





Abstract
Non-homologous end joining (NHEJ) is the main repair pathway for DNA double-strand breaks (DSBs) in cells with limited 5′ resection. To better understand how overhang polarity of chromosomal DSBs affects NHEJ, we made site-specific 5′-overhanging DSBs (5′ DSBs) in yeast using an optimized zinc finger nuclease at an efficiency that approached HO-induced 3′ DSB formation. When controlled for the extent of DSB formation, repair monitoring suggested that chromosomal 5′ DSBs were rejoined more efficiently than 3′ DSBs, consistent with a robust recruitment of NHEJ proteins to 5′ DSBs. Ligation-mediated qPCR revealed that Mre11-Rad50-Xrs2 rapidly modified 5′ DSBs and facilitated protection of 3′ DSBs, likely through recognition of overhang polarity by the Mre11 nuclease. Next-generation sequencing revealed that NHEJ at 5′ DSBs had a higher mutation frequency, and validated the differential requirement of Pol4 polymerase at 3′ and 5′ DSBs. The end processing enzyme Tdp1 did not impact joining fidelity at chromosomal 5′ DSBs as in previous plasmid studies, although Tdp1 was recruited to only 5′ DSBs in a Ku-independent manner. These results suggest distinct DSB handling based on overhang polarity that impacts NHEJ kinetics and fidelity through differential recruitment and action of DSB modifying enzymes.
INTRODUCTION
DNA double-strand breaks (DSBs) are highly cytotoxic DNA lesions that threaten the integrity of the genome and can trigger detrimental chromosomal rearrangements (1). However, DSBs are also essential substrates for normal cell activities such as meiotic and V(D)J recombination (2), processes with naturally diverse end structures.
DSBs are repaired by two major pathways. Homologous recombination (HR) repairs DSBs through use of an intact homologous donor sequence. HR and its subpathways are initiated by 5′ to 3′ resection of one strand of DNA by various endo/exonucleases (e.g. Mre11, Exo1 and Dna2 in yeast) to facilitate a homology search by the preserved and exposed 3′-terminated strand (3). In cells with limited or no resection, non-homologous end joining (NHEJ), which directly rejoins two ends of the broken DNA, is the dominant DSB repair pathway (4). The conserved actions of NHEJ in different kingdoms can be organized into three steps, using yeast NHEJ as an example: (i) rapid DSB binding by the Yku70/Yku80 (Ku) heterodimer, which recruits other repair factors and protects the DSBs from degradation, (ii) limited modification of the DSB ends, e.g. by the nuclease complex MRX (Mre1l-Rad50-Xrs2) and polymerase Pol4, a necessary step when direct religation of the ends is impossible and (iii) ligation by the specialized NHEJ ligase, DNA ligase IV (Dnl4-Lif1-Nej1). Notably, although these steps can be described in a logical order, the NHEJ reaction is likely dynamic with proteins rapidly binding and dissociating from the DSB, similar to other pathways like nucleotide excision repair (5).
Physiological and pathological DSBs often harbor diverse end structures, such as various combinations of overhang polarity, sequence, length and base lesions. The nature of NHEJ predicts that repair progression and fidelity will be affected by these local features of the two rejoining DSB ends, which demand a variety of enzymatic activities to resolve, e.g. nuclease cleavage, polymerization, phosphorylation and ligation (4). Previous studies of diverse end structures have mainly used in vitro approaches with recombinant proteins and cell-free extracts due to the ease of generating defined DSBs (6–8). In vivo rejoining of extrachromosomal DSBs can be studied by transforming cells with linearized plasmids (9). We reported a yeast system to study polymerase requirements of NHEJ using linearized plasmids ligated with various end structures (10,11). A similar approach was also used in recent studies of NHEJ end processing and ligation fidelity in human cell lines (12,13). These previous studies revealed a strong influence of DSB end structure on NHEJ in terms of repair protein usage, efficiency and accuracy, but the lack of an efficient system to generate diverse site-specific DSBs in chromosomes has limited further study. Engineered endonucleases such as zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) can generate chromosomal 5′-overhanging DSBs (5′ DSBs) at target genomic sites via dimerization of two FokI DNA cleavage domains (14). Although both ZFNs and TALENs have been used for genome editing in various organisms and cell types (14), their relatively slow cleavage in cells has impeded their application in studies of NHEJ reaction progression.
In this study, we investigate the impact of DSB overhang polarity and sequence content on chromosomal NHEJ by developing an efficient inducible system to generate site-specific 5′ DSBs in the yeast genome using an optimized ZFN. A variety of assays were used to compare the processing and rejoining of these 5′ DSBs to 3′ DSBs induced by the mega-endonuclease HO at the same locus. Results revealed differences in the cellular response to 5′ and 3′ DSBs, including very efficient recruitment of NHEJ factors to 5′ DSBs and an associated higher joining efficiency. This behavior was further associated with a more aggressive modification of 5′ DSB ends and more frequent mutations in repair joints. These and other observations demonstrate that overhang polarity can substantially impact NHEJ kinetics and fidelity through the differential recruitment and action of DSB modifying enzymes such as MRX, Pol4 and Tdp1.
MATERIALS AND METHODS
Yeast growth and manipulation
Haploid yeast strains (Supplementary Table S1) used in this study were isogenic derivatives of BY4741 (15). Gene disruptions and modified alleles were made using a polymerase chain reaction (PCR)-mediated technique (15) or a URA3 pop-in/pop-out method (16) and confirmed by PCR and sequencing. The chromosomal ZFN cut sites were made by replacing the 24-bp core HO recognition site in the ILV1 promoter in a previously described strain (17), with a new 24-bp sequence, consisting of two 9-bp ZFN recognition sites flanking a central 6-bp cut site. The recognition sites for the four ZFNs used are: 5′-GTTGGTGCT for StickyC.ZFNIII; 5′-GGGGAAGAA for QQR.ZFN; 5′-GAAGATGGT for GFP.ZFN1; 5′-GACGACGGC for GFP.ZFN2. The central cut sites for the selected GFP.ZFN2 were: 5′-GGATCC for ZFNcs(GATC); 5′-AAACAG for ZFNcs(AACA); 5′-TGAGAT for ZFNcs(GAGA). Codon optimization was done by GeneArt gene synthesis from Life Technologies (Supplementary File). Chromosomal integration of the GAL1-V10prm::GFP.ZFN2(CO) expression cassette was made by knocking out the native GAL1 locus with a LEU2 marker, and then replacing LEU2 with a PCR product carrying GAL1-V10prm::GFP.ZFN2(CO) and a URA3 maker. Yeast were grown at 30°C in either rich medium containing 1% yeast extract, 2% peptone and 40 μg/ml adenine (YPA) or synthetic defined (SD) medium with either 2% glucose, 2% galactose or 3% glycerol as the carbon source.
Survival assay
Cleavage activities of ZFN candidates were compared using a survival assay (17). Briefly, overnight cultures in selection medium or rich dextrose liquid medium (YPAD) were inoculated into YPA-glycerol and grown overnight to a final OD600 of 0.3–0.6. Galactose was then added to induce endonuclease expression. At varying time-points after induction aliquots were serially diluted, plated to YPAD and incubated at 30°C for 3 days. Survival rate was measured as the ratio of colony counts corrected for dilution at each time point to counts before galactose addition.
DSB-monitoring assay
Yeast cultures were handled as for survival assays, except that after induction of endonuclease expression by galactose, cells were frozen on dry ice at various time points. Genomic DNA was extracted from frozen samples using GeneClean (MP Biomedicals) and subjected to qPCR using the Agilent Mx3005P system with SYBR Select Master Mix (Life Technologies). Cut site alleles were amplified with primers 5′-AAAAAGCGCAGCGGGTAG and 5′-CTCAAAGCAGCAACAACAAAAGT. A control allele in the CAN1 locus was amplified with primers 5′-GTGGCCTTTGCTGTTTGC and 5′-CGAGATACGATTACTCCAGTTCC. The percentage of cells with intact or rejoined cut sites was calculated by normalizing the ΔCt between cut-site and control of a time-point to the 0-h time point.
Chromatin immunoprecipitation (ChIP)
A 13Myc sequence was inserted just before the stop codon at the C-terminus of the native chromosomal YKU80, DNL4, POL4, XRS2, EXO1 and TDP1 genes and ChIP performed as previously described (17). qPCR was carried out as for the DSB-monitoring assay. A segment of the ILV1 promoter region 131 bp from the cut site was amplified with primers 5′-AGTTCTTTCTTGTGTGAGTGCT and 5′-CACTTTGGACTGTTTACCTTGC. A control allele in the ACT1 locus was amplified with primers 5′-AGAGTTGCCCCAGAAGAACA and 5′-GGCTTGGATGGAAACGTAGA.
Ligation-mediated qPCR (LM-qPCR)
Linkers in the LM-qPCR assay were made by mixing two PAGE-purified synthetic oligonucleotides from IDT at 10 uM in annealing buffer (10 mM Tris, pH 7.5, 50 mM NaCl, 1 mM EDTA), heating at 95°C for 5 min, and slow cooling to RT for 2 h. Annealed linkers were stored at −20°C. Linkers with a 4-nt HO overhang used to study end processing kinetics were 5′-TGCACGGAGAAGGCTAGAGTAGATAGTTGAGTCGACAACA annealed to 5′-/5Phos/GTCGACTCAACTATCTAC/3ddC/ (/5Phos/: 5’ phosphorylation, /3ddC/:3' dideoxycytidine). Linkers with a 4-nt ZFN 5′-TGTT overhang used to study end processing kinetics were 5′-TGCACGGAGAAGGCTAGAGTAGATAGTTGAGTCGAC annealed to 5′-/5Phos/TGTTGTCGACTCAACTATCTAC/3ddC/. Linkers with 4-nt ZFN 5′-GATC overhang used to study putative Tdp1 intermediates were 5′-CTTGAGACGA/3ddC/ annealed to 5′-/5Phos/GATCGTCGTCTCAAGTCTAGCCTTCTCCGTGCA. Linkers with 5-nt 5′-GGATC overhang used to study putative Tdp1 intermediates were 5′-CTTGAGACGA/3ddC/ annealed to 5′-/5Phos/GGATCGTCGTCTCAAGTCTAGCCTTCTCCGTGCA.
T4 PNK (NEB) treatment of DNA substrates was carried out prior to ligation in the supplied buffer according to the manufacturer's instructions. Ligation reactions were carried out in a thermal cycler using the Promega Quick Ligation system with 10 nM of linkers and 3.5 µl of GeneClean purified genomic DNA at 16°C for 2 h, followed by heat inactivation at 95°C for 5 min. Reactions were diluted 8-fold with Milli-Q water and then subjected to qPCR as described for the DSB-monitoring assay. Linker ligation events were amplified with primers 5′-TGCACGGAGAAGGCTAGA and 5′-GAACTCAAAGCAGCAACAACA. A control allele in the CAN1 locus was amplified with primers 5′-GTGGCCTTTGCTGTTTGC and 5′-CGAGATACGATTACTCCAGTTCC. Ligation efficiency was calculated by correcting the qPCR signal of the linker ligation events for (i) the amount of genomic DNA used in the ligation reaction, by normalizing to the qPCR signal of the CAN1 control allele, (ii) the ratio of PCR template molecules between the ligation event and the control allele due to the linker and primer configurations (this ratio was 2 in the reported assays), (iii) the percentage of cells with a broken cut site, determined from the same sample by the DSB-monitoring assay above, and (iv) the ligation efficiency of linkers to restriction enzyme linearized plasmids mixed with genomic DNA (we used BstXI to generate the 4-nt HO overhang, BspMI to generate the 4-nt ZFN 5′-TGTT overhang, and BamHI to generate the 4-nt ZFN 5′-GATC overhang in linearized plasmids). Final data thus reveal the fraction of DSB ends (not cut-site alleles) in a sample that could be detected by LM-qPCR.
Monitoring 5′ resection by qPCR
The 5′ resection was monitored in the same samples used for DSB-monitoring and LM-qPCR assays by a qPCR protocol modified from previous studies (18,19). GeneClean purified genomic DNA was subjected to either PstI or mock digestion overnight at 37°C, followed by heat-inactivation for 5 min at 95°C. qPCR signals with primers 5′-TGTTAACACTGACGAAATCTGTG and 5′-AAGGGCACCAGATGGTTC, flanking a PstI site 1.2 kb from the DSB, were normalized between PstI-digested and mock treated samples as previously described (18,19) to calculate the percentage of ssDNA at this site.
Nuclease immunoblot
The chromosomal GAL1-HO and GAL1-V10-GFP.ZFN2(CO) alleles were modified by inserting a 3HA epitope tag just before the stop codon, similar to ChIP studies above. Expression was induced by galactose and repressed by glucose using a similar time course as LM-qPCR studies for each enzyme. Cells (1.5 × 108 cells per sample) were harvested at indicated time points and proteins purified by trichloroacetic acid precipitation. Western blots were developed using anti-HA monoclonal antibody (Sigma-Aldrich) at 1:1000 dilution and visualized using enhanced chemiluminescence (Pierce).
Next generation sequencing and joint analysis
Primers 5′-AGGGCAAAAAGAAAAAGCGCA and 5′-GTTTTATCAAGGAAGGTGACA were used to amplify the cut site allele in the ILV1 promoter, yielding a 389-bp product. One primer was close enough to the cut site that 101-nt Illumina reads crossed the cut site and any repair joints that did not delete the primer sequence. The same primers were able to amplify a control allele with an Arabidopsis sequence in the CAN1 locus. After a first round of 15 cycles, PCR products were subjected to 10 additional cycles using primers with the same 3′ end sequences and 5′ tails that included a 6-nt barcode to allow sample multiplexing and 4 random nucleotides to improve Illumina cluster detection (see Supplementary Table S2 for primer sequences). Sample indexing was achieved by combining barcodes in forward and reverse primers. Finally, Illumina adapter sequences were added with eight additional PCR cycles using primers 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT and 5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT. PCR reactions were optimized to have equal efficiency for the DSB and control alleles and ended prior to the saturation to prevent bias. Final PCR products from different samples were subjected to qPCR quantification of the control allele with primers 5′-GGCTGTCAACAGAGAAACTG and 5′-GCATCCGTGCTAGTTAGAGG and pooled to yield the same amount of control allele molecules for each sample. The resulting library was subjected to Illumina HiSeq2000 100-bp paired-end sequencing. Notably, in order to allocate the majority (∼80%) of final sequencing reads (∼150 million) to the cut site allele while having enough control reads for quantification, we intentionally mixed two strains with identical genotypes except that only one carried the control allele in the CAN1 locus, in a ratio of 1:40 for HO strains and 1:10 for ZFN strains.
Sequence reads were filtered for the presence of a recognizable sample barcode, allowing up to one mismatch, and for the PCR primer sequence. After trimming 3′ bases of unacceptably low quality, Needleman-Wunsch alignment was then performed for each sequence read to the starting alleles to assign it to either DSB or control. To identify joint types for DSB reads, the sequence surrounding the input DSB was pre-processed into a list of all possible blunt and microhomology joints. The sequence read that would be obtained for each joint was determined as well as its alignment string relative to the intact DSB allele. The alignment string for each sample read was compared to this list to determine joint identities. Mismatches more than two bases internal to the DSB end were ignored as presumptive sequencing errors. When the alignment string of a read matched more than one known joint it was called ‘ambiguous’. Rare reads that matched no joints were ‘unknown’. Joint counts, as well as aggregate counts of all joints of a similar class, were normalized to control read counts to allow comparison between samples.
Yeast two-hybrid
The yeast two-hybrid bait construct with full-length Tdp1 was created by gap repair in yeast and mated to our NHEJ two-hybrid prey array as previously described (20,21).
RESULTS
Efficient induction of chromosomal 5′ DSBs by ZFN
We selected the most active ZFN from four candidates using a survival assay (17). These ZFNs included StickyC-ZFNIII and QQR.ZFN, which promote site-specific mutagenesis in plants (22,23), and GFP.ZFN1 and GFP.ZFN2, engineered to target GFP. GFP.ZFN2 showed the best activity when expressed from an extrachromosomal plasmid (Supplementary Figure S1A). For further optimization, the GFP.ZFN2 coding sequence was integrated downstream of the native GAL1 promoter to allow control of its expression by carbon source; importantly, this construction made the cells gal1 and unable to ferment galactose. The GFP.ZFN2 cut site (ZFNcs) was placed in a nucleosome-free region of the ILV1 promoter as in our previously described HO 3′ DSB system (17,24). We optimized GFP.ZFN2 codon usage for yeast to yield GFP.ZFN2(CO), which improved cleavage activity, including a stronger non-specific cleavage (Supplementary Figure S1B). Indeed, we had difficulty constructing a viable GFP.ZFN2(CO) wild-type strain, presumably due to leaky expression in glucose. We therefore replaced the GFP.ZFN2(CO) GAL1 promoter with the mutated version GAL1-V10 (25), which allowed strain propagation while maintaining strong induction by galactose. For simplicity, ‘HO’ below represents chromosomal expression of HO endonuclease from the wild-type GAL1 promoter and ‘ZFN’ represents chromosomal expression of GFP.ZFN2(CO) from GAL1-V10, each with its cognate cut site at ILV1 (Figure 1A). Notably, DSBs induced by HO have a fixed 3′ overhang sequence of 5′-AACA, while ZFN 5′ overhang sequences can vary (Figure 1B). Unless otherwise specified, the ZFN overhang sequence used was 5′-GATC.
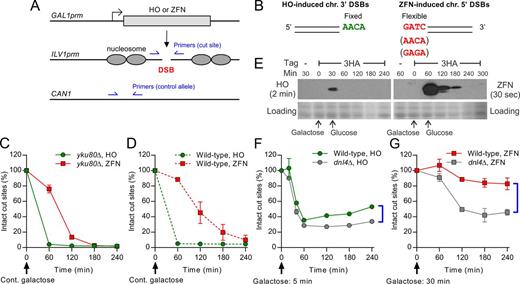
Induction of chromosomal 3′ and 5′ overhanging DSBs by HO and an optimized ZFN. (A) DSB induction paradigm for HO and ZFN. The coding sequence of the endonuclease was placed downstream of the native GAL1 promoter. The target cut site was placed into a nucleosome-free region of the native ILV1 promoter. Two pairs of qPCR primers for the DSB-monitoring assay are shown, amplifying the cut site and a control allele in CAN1. (B) Chromosomal 3′ and 5′ DSBs generated by HO and ZFN, respectively, showing overhang polarity and sequences used. (C and D) DSB-monitoring of yku80Δ and wild-type strains, respectively, under continuous galactose induction, for comparing DSB formation kinetics. (E) 3HA-tagged HO and ZFN were induced by galactose and repressed by changing to glucose. Expression levels over time were revealed by immunoblotting. Because HO levels were much lower, different exposure times were used for HO and ZFN. Fast Green protein staining of the blot is shown to judge loading. (F) DSB-monitoring of HO strains with 5-min galactose induction before changing to glucose to reveal repair by NHEJ. (G) DSB-monitoring of ZFN strains with 30-min galactose induction before changing to glucose, showing a higher degree of NHEJ at 5′ DSBs. Brackets in (D) and (E) highlight the extent of Dnl4-dependent NHEJ by comparing wild-type and dnl4Δ strains. Results are the mean ± standard deviation of three independent experiments.
We monitored the formation of site-specific DSBs in a time course using qPCR by normalizing cut-site signal to a control allele to determine the percentage of intact cut sites (Figure 1A). Results showed that our optimized ZFN rapidly induced DSBs in about 70% of haploid yeast alleles within the 1-h window from 60 to 120 min and nearly 100% by 3 h in a NHEJ-defective yku80Δ strain (Figure 1C). This activity was improved over previous ZFN and TALEN yeast systems (26,27) although less robust than HO (Figure 1C). The 1-h delay before rapid ZFN cleavage suggests a slower protein folding and/or DNA binding kinetics of the ZFN. Compared to yku80Δ, a wild-type ZFN strain showed slightly slower accumulation of DSBs presumably due to concurrent rejoining (Figure 1D). To further understand differences in enzyme kinetics we monitored tagged endonuclease proteins by western blot, which revealed a markedly lower peak expression of HO as compared to ZFN (Figure 1E), despite more efficient cutting by HO (Figure 1C and D). This is consistent with the short half-life of HO (28) and improved ZFN mRNA stability after codon optimization. ZFN enzyme is thus much less efficient in vivo than HO, making it difficult to judge the impact of the observed decay of each enzyme following glucose repression (Figure 1E). Together, these observations reveal a limitation that differences between 5′ and 3′ DSBs might sometimes reflect enzyme dynamics, including the timing of DSB formation and recleavage by persistent nuclease.
ZFN-induced 5′ DSBs are efficiently rejoined
To measure NHEJ efficiency at chromosomal DSBs, we compared DSB formation and repair in wild-type and NHEJ-defective dnl4Δ strains lacking the NHEJ ligase. The difference in the yield of intact cut sites between wild-type and dnl4Δ at late time points (60–240 min for HO and 120–240 min for ZFN) reflects repair by NHEJ. To avoid extensive recleavage of the cut sites, we only induced endonuclease expression for a short time (5 min for HO and 30 min for ZFN), which resulted in DSBs in about 60–70% of alleles for each enzyme in dnl4Δ strains (Figure 1F and G), indicating that only a small amount of endonuclease had been expressed. We observed about 20% more intact HO cut sites in wild-type as compared to dnl4Δ, a difference that became about 40% for ZFN cut sites (Figure 1F and G). These results suggest that ZFN-induced 5′ DSBs are efficiently rejoined, perhaps more efficiently than HO-induced 3′ DSBs if the kinetic differences between nucleases had a minimal effect due to the transient induction paradigm used.
NHEJ proteins are more robustly recruited to 5′ than 3′ DSBs
Several labs have monitored the recruitment of yeast DNA repair factors to HO 3′ DSBs using chromatin immunoprecipitation (ChIP) (24,29,30), which we extend here to 5′ DSBs at the same locus. Because HO induces more rapid DSB formation (Figure 1C and D), we expected more efficient recruitment of NHEJ proteins to HO- than to ZFN-induced DSBs. However, we instead observed much higher ChIP signal for all tested NHEJ proteins at ZFN-induced 5′ DSBs even at early time points, and also at late time points in which nearly all cells had DSBs under continuous galactose induction (Figure 2A–C and Supplementary Figure S2A). The fold difference in peak enrichment at 5′ versus 3′ DSBs was about 5-fold for Yku80 and Pol4 and 15-fold for Dnl4 and Xrs2. To determine whether this effect was an artifact resulting from ZFN bound to the cut site, we repeated the experiment with mutant ZFN-D450A, which binds DNA normally but is catalytically inactive (31). NHEJ proteins were not recruited to the cut site with ZFN-D450A (Figure 2A–D), indicating that recruitment with wild-type ZFN was specific to the induced DSBs. In addition, no Dnl4 ChIP signal was detected in a no-antibody control (Figure 2C). Consistent with Ku's role in recruiting NHEJ factors, deletion of YKU80 abolished recruitment of Pol4 and Dnl4 to 5′ DSBs (Figure 2B and C). In contrast to the NHEJ factors, ChIP signal of Exo1, a 5′ resection exonuclease, was similar between 5′ and 3′ DSBs (Supplementary Figure S2B). Collectively, these results suggest that more cells have stable recruitment of a proper NHEJ repair complex to 5′ than to 3′ DSBs, consistent with the higher NHEJ efficiency at 5′ DSBs above.

Recruitment of DNA repair factors to 5′ and 3′ DSBs. ChIP results of C-terminal 13Myc-tagged (A) Yku80, (B) Pol4, (C) Dnl4, (D) Tdp1, under continuous galactose induction. Yku80, Pol4 and Dnl4 NHEJ proteins showed consistently higher Ku-dependent recruitment to 5′ DSBs at the ILV1 locus, while Tdp1 showed recruitment to only 5′ DSBs in a manner suppressed by Ku. Results are the mean ± standard deviation of at least two independent experiments except those of negative controls (e.g. ZFN-D450A) which is from one experiment.
Robust modification of 5′ DSB ends
Limited modification of DSBs during NHEJ is obligatory at incompatible ends but can also occur at compatible ends in a fashion competitive with direct rejoining, where it results in mutations. To study the kinetics of even limited end modification, we used ligation-mediated qPCR (LM-qPCR). Here, any end modification, including base loss, addition, or changes in the chemical structure of terminal nucleotides, greatly reduces ligation efficiency of the DSB end to a linker with a complementary overhang. After normalizing for the fraction of still-broken cut-site alleles, quantification of linker ligation by qPCR therefore reveals the fraction of remaining DSB molecules without any such end modifications (see ‘Materials and Methods’ section). Linkers matching either the 4-nt 3′ or 5′ overhangs were ligated to HO- and ZFN-induced DSBs, respectively (Figures 3A and 4A). For best comparison, the ZFNcs used here had the same overhang sequence as HOcs, but with opposite polarity. Importantly, the extensive 5′ resection that initiates HR will also abolish LM-qPCR signal; we separately monitored this activity using a qPCR method that follows loss of internal restriction enzyme site cleavage upon transition to single-strandedness (Figure 3B) (18,19,32).

LM-qPCR and resection analysis of end modification at HO-induced 3′ DSBs. (A) LM-qPCR design to detect unmodified ends of HO-induced 3′ DSBs. (B) q-PCR assay to monitor 5′ resection. (C–F) Time-course of DSB formation, end modification and resection using the same real-time samples from (C) wild-type, (D) yku80Δ, (E) rad50Δ and (F) mre11-H125N cells. Yeast were subjected to either continuous galactose induction or induction for 30 min followed by transfer to glucose to repress further endonuclease expression. The loss of MRX increased modification of HO-induced 3′ DSB ends to an extent similar to the loss of Ku. Results are the mean ± standard deviation of two independent experiments.

LM-qPCR and resection analysis of end modification at ZFN-induced 5′ DSBs. (A) LM-qPCR design to detect unmodified ends of ZFN-induced 5′ DSBs. (B–D) Time-course of DSB formation, end modification and resection using the same real-time samples from (B) wild-type, (C) yku80Δ, (D) rad50Δ and (E) mre11-H125N cells. Yeast were subjected to either continuous galactose induction or induction for 60 min followed by transfer to glucose to repress further endonuclease expression. In contrast to Figure 3, loss of MRX or its nuclease activity significantly reduced modification of ZFN-induced 5′ DSB ends as revealed by LM-qPCR. Results are the mean ± standard deviation of two independent experiments.
In wild-type yeast, we detected substantial ligatable (unmodified) 3′ DSB ends after 30 min of HO induction, coinciding with the rapid formation of DSBs (Figure 3C). We split the culture at 30 min; one half maintained continuous DSB induction in galactose while the other was transferred to glucose to stop induction and allow repair (DSB-monitoring, Figure 3C). The kinetic profiles of end modification were very different in the two conditions. HO 3′ DSB ends were rapidly modified after changing to glucose, but largely preserved in galactose over the 6-h time course (LM-qPCR, Figure 3C). We recently reported a similar difference using Southern-blotting (24) that 5′ resection is inefficient in galactose in our gal1 strains due to their inability to ferment galactose, with resection promoted by changing to glucose (Resection, Figure 3C). Indeed, at 3′ DSBs most of the LM-qPCR signal loss over time appeared to correspond to resection, which was simply curtailed under continuous galactose. In contrast, we detected significantly more robust end modification of ZFN-induced 5′ DSBs, evident as reduced LM-qPCR signal even under continuous galactose when resection was minimal, as compared to HO-induced 3′ DSBs (compare LM-qPCR results in Figures 3C and 4B). These data suggest increased limited modification of 5′ DSB ends associated with NHEJ. Consistent with the role of Ku in protecting DSB ends, yku80Δ strains showed more robust end modification and resection at both 3′ and 5′ DSBs (Figures 3D and 4C). 5′ DSBs were again modified more robustly than 3′ DSBs in yku80Δ strains (compare LM-qPCR in Figures 3D and 4C).
MRX induces modification of 5′ DSBs and facilities protection of 3′ DSBs
The yeast MRX and orthologous mammalian MRN complexes have diverse roles in both NHEJ and HR (4). We first validated that MRX is required for yeast NHEJ of both chromosomal 3′ and 5′ DSBs (DSB-monitoring, Figures 3E and 4D). MRX possesses both endonuclease and 3′ exonuclease activity and is a DSB modifying enzyme. Consistently, loss of MRX in a rad50Δ ZFN strain resulted in a large increase of unmodified 5′ DSB ends (LM-qPCR, Figure 4D), suggesting that MRX is a major processing enzyme for 5′ DSBs and also validating the expected structure of the DSBs generated by the ZFN. Loss of MRX also led to a 10-fold hyper-recruitment of Yku80 to 5′ DSBs, which may further repress end modification by other enzymes (Supplementary Figure S2D). Consistent with observations from others (33), loss of MRX or Exo1 mildly increased Yku80 recruitment (∼2–3 fold) to 3′ DSBs (Supplementary Figure S2C). Nevertheless, we observed, in marked contrast to 5′ DSBs, that loss of MRX instead increased modification of HO-induced 3′ DSB ends to an extent similar to the loss of Ku (LM-qPCR, Figure 3D and E). We also noticed that the Yku80 recruited to 3′ DSBs in the absence of MRX was less stable and started dissociating after 2 h (Supplementary Figure S2C), suggesting that the physical presence of MRX may stabilize Ku and facilitate its function to protect 3′ DSB ends.
Importantly, the nuclease activity of MRX did not contribute substantially to 3′ DSB modification since the nuclease-dead mutant mre11-H125N (34) showed similar kinetics of end modification as wild-type (Figure 3F). However, again in contrast to 3′ DSBs, mre11-H125N conferred a large increase in the presence of unmodified 5′ DSBs as compared to wild-type, to a similar extent as rad50Δ (Figure 4E). Thus, the greater extent of limited modification of 5′ as compared to 3′ DSB ends depends on, and is likely catalyzed by, the Mre11 nuclease. Taken together, our results suggest that MRX has opposing roles in modification of 5′ and 3′ DSBs, promoting limited degradation of 5′ DSBs via the Mre11 nuclease while protecting 3′ DSBs.
Yeast Tdp1 is recruited to 5′ but not 3′ DSBs and is restricted by Ku
The tyrosyl-DNA phosphodiesterase Tdp1 is a general 3′ phosphoesterase capable of removing 3′-terminal lesions as well as nucleosides to yield a 3′ phosphate (35). Tdp1 was shown to suppress the formation of insertional mutations in plasmids carrying 5′ but not 3′ DSBs (36), suggesting that it controls repair fidelity by removing the 3′-terminal DSB nucleoside to temporarily inhibit undesirable filling of 5′ overhangs prior to rejoining (36). In exploring this potential role of Tdp1 at chromosomal 5′ DSBs, we first noted a small yet significant Tdp1 ChIP enrichment signal only at 5′ DSBs (Figure 2D), consistent with plasmid results. However, loss of Yku80 significantly increased Tdp1 recruitment (Figure 2D), in contrast to the reduced recruitment of NHEJ enzymes like Dnl4 and Pol4. A possible explanation for this difference is that DSB ends without Ku protection suffer damage that is repaired by Tdp1. More generally, Tdp1 and NHEJ may be competitive. Although Tdp1 loss did not significantly alter Yku80 or Pol4 recruitment (Figure 2A and B), Tdp1 overexpression did slightly but significantly impede NHEJ (Supplementary Figure S3A). We also detected no protein–protein interaction between Tdp1 and yeast NHEJ proteins (Supplementary Figure S3B).
We modified our LM-qPCR assay in an attempt to demonstrate the putative Tdp1 cleavage intermediate at chromosomal 5′ DSBs (Supplementary Figure S3C). As a control, we synthesized substrates with a 3′ recessed phosphate or hydroxyl in a 5-nt 5′ overhang (Supplementary Figure S3D). As expected, linkers with complementary 5-nt overhangs could efficiently ligate to the synthetic duplex with the 3′ hydroxyl, or to the duplex with the 3′ phosphate after pre-treatment with T4 PNK, a 3′ phosphatase (Supplementary Figure S3D). However, we could not detect the Tdp1 intermediate at 5′ DSBs in vivo; linker ligation efficiency was the same between the T4 PNK and mock treatment groups and unaffected by Tdp1 overexpression or loss of the 3′ phosphatase Tpp1 (Supplementary Figure S3E).
Quantitative next-generation sequencing reveals kinetics of NHEJ mutagenesis
We next subjected cut-site PCR products to next-generation sequencing (NGS) to analyze the appearance of different joint types as they formed in culture. This approach removes the need for cell outgrowth and reveals low frequency joint types. Similar to DSB-monitoring above (Figure 1A), the primers used to create sequenced products also amplified a control allele to allow normalization for cell number and thus determination of absolute amounts of broken and intact cut sites between samples (Figure 5A). To study the impact of both overhang polarity and sequence on NHEJ mutagenesis, we utilized strains harboring ZFNcs with either a 5′-GATC or a 5′-GAGA overhang, the latter containing a direct repeat sequence. As for LM-qPCR, we induced expression of HO for 30 min and ZFN for 60 min to generate enough DSBs and then allowed repair to occur.

High-throughput kinetic analysis of NHEJ joint types by NGS. (A) Sequencing library construction by simultaneous amplification of DSB and control alleles, with barcodes to allow sample pooling. (B) Time course of formation of all joint types in strains carrying HOcs, ZFNcs(GATC) and ZFNcs(GAGA), yielding results similar to qPCR-based DSB monitoring. (C) Similar to (B), except restricting plots to insertion joints to highlight strict Pol4 dependence only at 3′ DSBs. (D) Comparison of the distributions of mutagenic joints for strains carrying ZFNcs(GATC) and ZFNcs(GAGA) after 6-h of galactose induction, showing the mutagenic effect of repeated nucleotides in the overhang. NT, non-templated insertions, T, templated insertions. (E) Diagram of extensive recleavage to enrich mutagenic joints. (F) Mutation frequency of wild-type, pol4 Δ and dnl4Δ strains carrying HOcs, ZFNcs(GATC) and ZFNcs(GAGA), using samples after 6-h galactose induction. 5′ DSBs showed notably higher NHEJ-dependent mutagenesis. Results are the mean ± standard deviation of two independent experiments.
Joint types can be categorized into several groups: precise joints with no mutations and mutagenic joints with a gain (insertion) or loss (deletion) of DNA content. Importantly, total joint recovery in wild-type (Figure 5B), of which more than 97% is the unmodified precise joint, recapitulated the DSB-monitoring profiles using qPCR (DSB-monitoring in Figures 3 and 4), validating the NGS design. An open question is whether NHEJ mutations occur at the same time as precise rejoining or only much later when precise rejoining fails. We observed the formation of mutagenic joints with the same apparent kinetics as precise joints for 3′ DSBs, although precise joints were much more frequent (Supplementary Figure S4A and Figure 5B), arguing that precise and mutagenic joints form simultaneously in a competitive fashion.
We further observed that the frequency of mutagenic joints with ZFNcs(GAGA) was about 7-fold higher than with ZFNcs(GATC) (Supplementary Figure S4A). Of specific interest was an increase in insertional mutagenesis (Figure 5C) mediated in part by the predicted strong secondary annealing of the terminal GA and TC dinucleotides in the overhangs on the two sides of the ZFNcs(GAGA) DSB, leading to joint I(ga) with an extra GA (Supplementary Figure S4E). Meanwhile, the robust formation of insertion joints of this and various other templated and non-templated classes with ZFNcs(GAGA) corresponded to a lower absolute and relative frequency of deletion joints as compared to ZFNcs(GATC) (Supplementary Figure S4B and Figure 5D), possibly due to a competition between different mutagenic processes.
Pol4 is indispensable for insertional mutagenesis at 3′ but not 5′ DSBs
Polymerization of new nucleotides is obligatory in the formation of insertion joints. We previously showed that Pol4 is the main NHEJ DNA polymerase in yeast and absolutely required for gap-filling at plasmid and chromosomal 3′-DSBs (Figure 5C), but not plasmid 5′ DSBs (10,11). Examination of chromosomal 5′ DSBs completed this analysis by demonstrating that, although significantly reduced, insertion joints were still detectable for ZFN in absence of Pol4, in contrast to their complete loss at chromosomal 3′ DSBs (Figure 5C). Thus, other less preferred polymerases can catalyze insertions with limited efficiency at 5′ but not 3′ chromosomal DSBs when Pol4 is absent.
Importantly, although deletion joints do not strictly require gap-filling, base removal and resynthesis may sometimes occur. Indeed, we observed an involvement of Pol4 in the most frequent deletion joints (Supplementary Figure S4C). However, different joints showed very different extents of Pol4 dependence as revealed by the fold difference between wild-type and pol4Δ strains. This result demonstrates that formation of deletion joints likely involves multiple competing processes rather than representing just one mechanism.
Consistent with our LM-qPCR results of Tdp1 intermediates (Supplementary Figure S3E), we did not observe a significant difference in the frequency of insertional mutagenesis among wild-type, tdp1Δ and tpp1Δ strains in two NGS runs (Supplementary Figure S4D), in contrast to the predictions from previous plasmid studies of increased 5′ DSB insertion mutations in tdp1Δ yeast (36). Here it is notable that the overall frequency of insertion mutations was much lower for chromosomal DSBs than previously seen for plasmids.
NHEJ of chromosomal 5′ DSBs generates more frequent mutations than 3′ DSBs
Because we observed above that 5′ DSBs underwent more robust end modification, we hypothesized that we might see more frequent mutagenesis during NHEJ of 5′ DSBs. Here, direct comparison of rare mutation frequencies at 3′ and 5′ DSBs can be challenging and we avoided comparing their kinetic profiles in Supplementary Figure S4A. A more appropriate approach is to compare the frequency of mutagenic joints after extensive endonuclease induction, where recleavage of the precise joint promotes enrichment of mutagenic joints (Figure 5E). After 6 h of continuous galactose induction, we observed significantly more frequent mutations in ZFNcs as compared to HOcs in both wild-type and pol4Δ cells, but much less so in NHEJ-deficient dnl4Δ cells (Figure 5F). These results suggest that NHEJ of 5′ DSBs is more mutagenic as compared to 3′ DSBs.
Interestingly, the mutated I(ga) joint seen with ZFNcs(GAGA) was severely impeded in dnl4Δ and yku80Δ cells, as typically seen for overhang-to-overhang imprecise joining, but much less so in rad50Δ cells (Supplementary Figure S4E). Because I(ga) is formed by partial annealing of the two unmodified overhangs (Supplementary Figure S4E), residual I(ga) joints in rad50Δ cells suggest a possible reduced efficiency of end modification in the absence of MRX at 5′ DSBs, consistent with results from the LM-qPCR assay.
DISCUSSION
We developed a system to generate chromosomal 5′ DSBs in yeast by optimization of a ZFN and observed that their rejoining seems to be more efficient as compared to HO 3′ DSBs, consistent with a robust recruitment of NHEJ factors to 5′ DSBs. Further application of high-resolution LM-qPCR and quantitative NGS assays revealed increased local end modification and NHEJ-dependent mutagenesis at chromosomal 5′ DSBs. Although there is a limitation arising from differences in HO and ZFN enzyme dynamics, the combined data support a model in which overhang polarity of chromosomal DSBs impacts NHEJ kinetics and fidelity through differential recruitment and action of DSB modifying enzymes (Figure 6).

Impact of overhang polarity on kinetics and fidelity of yeast NHEJ. (A) At chromosomal 3′ DSBs engaging NHEJ with inactive 5′ resection, MRX is inefficient in supporting limited end modifications. Instead it stabilizes Ku, which recruits other NHEJ proteins such as LigIV and Pol4, resulting in mainly precise NHEJ. In the absence of MRX in rad50Δ cells, Ku and/or DSBs may be less stable, increasing susceptibility of DSB ends to limited modification by other enzymes. Loss of Ku abolishes recruitment of essential NHEJ proteins such as LigIV and also sensitizes DSBs to modification. (B) At chromosomal 5′ DSBs, MRX facilities efficient limited end modification even when resection is inactive. Combined with a more stable recruitment of NHEJ proteins, such as Ku and LigIV, 5′ DSBs are more efficiently repaired by NHEJ at the cost of more frequent mutagenesis. Loss of MRX significantly reduces end modification and leads to hyper-recruitment of Ku, which may further repress end modification. Similar to 3′ DSBs, loss of Ku in 5′ DSBs abolishes recruitment of essential NHEJ proteins and further sensitizes DSB ends to modification facilitated by MRX.
Induction of chromosomal 5′ DSBs by ZFN
Endonucleases commonly used to study DSB repair in yeast, such as HO and I-SceI, generate 3′ DSBs. Although recent reports used the genome editing nucleases ZFN, TALEN and CRISPR/Cas9 to study translocations and chromatin structure upon 5′ DSB induction (1,26,27,37,38), the activities of the nucleases are often insufficient for real-time monitoring of NHEJ. We report an optimized ZFN system to generate chromosomal 5′ DSBs with cleavage kinetics that approached the efficient HO system. ZFN cleavage initiated ∼60 min after endonuclease induction as compared to 15 min for HO, but then proceeded rapidly. We reason that the ZFN delay may result from a slower kinetics of expression, protein folding, accumulation and/or cut site targeting. We also noticed that the recovery of intact ZFN sites was slower after repressing endonuclease expression (DSB-monitoring, Figure 4B), suggesting that there was recleavage. Indeed, the ZFN almost certainly has a longer half-life than the short 10-min half-life of HO (28), consistent with the much lower peak level of HO than ZFN (Figure 1E). We paid careful attention to these phenomena to avoid bias in our analyses. It may be possible to improve ZFN properties by regulating either nuclear-cytoplasmic shuttling (39) or post-cleavage stability (40).
Chromosomal 5′ DSBs are joined efficiently at a cost of increased mutagenesis
Results with the ZFN and HO systems consistently indicate that overhang polarity of chromosomal DSBs impacts the kinetics and fidelity of yeast NHEJ. DSB monitoring suggested that chromosomal 5′ DSBs are processed and rejoined more efficiently as compared to 3′ DSBs at the same locus. At least two factors appear to contribute to this phenomenon (Figure 6). First, we surprisingly observed a more robust binding of NHEJ proteins to 5′ DSBs, including Yku80, Dnl4, Pol4 and Xrs2. This result might imply that more NHEJ proteins are bound per DSB end, but more likely reflects a more stable binding to 5′ DSBs and thus a greater fraction of time spent bound. Second, perhaps as a result of increased NHEJ protein residence time, LM-qPCR revealed that 5′ DSBs were more likely to undergo limited end modification that prevented linker ligation, even in cells prevented from executing robust HR resection. The differences between the 5′ and 3′ DSB systems noted above might influence results, but the consistency of findings using different methods gives confidence that chromosomal 5′ and 3′ DSBs are not handled equivalently. The consequence of these observations was that, somewhat paradoxically, more robust NHEJ action at 5′ DSBs was associated with a greater tendency toward mutation. Importantly, precise NHEJ was still by far the most frequent outcome even at 5′ DSBs (Figure 5 and Supplementary Figure S4). Nevertheless, the ∼50-fold higher mutation frequency in wild-type ZFN strains in a paradigm of prolonged nuclease expression (Figure 5F) implies that 5′ DSBs are rejoined more efficiently in part by exploring other joining possibilities through end modification.
Yeast NHEJ efficiency and fidelity have not previously been noted to be different for 3′ and 5′ DSBs in studies with transformed plasmids (10,36,41), although Mycobacteria showed more accurate rejoining of 3′ than 5′ DSBs (42) and plasmids with 5′ DSBs did show modestly higher joining efficiency in a human cell study (9). While differences in HO and ZFN dynamics might influence results, so too might differences in repair of native chromosomal versus non-physiological plasmid DSBs. Importantly, mutation frequency of 3′ DSBs in wild-type cells was significantly lower in our chromosomal system (about 0.1% in Figure 5F) than in typical plasmid assays (about 1–5%) (10,36,41), suggesting that artificially introduced plasmids may be prone to mutation and not an ideal basis for comparison. Further studies with better chromosomal systems and sensitive mutation detection will improve our understanding of differential DSB handling. Additionally, yeast plasmid and in vivo studies suggest that DSBs with blunt ends are repaired less efficiently by NHEJ (43,44). It would be valuable to test whether site-specific chromosomal blunt DSBs behave differently than 5′ or 3′ DSBs at the same locus using the recently developed ZF-PvuII DSB system (45).
Recognition of overhang polarity by DSB modifying enzymes
Differential handling of DSBs based on overhang polarity demands that NHEJ or other protein factors have polarity-dependent actions. Yeast Pol4 was shown to be required for gap-filling of 3′ DSBs and to be partially dispensable at extrachromosomal 5′ DSBs (10). Joint sequencing results here establish the same differential requirement of Pol4 at chromosomal 3′ and 5′ DSBs. The basis for this difference is revealed by the fact that template-dependent polymerization at annealed short 3′ DSB overhangs requires stabilization of a disrupted template strand within the polymerase active site, supported by a specialized Pol X family protein loop (46,47). In contrast, the recessed 3′ strand in a 5′ DSB might be filled independently of the other DSB end, thereby allowing the use of other NHEJ-unspecialized polymerases. Other studies have revealed a contribution of replicative yeast polymerases, especially Pol3 (Pol δ), to both NHEJ and microhomology-mediated end joining (48,49).
The lyase activity of human and yeast Ku supports the notion that Ku can recognize complex DSB structures as an end modifying enzyme (50,51). Studies of human DNA-PKcs also showed that it can recognize overhang polarity to regulate its kinase activity through the C-terminal domain of human Ku80 (52,53), although yeast lack DNA-PKcs and have a substantially shorter Yku80 C-terminal domain (21). While early biochemical efforts showed that Ku had similar binding affinity to linearized plasmids with different overhang polarities (54,55), our ChIP data showed a more efficient recruitment of Yku80 to chromosomal 5′ DSBs, raising the possibility that Ku binding to chromosomes might be affected by DSB structure.
Most importantly, our data indicate that MRX helps recognize overhang polarity and regulate the ensuing repair process. MRX has a central role in both NHEJ and the initiation of HR 5′ resection (4). Its roles in NHEJ appear to entail structural stabilization of DSBs and/or NHEJ repair complexes, and possibly NHEJ-associated end modification (56). Notably, it was shown in biochemical studies that human Mre11 and Mre11–Rad50 complex could degrade synthetic DNA duplexes with 5′ but not 3′ ends (57), and that Mre11-Rad50 had a preference for binding DNA substrates with 5′ ends (58). Instead of degrading 3′ DSBs directly, recombinant MRX and its mammalian ortholog MRN were shown to have DNA unwinding activity on 3′ DSBs more than 5′ DSBs (59,60), which can stimulate Exo1's flap endonuclease to produce 5′-recessed intermediates for resection (61,62). Indeed, Exo1 itself preferentially degrades 3′ DSBs in vitro (61,63). Our in vivo results are consistent with these biochemical data and suggest that MRX has opposite net effects on the processing of 5′ and 3′ DSBs. At 5′ DSBs, MRX appears to facilitate a more robust but limited modification of DSB ends in the absence of extensive resection in a manner facilitated, and likely catalyzed, by the Mre11 nuclease. At 3′ DSBs an early effect of MRX on stabilization of Ku and associated end preservation predominates (Figure 6). Intriguingly, consistent with our observations for 3′ DSBs, a recent study suggested that timely removal of MRX from DSBs by Sae2 is important for resection, presumably through dissociating Ku and the NHEJ complex (64).
The role of Tdp1 in 5′ DSB processing
With our chromosomal ZFN system, we were able to test predictions that Tdp1 enzyme activity suppresses insertional mutagenesis at 5′ DSBs via an intermediate that blocks 5′ overhang filling (36). Indeed, we detected Tdp1 recruitment only to 5′ DSBs. However, Tdp1 did not behave like other yeast NHEJ proteins, but appeared to compete with them, as suggested by the increased recruitment of Tdp1 in the absence of Ku. Moreover, we failed to detect the putative Tdp1 intermediate in cells by LM-qPCR or Tdp1-NHEJ protein interactions by two-hybrid. Consistently, we did not see a difference in the frequency of recovered insertion mutations between wild-type and tdp1Δ strains. The reason for this discrepancy between studies might include strain background differences, or a difference in the cellular processing of chromosomal versus plasmid DSBs. For example, transformed plasmids must traverse the cytoplasm, perhaps giving more opportunity for processing by Tdp1. Notably, a recent study showed that human Tdp1 can interact with XLF and Ku to promote DNA binding in vitro (65), so our results do not rule out Tdp1 function(s) in NHEJ, perhaps in a species specific manner.
We would like to thank John L. Nitiss and Tzvi Tzfira for their generous gifts of ZFN constructs, Kevin Lewis for the GAL1-V10 promoter used in our optimized ZFN system, and Vijay Jasti and Christine Canman for assistance with immunoblotting.
FUNDING
National Institutes of Health [CA102563 to T.E.W.]; University of Michigan Comprehensive Cancer Center Fund for Discovery [to T.E.W.]; Mary Sue and Kenneth Coleman Endowed Rackham Graduate and Rackham One Term Dissertation Fellowships [to Z.L.]. Funding for open access charge: NIH; Departmental discretionary funds.
Conflict of interest statement. None declared.
Present address:
Zhuobin Liang, Yale University School of Medicine, 333 Cedar Street, SHM C130A, New Haven, CT 06510, USA.
Sivakumar Nallasivam, Mycroarray, 5692 Plymouth Road, Ann Arbor, MI 48105, USA.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments