-Trap affinity resin was used to pull down GFP proteins. Immunoprecipitation was then analyzed by western immunoblotting with antibodies against GFP and Flag. (E) Purified full-length Phi1-GST recombinant protein and Snf21-GFP isolated from yeast cells were subjected to a GFP pull-down assay followed by western immunoblotting analysis with antibodies against GST and GFP. (F) Purified GST-tagged N (1–127 amino acids) and C (128–241 amino acids) terminal half of the Phi1 proteins and Snf21-GFP isolated from yeast cells were subjected to a GFP pull-down assay followed by western immunoblotting analysis with antibodies against GST and GFP. ChIP analysis of Snf21-GFP in phi1 mutant (G) and GFP-Phi1 in snf21ts mutant (H) using GFP-Trap affinity resin. As a control, wild-type cells in which target proteins were not tagged were used. (I) The DNA recovered from the IP in H was amplified using qPCR and SYBR Green with primers specific to the dh centromeric repeats normalized to that of the fbp1 gene. Relative enrichment (% input) of dh centromeric repeats was calculated. Data were expressed as means ± standard deviation in triplicate. Asterisks indicated statistical significance (P < .05, t-test unpaired) versus wild type.
To gain more insight into the molecular mechanism of Phi1 in chromosome segregation, a GFP-Trap pull-down experiment was performed to identify proteins associated with Phi1, and their identities were determined by MALDI-MS/MS analysis (Fig. 4C). In addition to Phi1 proteins, three peptides of Snf21 (the full-length size top one and two degradation products), an ATPase of the RSC complex [20], with 27.5% protein sequence coverage, were recovered in the lysate. The interaction between Phi1 and Snf21 was further confirmed by immunoprecipitation with the Snf21-Flag strain. As shown in Fig. 4D, Snf21 specifically interacted with Phi1, but not with the GFP control, suggesting that these proteins were in a stable complex. To assess whether the interaction was direct, an in vitro binding assay was performed. As shown in Fig. 4E and F, purified recombinant Phi1 specifically interacted with Snf21 through both of its N (1–127 amino acids) and C (128–241 amino acids) terminals, but not with the GST control in the pull-down experiment.
The interaction between Phi1 and Snf21 was of particular interest to us given that a function of Snf21 together with the cohesin loader in establishing sister chromatid cohesion had recently been suggested [20]. Phi1 might interact with Snf21 to associate with the centromere for cohesin loading. In line with the idea, as shown in Fig. 4G, Snf21 was preferentially enriched at the centromeric dh repeats, but not on the open reading frame of the fbp1 gene as determined by CHIP analysis. Similarly, Phi1 was associated with centromeres (Fig. 4H) in an Snf21-dependent manner, as the association of Phi1 was reduced in the thermosensitive snf21-36 mutant at its restrictive temperature of 36°C (Fig. 4I). Conversely, the association of Snf21 with centromeres was not affected by phi1 deletion (Fig. 4G). Together, these results suggested that Phi1 interacted with Snf21 to function in the centromere.
Phi1 bridges the interaction between cohesin and RSC complex
The results described above suggested a role of Phi1 together with Snf21 in centromere function. We hypothesized that Phi1 might function as an adapter to link cohesin to the Snf21–RSC complex for chromosome loading. In support of this idea, the interaction between Rad21 and Snf21, as demonstrated by immunoprecipitation, was compromised in phi1 mutant (Fig. 5A). Furthermore, we showed that Phi1 physically interacted with Rad21 in the pull-down experiment. As shown in Fig. 5B and C, purified recombinant Phi1 specifically interacted with Rad21 mediated through its N-terminal (1–127 amino acids), but not with the GST control. Rad21 stably interacted with Smc1 and Smc3. The results supported the conclusion that Phi1 interacted with cohesin. Together, these results suggested that Phi1 interacted with cohesin Rad21 and Snf21 ATPase of the RSC complex to bridge the interaction between these two complexes.
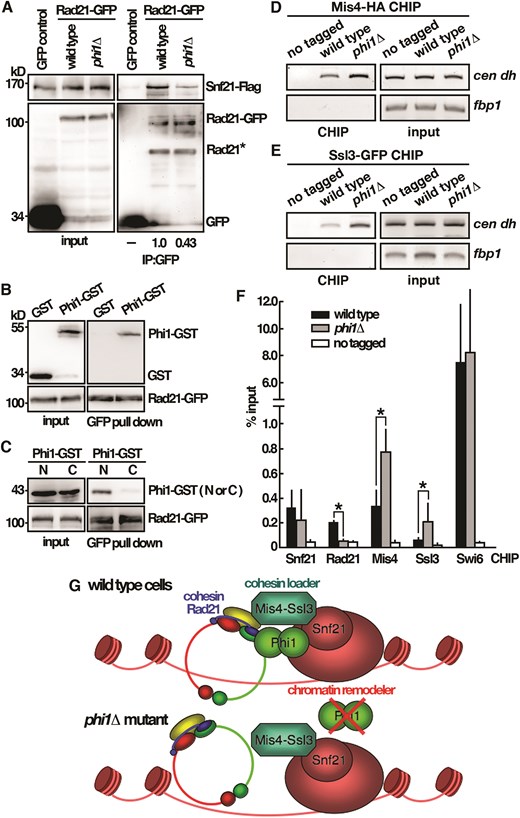
Phi1 bridges the interaction between cohesin and RSC complex. (A) Coimmunoprecipitation was performed with extracts prepared from Snf21-Flag tagged strains expressing Rad21-GFP or GFP control proteins. GFP-Trap affinity resin was used to pull down GFP proteins. Immunoprecipitation was then analyzed by western immunoblotting with antibodies against GFP and Flag. Rad21* indicated degradation products. (B) Purified full-length Phi1-GST recombinant protein and Rad21-GFP isolated from yeast cells were subjected to a GFP pull-down assay followed by western immunoblotting analysis with antibodies against GST and GFP. (C) Purified GST-tagged N (1–127 amino acids) and C (128–241 amino acids) terminal half of the Phi1 proteins and Rad21-GFP isolated from yeast cells were subjected to a GFP pull-down assay followed by western immunoblotting analysis with antibodies against GST and GFP. ChIP analysis of Mis4-HA (D) and Ssl3-GFP (E) in phi1 mutant using GFP-Trap affinity resin. As a control, wild-type cells in which target proteins were not tagged were used. (F) The DNA recovered from the IP with target proteins indicated was amplified using qPCR and SYBR Green with primers specific to the dh centromeric repeats normalized to that of the fbp1 gene. Relative enrichment (% input) of dh centromeric repeats was calculated. Data were expressed as means ± standard deviation in triplicate. Asterisks indicated statistical significance (P < .05, t-test unpaired) versus wild type. (G) Schematic representation of the Phi1 function in cohesin loading onto chromosome.
In Saccharomycescerevisiae, RSC acts as a chromatin receptor that recruits Scc2–Scc4 by a direct protein interaction with Sth1, the budding yeast homologue of Snf21, for cohesin loading [21]. The Scc4 and Sth1 interaction region FEDWF motif is conserved in Snf21 protein. Similarly, Snf21 physically interacts with Ssl3, the fission yeast orthologue of Scc4, in S. pombe [20]. To gain more insight into the function of Phi1 in cohesin loading, experiments were performed to determine the effect of deletion of phi1on the centromere association of Mis4 (Fig. 5D), the fission yeast orthologue of Scc2, and Ssl3 (Fig. 5E) by ChIP analysis analyzed by qPCR (Fig. 5F). In agreement with the centromeric silencing results, the association of Swi6 with centromeres, unlike Rad21, was not affected by the deletion of phi1. Intriguingly, in contrast to the reduced level of Rad21 associated with centromeres, in the absence of Phi1, a significant increase in the levels of Mis4 and Ssl3 associated with centromeres was observed in phi1 cells. These results suggested a function of Phi1 together with Mis4–Ssl3 for chromosome loading in a stepwise fashion. In the absence of Phi1, the loading of cohesin was compromised, and the loader of Mis4–Ssl3 was accumulated on centromeres (Fig. 5G). Based on these results, we hypothesized that Phi1 functioned in parallel with cohesin loader to promote chromosome loading.
The identified characteristic features of Phi1 are conserved in human homologue
Next, we wanted to see whether the identified characteristic features of Phi1 were evolutionarily conserved in humans. Unlike Phi1, the most closely related human homologues Apt1 and Apt2 both contained the conserved triple Ser, Asp, and His residues (Fig. 6A) and were positive for the palmitoyl hydrolase activity [22]. Experiments were performed to determine whether Apt1 and Apt2 interacted with Brg1, the human homologue of Snf21, and Rad21 in cohesin. Human HEK293T cells expressing GFP-tagged Apt1 or Apt2 were subjected to immunoprecipitation using specific monoclonal antibodies against Brg1 and Rad21. The presence of Apt1 and Apt2 in the precipitates was determined by an anti-GFP antibody. As shown in Fig. 6B, both Apt1 and Apt2 were able to coprecipitate with Brg1 and Rad21. Mutations of the active site that conserved serine residue to alanine in Apt1 and Apt2 did not affect the interaction with Brg1 and Rad21 suggesting that the interaction does not require enzyme activity.
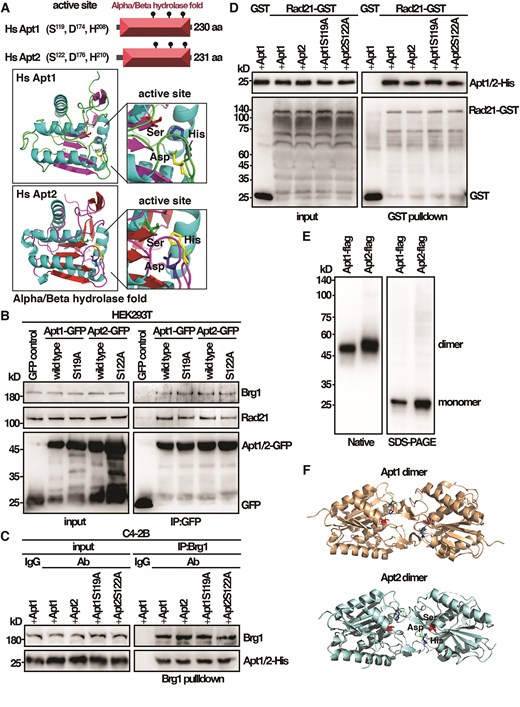
The identified characteristic features of Phi1 are conserved in human Apt1 and Apt2. (A) Schematic representation of the domain and AlphaFold-predicted structures of Apt1 and Apt2. (B) Coimmunoprecipitation was performed with extracts prepared from human HEK293T cells expressing Apt1 and Apt2 tagged with GFP. Immunoprecipitates were analyzed by western immunoblotting analysis using antibodies specific for Brg1, Rad21, and GFP. (C) Purified His-tagged Apt1 and Apt2 wild-type and mutant proteins from yeast and Brg1 proteins isolated from human C4-2B cells were subjected to a pull-down assay followed by western immunoblotting analysis with antibodies against His and Brg1. (D) Purified His-tagged Apt1 and Apt2 wild-type and mutant proteins were subjected to GST pull-down assay with GST-tagged human Rad21 from yeast followed by western immunoblotting analysis with antibodies against His and GST. (E) Western blot analysis of purified Apt1 and Apt2 Flag fusion proteins from yeast separated on native and SDS–PAGE gel detected by antibodies against Flag. (F) Schematic representation of Apt1 and Apt2 dimer structures.
To assess whether the interaction was direct, we performed an in vitro binding assay using purified His-tagged Apt1 and Apt2 wild-type and mutant proteins from yeast to interact with Brg1 proteins isolated from human C4-2B cells and GST-tagged human Rad21 from yeast. As shown in Fig. 6C and D, both Apt1 and Apt2 specifically interacted with Brg1 and Rad21, but not with the IgG and GST control. Similar results were obtained with the mutant Apt1 and Apt2 proteins that lost the enzyme activity. Together, these results suggested that similar to the inactive Phi1, Apt1 and Apt2 interacted with Brg1 and Rad21 in an enzymatic-independent manner.
The other line of evidence that Apt1 and Apt2 might function independently of their enzyme activity came from the native gel analysis of the Apt1 and Apt2 proteins. Like Phi1, Apt1 and Apt2 moved in a native gel twice the size than they separated in SDS–PAGE, suggesting a dimeric configuration (Fig. 6E). The crystal structures of Apt1 and Apt2 revealed a dimer with the active site buried in the dimer interface (Fig. 6F) [18]. Consequently, the access to the active site was restricted in the dimer to allow a substrate to reach the nucleophilic serine for its enzymatic function.
Co-depletion of Apt1 and Apt2 triggered chromosome segregation defects in C4-2B prostate cancer cells
Rad21 was located at the frequently amplified genomic 8q24 locus [23]. Similarly, Brg1 and Apt1 were also overexpressed in human prostate cancer [24]. To gain more insight into the function of the cohesin–Apt1–Brg1 complex and its role in tumorigenesis, we characterized these proteins in human prostate cancer cell lines. For this purpose, PC3 and C4-2B cancer cells were chosen because of their aggressive phenotype. We found that the RNA levels of Brg1 and Apt1 were upregulated in prostate cancer PC3 and C4-2B cells as compared with the normal prostate cells, RWPE1 (Fig. 7A). In addition, the core components of cohesin, Rad21, Smc1, and Smc3 RNA levels were also increased in C4-2B cells. Western blot analysis confirmed the upregulation of these proteins (Fig. 7B). Further to the increased protein level, immunofluorescence microscopy revealed a strong nuclear localization of Apt1 in PC3 and C4-2B cells (Fig. 7C). These results suggested a nuclear role of Apt1 with cohesin in these cells. Experiments were performed to explore their functions.

Rad21–Apt1–Brg1 complex was overexpressed in human prostate cancer. (A) Total RNA from cultures of human cells indicated was reverse transcribed using random primers and then PCR amplified with primer pairs specific for the genes of interest. The amount of RNA relative to the RWPE1 cells was expressed as means ± standard deviation in triplicate. Asterisks indicated statistical significance (P < .05, t-test unpaired) versus RWPE1 cells. (B) Total lysates of human cells indicated were subjected to western immunoblotting analysis with indicated antibodies. (C) Fluorescence micrographs showing Apt1 localization in cells indicated. Scale bar, 5 μm.
Cancer cells were constantly under the pressure of mitotic stress due to their difficult-to-replicate complicated genome and high metabolic oxidative rate, which both generated DNA damage to impair the progress of mitosis [25]. The upregulation of cohesin and nuclear Apt1 in C4-2B cells might contribute to improved stress adaptation. This was explored by downregulation of Apt1 and Apt2 by siRNA. As shown in Fig. 8A, we found that simultaneous depletion of Apt1 and Apt2 by siRNA led to growth defects in C4-2B cancer cells, and to a lesser extent, for the normal prostate RWPE1 and PC3 cancer cells. Silencing either one of Apt1 or Apt2 did not have a significant effect, suggesting that they had redundant functions and required co-depletion to give effects. Furthermore, co-treatment of the Apt1 inhibitor ML348 and the Apt2 inhibitor ML349 at the concentration of 10 μM (50 times over the concentration of their IC50) to inhibit the enzyme activity did not adversely affect the growth of C4-2B cells, suggesting that the lethality was not due to the presence of the Apt1 and Apt2 enzyme activity, and was most likely caused by the defects of the cohesin-related function identified in this study. We did not observe a dramatic effect of depletion of Apt1 and Apt2 in normal prostate RWPE1 cells, as in the case of yeast, which could be due to the functional redundancy of other pathways, such as the structure of heterochromatin and a surveillance system like a spindle checkpoint to safeguard the structure, whereas PC3 cancer cells might be less sensitive and not additive to the pathway.
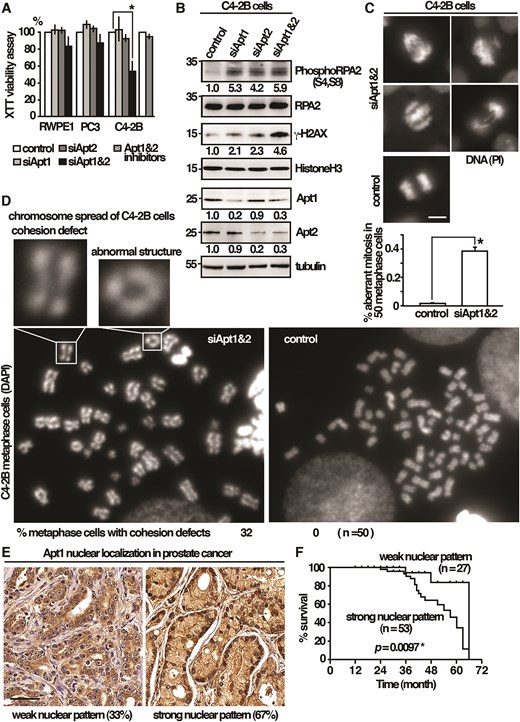
A pro-survival function of Apt1 and Apt2 against mitotic stress in cancer cells. (A) Human cells indicated were subjected to the XTT cell viability assay. Data were expressed as means ± standard deviation in triplicate. Asterisks indicated statistical significance (P < .05, t-test unpaired) versus control. (B) Total lysates of human cells indicated were subjected to western immunoblotting analysis with indicated antibodies. The relative level of protein of interest was indicated beneath each lane. Average of three independent experiments. (C) Fluorescence micrographs showing chromosome segregation defects in C4-2B cells co-depleted of Apt1 and Apt2. Scale bar, 5 μm. Percentage of cells with aberrant mitosis in late anaphase was given (n= 50) in triplicate. (D) Metaphase chromosome spreading assay of C4-2B cells co-depleted Apt1 and Apt2. Examples of premature separation of sister chromatids and abnormal ring-like chromosome structures are shown as indicated. (E) Representative Apt1 immunohistochemical staining of prostate cancer specimens (MPR1005sur prostate tissue microarray, TissurArray.com). Scale bar, 50 μm. (F) Kaplan–Meier curve showed the association between overall survival and Apt1 nuclear localization in 80 patients with malignant prostate cancer. Statistical significance was calculated using the log-rank test.
In addition to the canonical function in mitosis, cohesins also play vital roles in stabilizing the replication fork and promoting DNA repair [23]. The defects of the Apt1- and Apt2-related cohesin functions might affect DNA replication, leading to DNA damage to impair the progress of mitosis, which could be detrimental to certain cancer cells. In support of this idea, depletion of Apt1 or Apt2 induced phosphorylation of the replication stress marker RPA2 [26] and histone variant H2AX, a marker for DNA damage of double-strand breaks (Fig. 8B). Co-depletion of Apt1 and Apt2 further increased the levels of RPA2 and H2AX phosphorylation. These results suggested an overlapping function between Apt1 and Apt2. The lethality caused by DNA damage only occurred when both Apt1 and Apt2 were compromised to impair mitosis. In line with these results, aberrant mitosis, including chromosome unequal segregation, nondisjunction, and chromosome bridges, was significantly enriched in C4-2B cells when Apt1 and Apt2 were co-depleted (38% anaphase cells as compared to 2% in control cells, Fig. 8C). In support of a function in sister chromatid cohesion, premature separation of sister chromatids in Apt1 and Apt2 co-depleted C4-2B cells was revealed by metaphase chromosome spreading assay (32% metaphase cells) that was not seen in the control cells (Fig. 8D). Chromosomes with aberrant ring-like structures as a consequence of defective DNA repair [27] could also be found at low frequency (1% of chromosomes analyzed).
The results described above suggested a pro-survival function of Apt1 and Apt2 in C4-2B cells. We reasoned that the upregulation of Apt1 in cancer cells might be associated with a high mortality rate in clinical outcomes. To this end, we examined the protein expression of Apt1 in prostate cancer by IHC using commercially available tissue microarrays. In keeping with the results described above, a strong nuclear pattern of Apt1 was observed in a large proportion of samples (53/80) (Fig. 8E), which was associated with a significantly shorter overall survival (P= .0097) (Fig. 8F). These results suggested that strong nuclear localization of Apt1 could be a prognostic biomarker of clinical outcome.
Discussion
Palmitoyl hydrolase functions in cohesin loading onto chromosome
Protein S-palmitoylation enzymes palmitoylacyltransferases and palmitoyl hydrolase/fatty acyl protein thioesterases regulate diverse biological functions, including chromatin organization [13]. In this study, we described a cohesin-related function of palmitoyl hydrolase in S. pombe and humans. In the absence of Phi1, the responsible protein for the function in S. pombe, the recruitment of cohesin to centromeres was defective (Fig. 3). Consequently, in phi1 mutants, sister chromatids were precociously separated, leading to lagging chromosomes and activation of the spindle checkpoint during mitosis (Fig. 2).
Intriguingly, despite the structure similarity, unlike its human counterpart, Phi1 was inactive for the palmitoyl hydrolase activity (Fig. 1), suggesting that the cohesin-related function of Phi1 did not involve proteins modified by S-palmitoylation. In line with this idea, our study suggested that Phi1 was a part of the cohesin loading machinery (Fig. 5) and might have evolved to function specialized in this aspect, losing its enzyme activity. Cohesin loading onto chromosomes required the cohesin loader. The RSC acts as a docking platform that recruits the cohesin loader via a direct interaction. Our results suggested that Phi1 acted as an adaptor to bridge the interaction between cohesin Rad21 and the Snf21–RSC complex functioning together with the cohesin loader for chromosome loading in a stepwise fashion. In the absence of Phi1, the loading of cohesin was compromised, and the loader accumulated on centromeres.
Furthermore, we found that the identified characteristic features of Phi1 were conserved in its human homologues (Fig. 6). Similar to Phi1, Apt1 and Apt2 interacted with Brg1, the human homologue of Snf21, and Rad21 in the nucleus that does not require the enzyme activity. In support of an enzymatic independent function, the crystal structures of Apt1 and Apt2 revealed an inactive dimer conformation in which the active site was occluded with the dimer interface [18]. Little was known about the dimer and nuclear function of Apt1 and Apt2. It was tempting to speculate that cohesin loading might be the primary function of Apt1 and Apt2 dimers, which did not require the enzymatic activity, to provide a more stable space for protein–protein interaction. When performing its enzymatic function, the dimer would need to dissociate prior to interacting with its substrate. In line with the idea, Wnt5 signaling-mediated phosphorylation reduced Apt1 dimerization to increase Apt1 depalmitoylating activity [28]. Given the important role of protein S-palmitoylation and cohesin-related function in tumorigenesis, understanding how cells regulate and coordinate these two functions might help to create novel therapies against cancer.
A pro-survival function of Apt1 and Apt2 against mitotic stress in cancer cells
The results described above suggested a function of palmitoyl hydrolase in cohesin loading onto the chromosome. Considering the role of cohesin in a broad range of cellular processes that were hot spots in tumors, it was not surprising to see that Rad21, an important component in the cohesin complex, was among the most frequently dysregulated targets in human cancers. Amplification of Rad21 had been widely reported in human tumors (The Human Protein Atlas). Similarly, Apt1 and Brg1 were overexpressed in various cancers. Little was known about the cause and consequences of overexpressing these components in tumorigenesis. In this study, we identified a pro-survival function of Apt1 and Apt2 against mitotic stress in cancer cells through interaction with cohesin and chromatin remodelers that had not been described before. We found that prostate cancer C4-2B cells upregulated cohesin–Apt1–Brg1 complex to suppress mitotic catastrophe as co-depletion of Apt1 and Apt2 triggered aberrant mitosis in these cells. Given the vital roles of cohesin in DNA replication and repair, the mitotic catastrophe could be a consequence of defects in these processes due to their complicated genome and high metabolic oxidative rate. In support of this idea, a gain of rad21 copy number due to trisomy 8 was found to reduce replicative stress and support the growth of Ewing sarcoma cells [23]. Along this line, nuclear localization of elevated Apt1 in prostate cancer was associated with a high mortality rate in clinical outcomes, which could be attributed to the cohesin-related function of Apt1. Targeting cohesin and loader was an underexplored area of drug development, and our study might provide a strategy for such an approach.
Acknowledgements
We thank Y. Herry Sun for funding support and Frank Uhlmann for yeast strain.
Author contributions: Yi-Ting Wang (Investigation), Wan-Yi Hsiao (Investigation), Thanh-Vy Pham (Investigation), Bo-Ru Huang (Investigation), Shu-Dan Yeh (Supervision, Writing—review & editing), En Chi Hsu (Investigation, Writing—review & editing), and Shao-Win Wang (Conceptualization, Investigation, Writing—original draft, Writing—review & editing)
Supplementary data
Supplementary data is available at NAR online.
Conflict of interest
None declared.
Funding
This work was supported by the National Health Research Institutes (MG-112-PP-07) and National Science and Technology Council (113-2311-B-400-003) Taiwan. Thanh-Vy Pham was supported by NHRI-NCU Graduate Student Program. Funding to pay the Open Access publication charges for this article was provided by National Health Research Institutes.
Data availability
The data underlying this article are available in the article and in its online supplementary material.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments