





Abstract
The Saccharomyces cerevisiae genome encodes five sirtuins (Sir2 and Hst1–4), which constitute a conserved family of NAD-dependent histone deacetylases. Cells lacking any individual sirtuin display mild growth and gene silencing defects. However, hst3Δ hst4Δ double mutants are exquisitely sensitive to genotoxins, and hst3Δ hst4Δ sir2Δ mutants are inviable. Our published data also indicate that pharmacological inhibition of sirtuins prevents growth of several fungal pathogens, although the biological basis is unclear. Here, we present genome-wide fitness assays conducted with nicotinamide (NAM), a pan-sirtuin inhibitor. Our data indicate that NAM treatment causes yeast to solicit specific DNA damage response pathways for survival, and that NAM-induced growth defects are mainly attributable to inhibition of Hst3 and Hst4 and consequent elevation of histone H3 lysine 56 acetylation (H3K56ac). Our results further reveal that in the presence of constitutive H3K56ac, the Slx4 scaffolding protein and PP4 phosphatase complex play essential roles in preventing hyperactivation of the DNA damage-response kinase Rad53 in response to spontaneous DNA damage caused by reactive oxygen species. Overall, our data support the concept that chromosome-wide histone deacetylation by sirtuins is critical to mitigate growth defects caused by endogenous genotoxins.
INTRODUCTION
Post-translational modification of histones can directly influence chromatin structure, or serve as platforms for the recruitment of regulatory factors, thereby modulating DNA-associated processes (1). Acetylation of histone lysine residues is catalyzed by histone acetyltransferases (HATs), and reversed by histone deacetylases (HDACs). Sirtuins are an evolutionarily conserved family of HDACs that deacetylate lysines in a reaction that consumes nicotinamide adenine dinucleotide (NAD+) and releases nicotinamide and O-acetyl ADP ribose (2,3). These enzymes are found in archaea, eubacteria and eukaryotes (2) where they regulate key cellular pathways, e.g. metabolic processes, DNA replication and repair, telomere structure and function, gene expression and replicative lifespan (4).
The Saccharomyces cerevisiae genome contains five sirtuin genes: HST1–4 and SIR2 (5,6). Yeast Sir2 is the founding member of this family of enzymes, and was identified on the basis of its role in regulating gene silencing at the yeast mating loci (6), rDNA (7) and telomeres (8). These functions of Sir2 can be attributed in part to reversal of histone H4 lysine 16 acetylation (H4K16ac), an abundant and conserved modification of transcriptionally active chromatin (9,10). Sir2 activity influences replicative life-span by limiting recombination in rDNA and consequent formation of age-associated extrachromosomal ribosomal DNA circles (ERCs) (11,12). Hst1 (Homolog of Sir2) shares sequence similarity with Sir2 but presents divergent functions (13,14); this enzyme negatively regulates middle sporulation gene expression (15,16), and controls intracellular NAD+ levels and thiamine biosynthesis through transcriptional repression (17,18). Although Hst2 contains a nuclear export signal that mediates its cytosolic localization (19), it can deacetylate H4K16ac and influence cellular aging in the absence of Sir2 (20). Moreover overexpression of Hst2 results in rDNA and telomeric silencing that can compensate for sir2Δ defects, indicating that its functions partially overlap with those of Sir2 in the nucleus (21).
Yeast mutants lacking any one of the five sirtuins display relatively mild growth phenotypes (5). In contrast, hst3Δ hst4Δ double mutants grow poorly, and combining these two mutations with sir2Δ causes synthetic lethality via poorly understood mechanisms (5,22,23). Hst3 and Hst4 present remarkable selectivity for acetylated H3K56 in several fungal species, and exert partially redundant roles in deacetylating this residue (24–26). H3K56ac is catalyzed by the HAT Rtt109 and is found in virtually all newly-synthesized histone H3 deposited behind DNA replication forks in S phase (27–31). Hst3 and Hst4 are expressed in late S-G2/M and G1-G2/M, respectively, when they deacetylate nucleosomal H3K56ac genome-wide (25,32). Cells lacking both Hst3 and Hst4 present constitutively acetylated H3K56 throughout the cell cycle, and exhibit thermosensitivity, spontaneous DNA damage, and extreme sensitivity to genotoxin-induced replicative stress (22,23,25). These severe phenotypes are partially suppressed by mutations that prevent H3K56ac, e.g. H3K56R, suggesting that they are caused in large part by defective regulation of H3K56ac (22).
DNA lesions that impede the progression of replication forks activate a signaling cascade which is regulated by the apical kinase Mec1 (33). In response to genotoxic stress, Hst3 is targeted for proteasomal degradation in a Mec1-dependent manner, causing chromatin-borne H3K56ac to persist in G2/M (29,34). The fact that cells have evolved this capacity to preserve nucleosomal H3K56ac in response to replicative stress suggests that this modification may modulate certain aspects of the DNA damage response (DDR). Consistent with this, abnormal regulation of H3K56ac negatively influences homologous recombination-mediated sister chromatid exchange and break-induced replication (35–37). In addition, certain mutations in histone or DDR genes influence the severity of phenotypes caused by Hst3 and Hst4 deficiency (22,23). For example, the temperature and genotoxin sensitivity of hst3Δ hst4Δ mutants is suppressed by mutations abolishing H3K79 methylation, a histone modification known to promote Rad9 chromatin binding and subsequent activation of the Rad53 DDR kinase (23). These data suggest that DNA damage-induced signaling may contribute to the phenotypes of cells presenting constitutive H3K56ac, although the mechanisms remain poorly understood at the molecular level.
Nicotinamide (NAM) is a non-competitive pan-inhibitor of several NAD-dependent enzymes, including HDACs of the sirtuin family (2,38–39). Our previously published results indicate that NAM-induced sirtuin inhibition prevents growth of the pathogenic fungus Candida albicans by causing constitutive H3K56ac (24). To further understand this phenomenon, we performed genome-wide fitness assays to identify genes that influence growth of Saccharomyces cerevisiae in the presence of NAM. The data reveal that sirtuin-mediated deacetylation of H3K56ac promotes cell growth by preventing persistent activation of DNA damage-induced kinases in response to endogenous genotoxins.
MATERIALS AND METHODS
Yeast strains and growth conditions
Strains used in this study are listed in Table 1 and were generated and propagated using standard yeast genetics methods. Nicotinamide and methyl methanesulfonate (MMS) were purchased from Sigma-Aldrich.
Strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| BY4741 | BY4741 MATa ura3Δ0 leu2Δ0 his3Δ1 | (105) |
| BY4743 | BY4743 MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 LYS2/lys2Δ0 met15Δ0/MET15 ura3Δ0/ura3Δ0 | (105) |
| W303 | W303 MATa ade2–1 can1–100 his3–11,15 leu2–3,112 trp1–1 ura3–1 [psi+] rad5–535 | (106) |
| W5094–1C | W303 ADE2 RAD52-YFP RAD5 | (43) |
| HWY2493 | W303 ADE2 bar1Δ::LEU2 RFA1–8ala-YFP RAD5 | (23) |
| HWY297 | BY4741 rtt109Δ::KanMX | This study |
| ASY3111 | YBL574 hht1-hhf1Δ::LEU2 hht2-hhf2Δ::HIS3 [pCEN TRP1 HHT1-HHF1] | (107) |
| ASY3113 | YBL574 hht1-hhf1Δ::LEU2 hht2-hhf2Δ::HIS3 [pCEN TRP1 HHT-hhf1K16A] | (107) |
| HWY2949 | YBL574 hht1-hhf1Δ::LEU2 hht2-hhf2Δ::HIS3 [pCEN TRP1 HHT1-HHF1] rtt109Δ::URA3MX | This study |
| ASY3180 | BY4741 dpb4Δ::KanMx | This study |
| HWY2417 | BY4741 dun1Δ::KanMX | This study |
| HWY634 | BY4741 srs2Δ::KanMX | This study |
| ASY3188 | BY4741 tof1Δ::KanMX | This study |
| ASY3193 | BY4741 sae2Δ::KanMX | This study |
| HWY2477 | BY4741 mrc1Δ::KanMX | This study |
| HWY2460 | BY4741 mrc1Δ::KanMX rtt109Δ::URA3MX | This study |
| ASY1767 | BY4741 yku70Δ::KanMX | This study |
| ASY3164 | BY4741 yku70Δ::KanMX rtt109Δ::HPHMX | This study |
| EHY027 | BY4741 rad59Δ::KanMX | This study |
| EHY029 | BY4741 rad59Δ::KanMX rtt109Δ::URA3MX | This study |
| ASY2807 | BY4741 pol32Δ::KanMX | This study |
| ASY3159 | BY4741 pol32Δ::KanMX rtt109Δ::HPHMX | This study |
| HWY1608 | BY4741 slx4Δ::KanMX | This study |
| ASY1875 | BY4741 slx4Δ::HPHMX rtt109Δ::URA3MX | This study |
| HWY1610 | BY4741 rad1Δ::kanMX 25C10 | This study |
| HWY1609 | BY4741 slx1Δ::kanMX 10E7 | This study |
| ASY3147 | BY4741 mus81Δ::HPHMX | This study |
| HWY3228 | BY4741 mms4Δ::KanMX | This study |
| ASY2164 | BY4741 rtt101Δ::URA3MX | This study |
| ASY2168 | BY4741 rtt101Δ::URA3MX slx4Δ::HPHMX | This study |
| ASY2166 | BY4741 rtt107Δ::KanMX | This study |
| ASY2163 | BY4741 rtt107Δ::KanMX slx4Δ::HPHMX | This study |
| HWY525 | BY4741 rtt107Δ::KanMX | This study |
| HWY530 | BY4741 rtt107Δ::KanMX rtt109Δ::URA3MX | This study |
| ASY1763 | BY4741 psy2Δ::KanMX | This study |
| ASY1764 | BY4741 psy4Δ::KanMX | This study |
| ASY1765 | BY4741 pph3Δ::KanMX | This study |
| ASY1840 | BY4741 pph3Δ::HPHMX rtt109Δ::URA3MX | This study |
| EHY047 | BY4741 rad9Δ::KanMX | This study |
| ASY2796 | BY4741 rad9Δ::KanMX pph3Δ::HPHMX | This study |
| ASY3516 | BY4741 slx4Δ::KanMX rad9Δ::HPHMX | This study |
| EHY071 | BY4741 dot1Δ::KanMX | This study |
| ERY3386 | BY4741 slx4Δ::KanMX dot1Δ::URA3MX | This study |
| ERY3389 | BY4741 pph3Δ::HPHMX dot1Δ::URA3MX | This study |
| FY406 MATa hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-HTA1] | (107) | |
| ERY3394 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-HTA1] pph3Δ::HPHMX | This study |
| ERY3396 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-hta1S128A] pph3Δ::HPHMX | This study |
| HWY2878 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-HTA1] slx4Δ::KanMX | This study |
| HWY2879 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-hta1S128A] slx4Δ::KanMX | This study |
| HWY1936 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-hta1S128A] | This study |
| ASY2766 | W303 ADE2 RAD52-YFP RAD5 slx4Δ::HPHMX | This study |
| ASY2764 | W303 ADE2 RAD52-YFP RAD5 pph3Δ::HPHMX | This study |
| Y2573 | W303 dbf4Δ::TRP1 his3::PDBF4-dbf4–4A::HIS3 sld3–38A-10his-13MYC::KanMX4 | (87) |
| ERY3414 | W303 dbf4Δ::TRP1 his3::PDBF4-dbf4–4A::HIS3 sld3–38A-10his-13MYC::KanMX4 slx4Δ::HPHMX | This study |
| ERY3415 | W303 dbf4Δ::TRP1 his3::PDBF4-dbf4–4A::HIS3 sld3–38A-10his-13MYC::KanMX4 pph3Δ::HPHMx | This study |
| ASY2798 | W303 pph3Δ::HPHMX | This study |
| HWY2882 | W303 slx4Δ::HPHMX | This study |
| HWY2942 | BY4741 slx4Δ::KanMX dot1Δ::URA3MX rev3Δ::HIS3MX | This study |
| ASY3534 | BY4741 pph3Δ::HPHMX dot1Δ::URA3MX rev3Δ::KanMX | This study |
| ASY3667 | BY4741 pol30Δ::KANMX trp1Δ::KANMX slx4Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-POL30-TRP1] | ||
| ASY3668 | BY4741 pol30Δ::KANMX trp1Δ::KANMX slx4Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-pol30-K164R-TRP1] | ||
| ASY3669 | BY4741 pol30Δ::KANMX trp1Δ::KANMX pph3Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-POL30-TRP1] | ||
| ASY3670 | BY4741 pol30Δ::KANMX trp1Δ::KANMX pph3Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-pol30-K164R-TRP1] | ||
| HWY630 | BY4741 rad18Δ::KanMX | This study |
| HWY636 | BY4741 mms2Δ::KanMX | This study |
| ASY3522 | BY4741 slx4Δ::KanMX dot1Δ::URA3 rad18Δ::HIS3MX | This study |
| HWY2939 | BY4741 slx4Δ::kanMX dot1Δ::URA3MX mms2Δ::HIS3MX | This study |
| ASY3651 | BY4741 pph3Δ::KanMX rad18Δ::HIS3MX rad9Δ::URA3MX | This study |
| ASY3654 | BY4741 pph3Δ::KanMX mms2Δ::HPHMX rad9Δ::URA3MX | This study |
| ASY3519 | BY4741 pph3Δ::HPHMX rev3Δ::KanMX | This study |
| HWY2940 | BY4741 slx4Δ::KANMX rev3Δ::HIS3MX | This study |
| ICY703 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 [pCEN URA3 HST3] | (25) |
| ASY3537 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 pph3Δ::HPHMX [pCEN URA3 HST3] | This study |
| ASY3657 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 rtt107Δ::HPHMX [pCEN URA3 HST3] | This study |
| ASY2156 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 slx4Δ::KanMX [pCEN URA3 HST3] | This study |
| ASY3675 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 slx4Δ::KanMX rtt107Δ::HPHMX | This study |
| [pCEN URA3 HST3] | ||
| ASY3678 | BY4741 srl4Δ::KanMX | This study |
| ASY3679 | BY4741 him1Δ::KanMX | This study |
| ASY3680 | BY4741 hug1Δ::KanMX | This study |
| ICY1164 | MATa his3D200 leu2Δ1 lys2Δ202 trp1Δ63 ura3–52 bar1Δ::hygMX hst4Δ::TRP1 hst3::td-HST3- 13MYC::KanMX4 | (25) |
| ASY3139 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 sml1Δ::KanMX [pCEN URA3 HST3] | This study |
| ASY3143 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 sml1Δ::KanMX RAD53–3HA::HPHMX | This study |
| [pCEN URA3 HST3] | ||
| ASY3682 | BY4741 sml1Δ::KanMX slx4Δ::HIS3MX | This study |
| ASY3684 | BY4741 sml1Δ::KanMX pph3Δ::HIS3MX | This study |
| ASY3718 | BY4741 sml1Δ::KanMX slx4Δ::HIS3MX RAD53–3HA::HPHMX | This study |
| ASY3720 | BY4741 sml1Δ::KanMX pph3Δ::HIS3MX RAD53–3HA::HPHMX | This study |
| ASY4003 | BY4741 rev3Δ::KanMX dot1Δ::URA3MX | This study |
| ASY4014 | BY4741 rad9Δ::KanMX rad18Δ::HPHMX | This study |
| ASY4020 | BY4741 dot1Δ::KanMX rad18Δ::HPHMX | This study |
| ASY4023 | W303 RAD5 pph3Δ::HPHMX | This study |
| ASY4024 | W303 rad5–535 | This study |
| ASY4025 | W303 RAD5 | This study |
| ASY4026 | W303 rad5–535 pph3Δ::HPHMX | This study |
| ASY4027 | W303 RAD5 slx4Δ::HPHMX | This study |
| ASY4029 | W303 rad5–535 slx4Δ::HPHMX | This study |
| Strain | Genotype | Reference |
|---|---|---|
| BY4741 | BY4741 MATa ura3Δ0 leu2Δ0 his3Δ1 | (105) |
| BY4743 | BY4743 MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 LYS2/lys2Δ0 met15Δ0/MET15 ura3Δ0/ura3Δ0 | (105) |
| W303 | W303 MATa ade2–1 can1–100 his3–11,15 leu2–3,112 trp1–1 ura3–1 [psi+] rad5–535 | (106) |
| W5094–1C | W303 ADE2 RAD52-YFP RAD5 | (43) |
| HWY2493 | W303 ADE2 bar1Δ::LEU2 RFA1–8ala-YFP RAD5 | (23) |
| HWY297 | BY4741 rtt109Δ::KanMX | This study |
| ASY3111 | YBL574 hht1-hhf1Δ::LEU2 hht2-hhf2Δ::HIS3 [pCEN TRP1 HHT1-HHF1] | (107) |
| ASY3113 | YBL574 hht1-hhf1Δ::LEU2 hht2-hhf2Δ::HIS3 [pCEN TRP1 HHT-hhf1K16A] | (107) |
| HWY2949 | YBL574 hht1-hhf1Δ::LEU2 hht2-hhf2Δ::HIS3 [pCEN TRP1 HHT1-HHF1] rtt109Δ::URA3MX | This study |
| ASY3180 | BY4741 dpb4Δ::KanMx | This study |
| HWY2417 | BY4741 dun1Δ::KanMX | This study |
| HWY634 | BY4741 srs2Δ::KanMX | This study |
| ASY3188 | BY4741 tof1Δ::KanMX | This study |
| ASY3193 | BY4741 sae2Δ::KanMX | This study |
| HWY2477 | BY4741 mrc1Δ::KanMX | This study |
| HWY2460 | BY4741 mrc1Δ::KanMX rtt109Δ::URA3MX | This study |
| ASY1767 | BY4741 yku70Δ::KanMX | This study |
| ASY3164 | BY4741 yku70Δ::KanMX rtt109Δ::HPHMX | This study |
| EHY027 | BY4741 rad59Δ::KanMX | This study |
| EHY029 | BY4741 rad59Δ::KanMX rtt109Δ::URA3MX | This study |
| ASY2807 | BY4741 pol32Δ::KanMX | This study |
| ASY3159 | BY4741 pol32Δ::KanMX rtt109Δ::HPHMX | This study |
| HWY1608 | BY4741 slx4Δ::KanMX | This study |
| ASY1875 | BY4741 slx4Δ::HPHMX rtt109Δ::URA3MX | This study |
| HWY1610 | BY4741 rad1Δ::kanMX 25C10 | This study |
| HWY1609 | BY4741 slx1Δ::kanMX 10E7 | This study |
| ASY3147 | BY4741 mus81Δ::HPHMX | This study |
| HWY3228 | BY4741 mms4Δ::KanMX | This study |
| ASY2164 | BY4741 rtt101Δ::URA3MX | This study |
| ASY2168 | BY4741 rtt101Δ::URA3MX slx4Δ::HPHMX | This study |
| ASY2166 | BY4741 rtt107Δ::KanMX | This study |
| ASY2163 | BY4741 rtt107Δ::KanMX slx4Δ::HPHMX | This study |
| HWY525 | BY4741 rtt107Δ::KanMX | This study |
| HWY530 | BY4741 rtt107Δ::KanMX rtt109Δ::URA3MX | This study |
| ASY1763 | BY4741 psy2Δ::KanMX | This study |
| ASY1764 | BY4741 psy4Δ::KanMX | This study |
| ASY1765 | BY4741 pph3Δ::KanMX | This study |
| ASY1840 | BY4741 pph3Δ::HPHMX rtt109Δ::URA3MX | This study |
| EHY047 | BY4741 rad9Δ::KanMX | This study |
| ASY2796 | BY4741 rad9Δ::KanMX pph3Δ::HPHMX | This study |
| ASY3516 | BY4741 slx4Δ::KanMX rad9Δ::HPHMX | This study |
| EHY071 | BY4741 dot1Δ::KanMX | This study |
| ERY3386 | BY4741 slx4Δ::KanMX dot1Δ::URA3MX | This study |
| ERY3389 | BY4741 pph3Δ::HPHMX dot1Δ::URA3MX | This study |
| FY406 MATa hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-HTA1] | (107) | |
| ERY3394 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-HTA1] pph3Δ::HPHMX | This study |
| ERY3396 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-hta1S128A] pph3Δ::HPHMX | This study |
| HWY2878 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-HTA1] slx4Δ::KanMX | This study |
| HWY2879 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-hta1S128A] slx4Δ::KanMX | This study |
| HWY1936 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-hta1S128A] | This study |
| ASY2766 | W303 ADE2 RAD52-YFP RAD5 slx4Δ::HPHMX | This study |
| ASY2764 | W303 ADE2 RAD52-YFP RAD5 pph3Δ::HPHMX | This study |
| Y2573 | W303 dbf4Δ::TRP1 his3::PDBF4-dbf4–4A::HIS3 sld3–38A-10his-13MYC::KanMX4 | (87) |
| ERY3414 | W303 dbf4Δ::TRP1 his3::PDBF4-dbf4–4A::HIS3 sld3–38A-10his-13MYC::KanMX4 slx4Δ::HPHMX | This study |
| ERY3415 | W303 dbf4Δ::TRP1 his3::PDBF4-dbf4–4A::HIS3 sld3–38A-10his-13MYC::KanMX4 pph3Δ::HPHMx | This study |
| ASY2798 | W303 pph3Δ::HPHMX | This study |
| HWY2882 | W303 slx4Δ::HPHMX | This study |
| HWY2942 | BY4741 slx4Δ::KanMX dot1Δ::URA3MX rev3Δ::HIS3MX | This study |
| ASY3534 | BY4741 pph3Δ::HPHMX dot1Δ::URA3MX rev3Δ::KanMX | This study |
| ASY3667 | BY4741 pol30Δ::KANMX trp1Δ::KANMX slx4Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-POL30-TRP1] | ||
| ASY3668 | BY4741 pol30Δ::KANMX trp1Δ::KANMX slx4Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-pol30-K164R-TRP1] | ||
| ASY3669 | BY4741 pol30Δ::KANMX trp1Δ::KANMX pph3Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-POL30-TRP1] | ||
| ASY3670 | BY4741 pol30Δ::KANMX trp1Δ::KANMX pph3Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-pol30-K164R-TRP1] | ||
| HWY630 | BY4741 rad18Δ::KanMX | This study |
| HWY636 | BY4741 mms2Δ::KanMX | This study |
| ASY3522 | BY4741 slx4Δ::KanMX dot1Δ::URA3 rad18Δ::HIS3MX | This study |
| HWY2939 | BY4741 slx4Δ::kanMX dot1Δ::URA3MX mms2Δ::HIS3MX | This study |
| ASY3651 | BY4741 pph3Δ::KanMX rad18Δ::HIS3MX rad9Δ::URA3MX | This study |
| ASY3654 | BY4741 pph3Δ::KanMX mms2Δ::HPHMX rad9Δ::URA3MX | This study |
| ASY3519 | BY4741 pph3Δ::HPHMX rev3Δ::KanMX | This study |
| HWY2940 | BY4741 slx4Δ::KANMX rev3Δ::HIS3MX | This study |
| ICY703 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 [pCEN URA3 HST3] | (25) |
| ASY3537 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 pph3Δ::HPHMX [pCEN URA3 HST3] | This study |
| ASY3657 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 rtt107Δ::HPHMX [pCEN URA3 HST3] | This study |
| ASY2156 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 slx4Δ::KanMX [pCEN URA3 HST3] | This study |
| ASY3675 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 slx4Δ::KanMX rtt107Δ::HPHMX | This study |
| [pCEN URA3 HST3] | ||
| ASY3678 | BY4741 srl4Δ::KanMX | This study |
| ASY3679 | BY4741 him1Δ::KanMX | This study |
| ASY3680 | BY4741 hug1Δ::KanMX | This study |
| ICY1164 | MATa his3D200 leu2Δ1 lys2Δ202 trp1Δ63 ura3–52 bar1Δ::hygMX hst4Δ::TRP1 hst3::td-HST3- 13MYC::KanMX4 | (25) |
| ASY3139 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 sml1Δ::KanMX [pCEN URA3 HST3] | This study |
| ASY3143 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 sml1Δ::KanMX RAD53–3HA::HPHMX | This study |
| [pCEN URA3 HST3] | ||
| ASY3682 | BY4741 sml1Δ::KanMX slx4Δ::HIS3MX | This study |
| ASY3684 | BY4741 sml1Δ::KanMX pph3Δ::HIS3MX | This study |
| ASY3718 | BY4741 sml1Δ::KanMX slx4Δ::HIS3MX RAD53–3HA::HPHMX | This study |
| ASY3720 | BY4741 sml1Δ::KanMX pph3Δ::HIS3MX RAD53–3HA::HPHMX | This study |
| ASY4003 | BY4741 rev3Δ::KanMX dot1Δ::URA3MX | This study |
| ASY4014 | BY4741 rad9Δ::KanMX rad18Δ::HPHMX | This study |
| ASY4020 | BY4741 dot1Δ::KanMX rad18Δ::HPHMX | This study |
| ASY4023 | W303 RAD5 pph3Δ::HPHMX | This study |
| ASY4024 | W303 rad5–535 | This study |
| ASY4025 | W303 RAD5 | This study |
| ASY4026 | W303 rad5–535 pph3Δ::HPHMX | This study |
| ASY4027 | W303 RAD5 slx4Δ::HPHMX | This study |
| ASY4029 | W303 rad5–535 slx4Δ::HPHMX | This study |
| Strain | Genotype | Reference |
|---|---|---|
| BY4741 | BY4741 MATa ura3Δ0 leu2Δ0 his3Δ1 | (105) |
| BY4743 | BY4743 MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 LYS2/lys2Δ0 met15Δ0/MET15 ura3Δ0/ura3Δ0 | (105) |
| W303 | W303 MATa ade2–1 can1–100 his3–11,15 leu2–3,112 trp1–1 ura3–1 [psi+] rad5–535 | (106) |
| W5094–1C | W303 ADE2 RAD52-YFP RAD5 | (43) |
| HWY2493 | W303 ADE2 bar1Δ::LEU2 RFA1–8ala-YFP RAD5 | (23) |
| HWY297 | BY4741 rtt109Δ::KanMX | This study |
| ASY3111 | YBL574 hht1-hhf1Δ::LEU2 hht2-hhf2Δ::HIS3 [pCEN TRP1 HHT1-HHF1] | (107) |
| ASY3113 | YBL574 hht1-hhf1Δ::LEU2 hht2-hhf2Δ::HIS3 [pCEN TRP1 HHT-hhf1K16A] | (107) |
| HWY2949 | YBL574 hht1-hhf1Δ::LEU2 hht2-hhf2Δ::HIS3 [pCEN TRP1 HHT1-HHF1] rtt109Δ::URA3MX | This study |
| ASY3180 | BY4741 dpb4Δ::KanMx | This study |
| HWY2417 | BY4741 dun1Δ::KanMX | This study |
| HWY634 | BY4741 srs2Δ::KanMX | This study |
| ASY3188 | BY4741 tof1Δ::KanMX | This study |
| ASY3193 | BY4741 sae2Δ::KanMX | This study |
| HWY2477 | BY4741 mrc1Δ::KanMX | This study |
| HWY2460 | BY4741 mrc1Δ::KanMX rtt109Δ::URA3MX | This study |
| ASY1767 | BY4741 yku70Δ::KanMX | This study |
| ASY3164 | BY4741 yku70Δ::KanMX rtt109Δ::HPHMX | This study |
| EHY027 | BY4741 rad59Δ::KanMX | This study |
| EHY029 | BY4741 rad59Δ::KanMX rtt109Δ::URA3MX | This study |
| ASY2807 | BY4741 pol32Δ::KanMX | This study |
| ASY3159 | BY4741 pol32Δ::KanMX rtt109Δ::HPHMX | This study |
| HWY1608 | BY4741 slx4Δ::KanMX | This study |
| ASY1875 | BY4741 slx4Δ::HPHMX rtt109Δ::URA3MX | This study |
| HWY1610 | BY4741 rad1Δ::kanMX 25C10 | This study |
| HWY1609 | BY4741 slx1Δ::kanMX 10E7 | This study |
| ASY3147 | BY4741 mus81Δ::HPHMX | This study |
| HWY3228 | BY4741 mms4Δ::KanMX | This study |
| ASY2164 | BY4741 rtt101Δ::URA3MX | This study |
| ASY2168 | BY4741 rtt101Δ::URA3MX slx4Δ::HPHMX | This study |
| ASY2166 | BY4741 rtt107Δ::KanMX | This study |
| ASY2163 | BY4741 rtt107Δ::KanMX slx4Δ::HPHMX | This study |
| HWY525 | BY4741 rtt107Δ::KanMX | This study |
| HWY530 | BY4741 rtt107Δ::KanMX rtt109Δ::URA3MX | This study |
| ASY1763 | BY4741 psy2Δ::KanMX | This study |
| ASY1764 | BY4741 psy4Δ::KanMX | This study |
| ASY1765 | BY4741 pph3Δ::KanMX | This study |
| ASY1840 | BY4741 pph3Δ::HPHMX rtt109Δ::URA3MX | This study |
| EHY047 | BY4741 rad9Δ::KanMX | This study |
| ASY2796 | BY4741 rad9Δ::KanMX pph3Δ::HPHMX | This study |
| ASY3516 | BY4741 slx4Δ::KanMX rad9Δ::HPHMX | This study |
| EHY071 | BY4741 dot1Δ::KanMX | This study |
| ERY3386 | BY4741 slx4Δ::KanMX dot1Δ::URA3MX | This study |
| ERY3389 | BY4741 pph3Δ::HPHMX dot1Δ::URA3MX | This study |
| FY406 MATa hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-HTA1] | (107) | |
| ERY3394 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-HTA1] pph3Δ::HPHMX | This study |
| ERY3396 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-hta1S128A] pph3Δ::HPHMX | This study |
| HWY2878 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-HTA1] slx4Δ::KanMX | This study |
| HWY2879 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-hta1S128A] slx4Δ::KanMX | This study |
| HWY1936 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-hta1S128A] | This study |
| ASY2766 | W303 ADE2 RAD52-YFP RAD5 slx4Δ::HPHMX | This study |
| ASY2764 | W303 ADE2 RAD52-YFP RAD5 pph3Δ::HPHMX | This study |
| Y2573 | W303 dbf4Δ::TRP1 his3::PDBF4-dbf4–4A::HIS3 sld3–38A-10his-13MYC::KanMX4 | (87) |
| ERY3414 | W303 dbf4Δ::TRP1 his3::PDBF4-dbf4–4A::HIS3 sld3–38A-10his-13MYC::KanMX4 slx4Δ::HPHMX | This study |
| ERY3415 | W303 dbf4Δ::TRP1 his3::PDBF4-dbf4–4A::HIS3 sld3–38A-10his-13MYC::KanMX4 pph3Δ::HPHMx | This study |
| ASY2798 | W303 pph3Δ::HPHMX | This study |
| HWY2882 | W303 slx4Δ::HPHMX | This study |
| HWY2942 | BY4741 slx4Δ::KanMX dot1Δ::URA3MX rev3Δ::HIS3MX | This study |
| ASY3534 | BY4741 pph3Δ::HPHMX dot1Δ::URA3MX rev3Δ::KanMX | This study |
| ASY3667 | BY4741 pol30Δ::KANMX trp1Δ::KANMX slx4Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-POL30-TRP1] | ||
| ASY3668 | BY4741 pol30Δ::KANMX trp1Δ::KANMX slx4Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-pol30-K164R-TRP1] | ||
| ASY3669 | BY4741 pol30Δ::KANMX trp1Δ::KANMX pph3Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-POL30-TRP1] | ||
| ASY3670 | BY4741 pol30Δ::KANMX trp1Δ::KANMX pph3Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-pol30-K164R-TRP1] | ||
| HWY630 | BY4741 rad18Δ::KanMX | This study |
| HWY636 | BY4741 mms2Δ::KanMX | This study |
| ASY3522 | BY4741 slx4Δ::KanMX dot1Δ::URA3 rad18Δ::HIS3MX | This study |
| HWY2939 | BY4741 slx4Δ::kanMX dot1Δ::URA3MX mms2Δ::HIS3MX | This study |
| ASY3651 | BY4741 pph3Δ::KanMX rad18Δ::HIS3MX rad9Δ::URA3MX | This study |
| ASY3654 | BY4741 pph3Δ::KanMX mms2Δ::HPHMX rad9Δ::URA3MX | This study |
| ASY3519 | BY4741 pph3Δ::HPHMX rev3Δ::KanMX | This study |
| HWY2940 | BY4741 slx4Δ::KANMX rev3Δ::HIS3MX | This study |
| ICY703 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 [pCEN URA3 HST3] | (25) |
| ASY3537 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 pph3Δ::HPHMX [pCEN URA3 HST3] | This study |
| ASY3657 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 rtt107Δ::HPHMX [pCEN URA3 HST3] | This study |
| ASY2156 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 slx4Δ::KanMX [pCEN URA3 HST3] | This study |
| ASY3675 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 slx4Δ::KanMX rtt107Δ::HPHMX | This study |
| [pCEN URA3 HST3] | ||
| ASY3678 | BY4741 srl4Δ::KanMX | This study |
| ASY3679 | BY4741 him1Δ::KanMX | This study |
| ASY3680 | BY4741 hug1Δ::KanMX | This study |
| ICY1164 | MATa his3D200 leu2Δ1 lys2Δ202 trp1Δ63 ura3–52 bar1Δ::hygMX hst4Δ::TRP1 hst3::td-HST3- 13MYC::KanMX4 | (25) |
| ASY3139 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 sml1Δ::KanMX [pCEN URA3 HST3] | This study |
| ASY3143 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 sml1Δ::KanMX RAD53–3HA::HPHMX | This study |
| [pCEN URA3 HST3] | ||
| ASY3682 | BY4741 sml1Δ::KanMX slx4Δ::HIS3MX | This study |
| ASY3684 | BY4741 sml1Δ::KanMX pph3Δ::HIS3MX | This study |
| ASY3718 | BY4741 sml1Δ::KanMX slx4Δ::HIS3MX RAD53–3HA::HPHMX | This study |
| ASY3720 | BY4741 sml1Δ::KanMX pph3Δ::HIS3MX RAD53–3HA::HPHMX | This study |
| ASY4003 | BY4741 rev3Δ::KanMX dot1Δ::URA3MX | This study |
| ASY4014 | BY4741 rad9Δ::KanMX rad18Δ::HPHMX | This study |
| ASY4020 | BY4741 dot1Δ::KanMX rad18Δ::HPHMX | This study |
| ASY4023 | W303 RAD5 pph3Δ::HPHMX | This study |
| ASY4024 | W303 rad5–535 | This study |
| ASY4025 | W303 RAD5 | This study |
| ASY4026 | W303 rad5–535 pph3Δ::HPHMX | This study |
| ASY4027 | W303 RAD5 slx4Δ::HPHMX | This study |
| ASY4029 | W303 rad5–535 slx4Δ::HPHMX | This study |
| Strain | Genotype | Reference |
|---|---|---|
| BY4741 | BY4741 MATa ura3Δ0 leu2Δ0 his3Δ1 | (105) |
| BY4743 | BY4743 MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 LYS2/lys2Δ0 met15Δ0/MET15 ura3Δ0/ura3Δ0 | (105) |
| W303 | W303 MATa ade2–1 can1–100 his3–11,15 leu2–3,112 trp1–1 ura3–1 [psi+] rad5–535 | (106) |
| W5094–1C | W303 ADE2 RAD52-YFP RAD5 | (43) |
| HWY2493 | W303 ADE2 bar1Δ::LEU2 RFA1–8ala-YFP RAD5 | (23) |
| HWY297 | BY4741 rtt109Δ::KanMX | This study |
| ASY3111 | YBL574 hht1-hhf1Δ::LEU2 hht2-hhf2Δ::HIS3 [pCEN TRP1 HHT1-HHF1] | (107) |
| ASY3113 | YBL574 hht1-hhf1Δ::LEU2 hht2-hhf2Δ::HIS3 [pCEN TRP1 HHT-hhf1K16A] | (107) |
| HWY2949 | YBL574 hht1-hhf1Δ::LEU2 hht2-hhf2Δ::HIS3 [pCEN TRP1 HHT1-HHF1] rtt109Δ::URA3MX | This study |
| ASY3180 | BY4741 dpb4Δ::KanMx | This study |
| HWY2417 | BY4741 dun1Δ::KanMX | This study |
| HWY634 | BY4741 srs2Δ::KanMX | This study |
| ASY3188 | BY4741 tof1Δ::KanMX | This study |
| ASY3193 | BY4741 sae2Δ::KanMX | This study |
| HWY2477 | BY4741 mrc1Δ::KanMX | This study |
| HWY2460 | BY4741 mrc1Δ::KanMX rtt109Δ::URA3MX | This study |
| ASY1767 | BY4741 yku70Δ::KanMX | This study |
| ASY3164 | BY4741 yku70Δ::KanMX rtt109Δ::HPHMX | This study |
| EHY027 | BY4741 rad59Δ::KanMX | This study |
| EHY029 | BY4741 rad59Δ::KanMX rtt109Δ::URA3MX | This study |
| ASY2807 | BY4741 pol32Δ::KanMX | This study |
| ASY3159 | BY4741 pol32Δ::KanMX rtt109Δ::HPHMX | This study |
| HWY1608 | BY4741 slx4Δ::KanMX | This study |
| ASY1875 | BY4741 slx4Δ::HPHMX rtt109Δ::URA3MX | This study |
| HWY1610 | BY4741 rad1Δ::kanMX 25C10 | This study |
| HWY1609 | BY4741 slx1Δ::kanMX 10E7 | This study |
| ASY3147 | BY4741 mus81Δ::HPHMX | This study |
| HWY3228 | BY4741 mms4Δ::KanMX | This study |
| ASY2164 | BY4741 rtt101Δ::URA3MX | This study |
| ASY2168 | BY4741 rtt101Δ::URA3MX slx4Δ::HPHMX | This study |
| ASY2166 | BY4741 rtt107Δ::KanMX | This study |
| ASY2163 | BY4741 rtt107Δ::KanMX slx4Δ::HPHMX | This study |
| HWY525 | BY4741 rtt107Δ::KanMX | This study |
| HWY530 | BY4741 rtt107Δ::KanMX rtt109Δ::URA3MX | This study |
| ASY1763 | BY4741 psy2Δ::KanMX | This study |
| ASY1764 | BY4741 psy4Δ::KanMX | This study |
| ASY1765 | BY4741 pph3Δ::KanMX | This study |
| ASY1840 | BY4741 pph3Δ::HPHMX rtt109Δ::URA3MX | This study |
| EHY047 | BY4741 rad9Δ::KanMX | This study |
| ASY2796 | BY4741 rad9Δ::KanMX pph3Δ::HPHMX | This study |
| ASY3516 | BY4741 slx4Δ::KanMX rad9Δ::HPHMX | This study |
| EHY071 | BY4741 dot1Δ::KanMX | This study |
| ERY3386 | BY4741 slx4Δ::KanMX dot1Δ::URA3MX | This study |
| ERY3389 | BY4741 pph3Δ::HPHMX dot1Δ::URA3MX | This study |
| FY406 MATa hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-HTA1] | (107) | |
| ERY3394 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-HTA1] pph3Δ::HPHMX | This study |
| ERY3396 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-hta1S128A] pph3Δ::HPHMX | This study |
| HWY2878 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-HTA1] slx4Δ::KanMX | This study |
| HWY2879 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-hta1S128A] slx4Δ::KanMX | This study |
| HWY1936 | FY406 hta1-htb1Δ::LEU2 hta2-htb2Δ::TRP1 [pCEN HIS3 HTB1-hta1S128A] | This study |
| ASY2766 | W303 ADE2 RAD52-YFP RAD5 slx4Δ::HPHMX | This study |
| ASY2764 | W303 ADE2 RAD52-YFP RAD5 pph3Δ::HPHMX | This study |
| Y2573 | W303 dbf4Δ::TRP1 his3::PDBF4-dbf4–4A::HIS3 sld3–38A-10his-13MYC::KanMX4 | (87) |
| ERY3414 | W303 dbf4Δ::TRP1 his3::PDBF4-dbf4–4A::HIS3 sld3–38A-10his-13MYC::KanMX4 slx4Δ::HPHMX | This study |
| ERY3415 | W303 dbf4Δ::TRP1 his3::PDBF4-dbf4–4A::HIS3 sld3–38A-10his-13MYC::KanMX4 pph3Δ::HPHMx | This study |
| ASY2798 | W303 pph3Δ::HPHMX | This study |
| HWY2882 | W303 slx4Δ::HPHMX | This study |
| HWY2942 | BY4741 slx4Δ::KanMX dot1Δ::URA3MX rev3Δ::HIS3MX | This study |
| ASY3534 | BY4741 pph3Δ::HPHMX dot1Δ::URA3MX rev3Δ::KanMX | This study |
| ASY3667 | BY4741 pol30Δ::KANMX trp1Δ::KANMX slx4Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-POL30-TRP1] | ||
| ASY3668 | BY4741 pol30Δ::KANMX trp1Δ::KANMX slx4Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-pol30-K164R-TRP1] | ||
| ASY3669 | BY4741 pol30Δ::KANMX trp1Δ::KANMX pph3Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-POL30-TRP1] | ||
| ASY3670 | BY4741 pol30Δ::KANMX trp1Δ::KANMX pph3Δ::HPHMX rad9Δ::HIS3MX | This study |
| [pCEN-pol30-K164R-TRP1] | ||
| HWY630 | BY4741 rad18Δ::KanMX | This study |
| HWY636 | BY4741 mms2Δ::KanMX | This study |
| ASY3522 | BY4741 slx4Δ::KanMX dot1Δ::URA3 rad18Δ::HIS3MX | This study |
| HWY2939 | BY4741 slx4Δ::kanMX dot1Δ::URA3MX mms2Δ::HIS3MX | This study |
| ASY3651 | BY4741 pph3Δ::KanMX rad18Δ::HIS3MX rad9Δ::URA3MX | This study |
| ASY3654 | BY4741 pph3Δ::KanMX mms2Δ::HPHMX rad9Δ::URA3MX | This study |
| ASY3519 | BY4741 pph3Δ::HPHMX rev3Δ::KanMX | This study |
| HWY2940 | BY4741 slx4Δ::KANMX rev3Δ::HIS3MX | This study |
| ICY703 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 [pCEN URA3 HST3] | (25) |
| ASY3537 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 pph3Δ::HPHMX [pCEN URA3 HST3] | This study |
| ASY3657 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 rtt107Δ::HPHMX [pCEN URA3 HST3] | This study |
| ASY2156 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 slx4Δ::KanMX [pCEN URA3 HST3] | This study |
| ASY3675 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 slx4Δ::KanMX rtt107Δ::HPHMX | This study |
| [pCEN URA3 HST3] | ||
| ASY3678 | BY4741 srl4Δ::KanMX | This study |
| ASY3679 | BY4741 him1Δ::KanMX | This study |
| ASY3680 | BY4741 hug1Δ::KanMX | This study |
| ICY1164 | MATa his3D200 leu2Δ1 lys2Δ202 trp1Δ63 ura3–52 bar1Δ::hygMX hst4Δ::TRP1 hst3::td-HST3- 13MYC::KanMX4 | (25) |
| ASY3139 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 sml1Δ::KanMX [pCEN URA3 HST3] | This study |
| ASY3143 | FY833 hst3Δ::HIS3 hst4Δ::TRP1 sml1Δ::KanMX RAD53–3HA::HPHMX | This study |
| [pCEN URA3 HST3] | ||
| ASY3682 | BY4741 sml1Δ::KanMX slx4Δ::HIS3MX | This study |
| ASY3684 | BY4741 sml1Δ::KanMX pph3Δ::HIS3MX | This study |
| ASY3718 | BY4741 sml1Δ::KanMX slx4Δ::HIS3MX RAD53–3HA::HPHMX | This study |
| ASY3720 | BY4741 sml1Δ::KanMX pph3Δ::HIS3MX RAD53–3HA::HPHMX | This study |
| ASY4003 | BY4741 rev3Δ::KanMX dot1Δ::URA3MX | This study |
| ASY4014 | BY4741 rad9Δ::KanMX rad18Δ::HPHMX | This study |
| ASY4020 | BY4741 dot1Δ::KanMX rad18Δ::HPHMX | This study |
| ASY4023 | W303 RAD5 pph3Δ::HPHMX | This study |
| ASY4024 | W303 rad5–535 | This study |
| ASY4025 | W303 RAD5 | This study |
| ASY4026 | W303 rad5–535 pph3Δ::HPHMX | This study |
| ASY4027 | W303 RAD5 slx4Δ::HPHMX | This study |
| ASY4029 | W303 rad5–535 slx4Δ::HPHMX | This study |
Growth assays in 96 well plates
Cells were grown overnight in YPD in a humid chamber at 30°C. Cells were then diluted to OD600 0.0005 in 100 μl YPD containing nicotinamide in flat-bottomed 96 well plates. Plates were incubated for 48 h at 30°C in a humid chamber and OD630 was measured using a Biotek EL800 plate reader equipped with Gen5 version 1.05 software (Biotek instruments). OD630 from blank wells (YPD) was subtracted from OD630 readings and growth was normalized to untreated controls for each strain. Experiments were performed at least in triplicate and error bars represent the standard error of the mean of normalized growth. To calculate population doubling time, cells were grown overnight in YPD at 30°C. Cells were then diluted to OD600 0.01 in 100 μl YPD with or without 20 mM NAM in flat-bottomed 96 well plates. Cells were incubated for 48 h at room temperature with shaking in a Biotek ELX808 and OD630 readings taken every 30 min. OD630 readings were plotted on a graph, and exponential regression was used to calculate doubling times.
Cell synchronization and treatment with MMS
Cells were grown overnight in YPD medium at 25°C and arrested in G1 at 30°C in YPD containing 5 μg/ml α-factor for 90 min, followed by addition of a second dose of 5 μg/ml α-factor for 75 min. Cells were released into the cell cycle by washing them once with YPD and resuspending them in medium containing 50 μg/ml pronase (Sigma-Aldrich, P6911–1G) and methyl methanesulfonate (MMS).
Measurement of DNA content by flow cytometry
Cells were fixed with 70% ethanol prior to flow cytometry analysis, and DNA content was determined using Sytox Green (Invitrogen) as described (40). Flow cytometry was performed on a FACS Calibur instrument using the Cell Quest software. Graphs were generated using FlowJo 7.6.5 (FlowJo, LLC).
Immunoblots
Whole-cell lysates were prepared for SDS–polyacrylamide gel electrophoresis using an alkaline cell lysis (41) or standard glass beads/trichloroacetic acid precipitation methods. SDS-PAGE and protein transfers were performed using standard molecular biology protocols. Monoclonal anti-myc antibody (9E10) was purchased from Sigma-Aldrich. The antibody against histone H2A was purchased from Active Motif (Cat. No 39236). Anti-H3 (AV100), anti-H3K56ac (AV105) and anti-phosphorylated H2A (AV137) antibodies (29) were kindly provided by Dr Alain Verreault (Université de Montréal, Canada).
Rad53 autophosphorylation assays
Protein samples were prepared by the glass beads/tricholoroacetic acid precipitation method, resolved by SDS-PAGE and transferred to PVDF membranes using standard Towbin buffer (25 mM Tris and 192 mM glycine) without methanol or SDS at 0.8 mA cm−2 for 2 h in a Bio-Rad SD semi-dry transfer apparatus. Membranes were then processed as previously described (42).
Fluorescence microscopy
Cell samples were fixed using formaldehyde as described (43) and examined using a Zeiss Z2 Imager fluorescence microscope equipped with the AxioVision software or a DeltaVision fluorescence microscope equipped with SoftWorx (GE Healthcare). Images were analyzed with Image J 1.46E.
Determination of intracellular ROS by dihydrorhodamine 123 Staining
Intracellular Reactive Oxygen Species (ROS) levels were monitored as described (44). Briefly, cells were washed with water, resuspended in PBS + 15 μg/ml dihydrorhodamine 123 and incubated for 90 min at 30°C with shaking. Cell pellets were washed twice with Phosphate Buffered Saline (PBS) and fluorescence was measured in 10 000 cells using a FACS Calibur Flow cytometer equipped with the Cell Quest software. FlowJo 7.6.5 (FlowJo, LLC) was used for statistical analysis of the fluorescence distribution in the queried samples.
Fitness assays
Genome-wide fitness assays were performed as described (45). Briefly, duplicate pools of strains from the ‘barcoded’ BY4743 homozygous diploid mutant collection were incubated in YPD with or without 20 mM NAM at 30°C. Cells were collected at 0 generation (population doubling) to assess initial strain representation, and after 5 and 20 generations of exponential growth. Genomic DNA was extracted using the Zymo Research YeaStar kit as described (46). For each sample, two PCR reactions were performed (to amplify uptag and downtag sequences). Amplified DNAs were combined, and used to probe high-density oligonucleotide Affymetrix TAG4 DNA microarrays (47). These arrays contain at least five replicate features for each up- and downtags that are dispersed across the array so that outlier features can be discarded before calculating average intensity values for each tag. Hybridization, washing, staining and scanning were performed as described (46). After removal of outliers, intensity values for each tag were calculated by averaging unmasked replicates. Methods for outlier masking, correction for saturation, normalization and calculating sensitivity scores are described in detail elsewhere (45,46). Z-scores for each deletion mutant were calculated after 5 and 20 generations using the equation (X-mean/standard deviation). Gene ontology (GO) analysis was performed using the GO Term Finder tool of the Saccharomyces Genome Database (48,49). P-values ≤ 0.01 were considered significant. Redundant GO terms were removed using the REViGO web server (50).
Venn diagrams
Venn diagrams were generated using genes whose mutation was associated with an absolute z-score of over 2.58 (P-value < 0.01) from our data sets and those of Hillenmeyer et al. (51). P-values associated with the overlap in data sets were calculated using a hypergeometric function, taking into account that 4767 mutants were included in our analysis.
RNA profiling assay, microarray hybridization and data analysis
BY4743 cells were grown as described for fitness assays, in the absence or presence of 20 mM NAM for 1, 5 and 20 generations. Three independent biological replicates were processed, and 50 OD600nm units of cells were harvested at each time point. Cells were pelleted and washed with diethylpyrocarbonate (DEPC)-treated water. Cells were centrifuged at 3000 rpm for 3 min at room temperature, and cell pellets were snap-frozen in liquid nitrogen and stored at −80°C. Total RNA was extracted using a ‘hot phenol’ protocol (52) and purified using a QIAGEN RNeasy mini kit. Total RNA was quantified using a NanoDrop Spectrophotometer ND-1000 (NanoDrop Technologies, Inc.) and its integrity assessed using a 2100 Bioanalyzer (Agilent Technologies). Three independent RNA preparations were prepared for each time point (1, 5 or 20 generation) and NAM concentration (0 or 20 mM). Double stranded cDNA was synthesized from 100 ng of total RNA and in vitro transcription was performed to produce biotin-labeled cRNA using the Affymetrix Gene Chip 3′ IVT Express reagent kit (Affymetrix). After fragmentation, 5 μg of cRNA was hybridized on Yeast Genome 2.0 arrays (Affymetrix) and incubated at 45°C in a Genechip® Hybridization oven 640 (Affymetrix) for 16 h at 60 rpm. GeneChips were then washed in a GeneChips® Fluidics Station 450 (Affymetrix) using Affymetrix Hybridization Wash and Stain kit. Microarrays were scanned on a GeneChip® scanner 3000 (Affymetrix), and data were analyzed using the Partek Genomic Suite. GO analysis was performed using the GO Term Finder tool of the Saccharomyces Genome Database (48,49). P-values ≤ 0.005 were considered significant. Redundant GO terms were removed using the REViGO web server (50).
Gene set enrichment analysis (GSEA)
Fitness assay and RNA profiling data were analyzed as previously described (53).
RESULTS
Nicotinamide is genotoxic in Saccharomyces cerevisiae
We investigated the consequences of NAM-induced inhibition of sirtuins in fungi using S. cerevisiae as a model. Addition of 20, 50 or 100 mM NAM inhibited cell proliferation in a dose-dependent manner (Figure 1A). As reported, NAM exposure elevated H3K56ac levels, which is consistent with inhibition of Hst3 and Hst4 (Figure 1B) (25). As assessed by in situ autophosphorylation assay (Figure 1B), NAM also caused activation of Rad53, a critical DDR kinase acting downstream of Mec1 (42). In contrast, we did not detect NAM-induced modulation of H4K16ac, a well-known target of Sir2 and Hst2 in yeast (9–10,20). Exposure to 20 mM NAM also caused formation of Rad52-YFP and Rfa1-YFP foci, which are hallmarks of DNA damage and repair in yeast (Figure 1C) (54). We conclude that NAM either causes DNA lesions in yeast, or prevents the repair of endogenous DNA damage, leading to elevated DDR marks. Intriguingly, we found that 100 mM NAM did not induce Rad53 activation as strongly as 20 or 50 mM (Figure 1B), suggesting that growth inhibition caused by high NAM concentrations may not result solely from DNA damage.
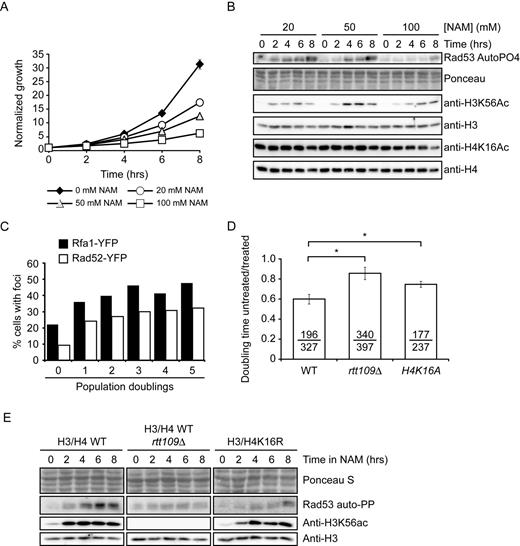
Exposure to NAM causes DNA damage in yeast. (A) NAM inhibits cell proliferation in a dose-dependent manner. Cells were grown in YPD containing the indicated concentrations of NAM. Cell growth was monitored by OD630 measurements. (B) NAM activates the DNA damage response kinase Rad53 and causes H3K56 hyperacetylation. Exponentially growing yeast cells were treated with NAM and samples were collected at the indicated time for immunoblotting and Rad53 autophosphorylation assays. (C) NAM causes the formation of Rad52-YFP and Rfa1-YFP foci. Exponentially growing yeasts were treated with 20 mM NAM and samples were examined by microscopy at the indicated time points. (D) NAM-induced growth defects result from H3K56 and H4K16 acetylation. Doubling times for strains of indicated genotypes were measured in YPD or YPD + 20 mM NAM, and values are represented as a ratio of the doubling time in NAM versus YPD. Error bars: standard error of the mean (3–6 experiments). Doubling time with/without NAM are indicated (in minutes; untreated/treated). (E) Lack of H3K56ac or H4K16ac attenuates NAM-induced activation of Rad53. Exponentially growing yeasts were treated with 20 mM NAM and processed as in (C). NAM: nicotinamide, * : P-value < 0.05 as calculated by unpaired one-tailed Student's t-test.
Published data clearly indicates that most, if not all, phenotypes observed in hst3Δ hst4Δ cells are due to increased H3K56ac (22,24–25). Consistent with a central role for this modification in causing NAM-induced DNA damage, lack of the H3K56 acetyltransferase Rtt109 abrogated growth inhibition and Rad53 activation caused by 20 mM NAM (Figure 1D–E). Although we did not observe any obvious increase in H4K16 acetylation levels by immunoblot assays (Figure 1B), it remains possible that subtle or localized increase in levels of this modification may contribute to NAM-induced DNA damage induction and growth inhibition. Consistently, we found that mutation of H4K16 to non-acetylatable arginine or alanine residues (H4K16R/A) improved growth in NAM-containing medium (Figure 1D) and reduced Rad53 activation (Figure 1E). We conclude that H3K56 hyperacetylation plays a dominant role in NAM-induced genotoxicity, but that other factors including H4K16ac may also contribute.
A genome-wide screen for Saccharomyces cerevisiae genes that modulate fitness in the presence of nicotinamide
To investigate the basis of NAM-induced inhibition of cell proliferation, we used two genome-wide approaches: a fitness assay to identify genes whose deletion results in sensitivity or resistance to NAM, and mRNA profiling to identify genes that are up- or down-regulated in response to NAM. Fitness assays using ‘barcoded’ yeast deletion collections have been used to study the genetic networks that respond to various stimuli (51,55). Briefly, strains of the ‘barcoded’ yeast homozygote diploid non-essential gene deletion collection were pooled and grown in the absence or presence of 20 mM NAM for 5 and 20 population doublings. Genomic DNA from treated and untreated cells was extracted, and DNA barcode sequences were amplified by PCR and hybridized to microarrays to assess their relative abundance (see Materials and Methods). Z-scores were derived from the comparison between treated and non-treated samples, and scores above or below ±2.58 (99% cumulative percentage) were further considered for analysis. Using these criteria, we found that more genes conferred fitness defects than advantage after 5 (84 versus 12, respectively) or 20 (467 versus 342, respectively) generations in the presence of NAM (Figure 2A–B and Supplementary Table S1). GO-term analysis of genes whose mutation caused significant fitness defects in NAM at 5 generations revealed an obvious enrichment for processes related to the DDR and replicative stress (Table 2). This was also the case at 20 generations, although other cellular processes were also identified that may be linked to reduced cell proliferation associated with long term growth in NAM (e.g. catabolic processes). Interestingly, the set of genes whose deletion conferred growth disadvantage in NAM significantly overlaps with published fitness analyses of pools of deletion strains treated with either of two genotoxic drugs: camptothecin (CPT) or methyl methanesulfonate (MMS) (Figure 2C–D) (51). We also applied Gene Set Enrichment Analysis (GSEA) (53) to correlate our fitness assays with available S. cerevisiae genome annotations including GO terms, protein–protein complexes (56) and genetic interactions (57). This analysis indicated a strong overlap between our data set and others describing physical or genetic interactions with genes involved in DNA repair, DNA damage signaling, and DNA replication (Figure 2F and Supplementary Table S2). These observations confirm that growth of S. cerevisiae cells in the presence of NAM strongly solicits cellular DDR pathways.
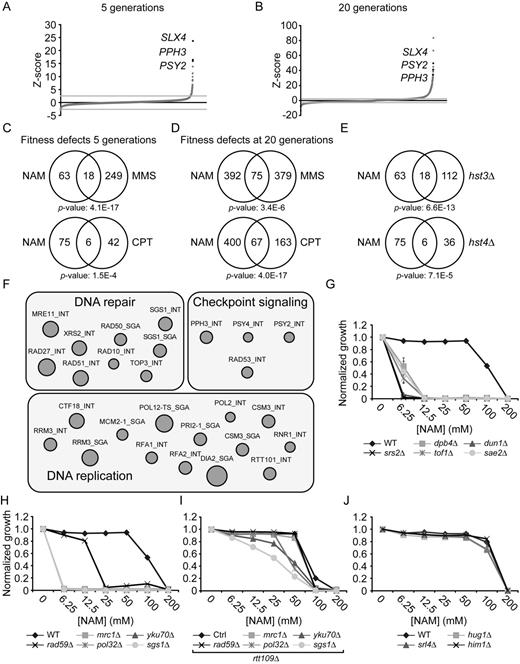
Genome-wide response to NAM-induced sirtuin inhibition. (A–B) Graphical representation of results from NAM fitness assays at 5 (A) and 20 (B) generations. Mutants were plotted according to their Z-score from lowest to highest. (C–D) Growth in NAM, methyl methanesulfonate (MMS) and camptothecin (CPT) share similar genetic requirements. Fitness assays data sets were compared and Venn diagrams were generated as described in Materials and Methods. Statistically significant results from the NAM fitness test were compared to published fitness assays in which cells were treated with either CPT or MMS for 5 (C) or 20 (D) generations. (E) Genes whose mutation reduces fitness in NAM overlap with those presenting negative genetic interaction with HST3 and HST4. (F) Gene set enrichment analysis was performed on statistically significant positive hits from the NAM fitness assay (see text for details). (G–I) Validation of fitness assays results using haploid deletion strains. Cells were grown in 96 well plates and OD630 readings were acquired as described in Materials and Methods. (J) Mutation of DNA damage response genes that are overexpressed in response to NAM do not influence growth in NAM-containing medium.
GO term analysis of fitness test data
| GO-Term ID | Description | Cluster frequency | Background frequency | P-value | Generation | Fitness |
|---|---|---|---|---|---|---|
| 0006974 | Cellular response to DNA damage stimulus | 23/81 | 299/7163 | 3.29E-11 | 5 | − |
| 0006259 | DNA metabolic process | 26/81 | 448/7163 | 4.13E-10 | 5 | − |
| 0006950 | Response to stress | 28/81 | 655/7163 | 7.02E-09 | 5 | − |
| 0050896 | Response to stimulus | 35/81 | 996/7163 | 1.30E-08 | 5 | − |
| 0006310 | DNA recombination | 14/81 | 181/7163 | 4.70E-06 | 5 | − |
| 0051052 | Regulation of DNA metabolic process | 11/81 | 100/7163 | 5.42E-06 | 5 | − |
| 0006260 | DNA replication | 12/81 | 153/7163 | 5.16E-05 | 5 | − |
| 0051053 | Negative regulation of DNA metabolic process | 7/81 | 36/7163 | 5.46E-05 | 5 | − |
| 0065007 | Biological regulation | 38/81 | 1536/7163 | 9.81E-05 | 5 | − |
| 0090304 | Nucleic acid metabolic process | 38/81 | 1573/7163 | 1.70E-04 | 5 | − |
| 0019222 | Regulation of metabolic process | 28/81 | 958/7163 | 3.50E-04 | 5 | − |
| 0006261 | DNA-dependent DNA replication | 10/81 | 128/7163 | 6.80E-04 | 5 | − |
| 0043170 | Macromolecule metabolic process | 54/81 | 2946/7163 | 8.20E-04 | 5 | − |
| 0044260 | Cellular macromolecule metabolic process | 53/81 | 2870/7163 | 9.60E-04 | 5 | − |
| 0007530 | Sex determination | 6/81 | 35/7163 | 1.03E-03 | 5 | − |
| 0022616 | DNA strand elongation | 6/81 | 37/7163 | 1.45E-03 | 5 | − |
| 0007049 | Cell cycle | 21/81 | 637/7163 | 2.71E-03 | 5 | − |
| 0044237 | Cellular metabolic process | 61/81 | 3724/7163 | 3.42E-03 | 5 | − |
| 0006139 | Nucleobase-containing compound metabolic process | 38/81 | 1773/7163 | 4.01E-03 | 5 | − |
| 0071897 | DNA biosynthetic process | 5/81 | 27/7163 | 5.22E-03 | 5 | − |
| 0044238 | Primary metabolic process | 58/81 | 3499/7163 | 7.11E-03 | 5 | − |
| 0071704 | Organic substance metabolic process | 60/81 | 3708/7163 | 8.45E-03 | 5 | − |
| 0065007 | Biological regulation | 163/467 | 1536/7163 | 1.22E-09 | 20 | − |
| 0022402 | Cell cycle process | 79/467 | 594/7163 | 2.28E-06 | 20 | − |
| 0007049 | Cell cycle | 80/467 | 637/7163 | 6.80E-06 | 20 | − |
| 0051276 | Chromosome organization | 64/467 | 495/7163 | 5.15E-05 | 20 | − |
| 0050896 | Response to stimulus | 103/467 | 996/7163 | 6.29E-05 | 20 | − |
| 0006974 | Cellular response to DNA damage stimulus | 45/467 | 299/7163 | 8.15E-05 | 20 | − |
| 0048285 | Organelle fission | 52/467 | 390/7163 | 8.16E-05 | 20 | − |
| 0006259 | DNA metabolic process | 59/467 | 448/7163 | 9.67E-05 | 20 | − |
| 0006950 | Response to stress | 73/467 | 655/7163 | 2.20E-04 | 20 | − |
| 0044699 | Single-organism process | 286/467 | 3588/7163 | 2.70E-04 | 20 | − |
| 0009057 | Macromolecule catabolic process | 53/467 | 392/7163 | 4.30E-04 | 20 | − |
| 0009056 | Catabolic process | 79/467 | 710/7163 | 8.20E-04 | 20 | − |
| 0044248 | Cellular catabolic process | 74/467 | 657/7163 | 1.24E-03 | 20 | − |
| 2000113 | Negative regulation of cellular macromolecule biosynthetic process | 42/467 | 302/7163 | 2.04E-03 | 20 | − |
| 0044265 | Cellular macromolecule catabolic process | 49/467 | 370/7163 | 2.36E-03 | 20 | − |
| 0044763 | Single-organism cellular process | 254/467 | 3165/7163 | 4.14E-03 | 20 | − |
| 0044767 | Single-organism developmental process | 38/467 | 268/7163 | 4.48E-03 | 20 | − |
| 0032502 | Developmental process | 38/467 | 271/7163 | 5.87E-03 | 20 | − |
| 0007533 | Mating type switching | 10/467 | 28/7163 | 6.95E-03 | 20 | − |
| 0010526 | Negative regulation of transposition, RNA-mediated | 3/12 | 8/7163 | 2.06E-05 | 5 | + |
| 0031297 | Replication fork processing | 2/12 | 4/7163 | 1.59E-03 | 5 | + |
| 0007005 | Mitochondrion organization | 40/342 | 390/7163 | 2.35E-03 | 20 | + |
| 0017182 | Peptidyl-diphthamide metabolic process | 5/342 | 7/7163 | 4.25E-03 | 20 | + |
| GO-Term ID | Description | Cluster frequency | Background frequency | P-value | Generation | Fitness |
|---|---|---|---|---|---|---|
| 0006974 | Cellular response to DNA damage stimulus | 23/81 | 299/7163 | 3.29E-11 | 5 | − |
| 0006259 | DNA metabolic process | 26/81 | 448/7163 | 4.13E-10 | 5 | − |
| 0006950 | Response to stress | 28/81 | 655/7163 | 7.02E-09 | 5 | − |
| 0050896 | Response to stimulus | 35/81 | 996/7163 | 1.30E-08 | 5 | − |
| 0006310 | DNA recombination | 14/81 | 181/7163 | 4.70E-06 | 5 | − |
| 0051052 | Regulation of DNA metabolic process | 11/81 | 100/7163 | 5.42E-06 | 5 | − |
| 0006260 | DNA replication | 12/81 | 153/7163 | 5.16E-05 | 5 | − |
| 0051053 | Negative regulation of DNA metabolic process | 7/81 | 36/7163 | 5.46E-05 | 5 | − |
| 0065007 | Biological regulation | 38/81 | 1536/7163 | 9.81E-05 | 5 | − |
| 0090304 | Nucleic acid metabolic process | 38/81 | 1573/7163 | 1.70E-04 | 5 | − |
| 0019222 | Regulation of metabolic process | 28/81 | 958/7163 | 3.50E-04 | 5 | − |
| 0006261 | DNA-dependent DNA replication | 10/81 | 128/7163 | 6.80E-04 | 5 | − |
| 0043170 | Macromolecule metabolic process | 54/81 | 2946/7163 | 8.20E-04 | 5 | − |
| 0044260 | Cellular macromolecule metabolic process | 53/81 | 2870/7163 | 9.60E-04 | 5 | − |
| 0007530 | Sex determination | 6/81 | 35/7163 | 1.03E-03 | 5 | − |
| 0022616 | DNA strand elongation | 6/81 | 37/7163 | 1.45E-03 | 5 | − |
| 0007049 | Cell cycle | 21/81 | 637/7163 | 2.71E-03 | 5 | − |
| 0044237 | Cellular metabolic process | 61/81 | 3724/7163 | 3.42E-03 | 5 | − |
| 0006139 | Nucleobase-containing compound metabolic process | 38/81 | 1773/7163 | 4.01E-03 | 5 | − |
| 0071897 | DNA biosynthetic process | 5/81 | 27/7163 | 5.22E-03 | 5 | − |
| 0044238 | Primary metabolic process | 58/81 | 3499/7163 | 7.11E-03 | 5 | − |
| 0071704 | Organic substance metabolic process | 60/81 | 3708/7163 | 8.45E-03 | 5 | − |
| 0065007 | Biological regulation | 163/467 | 1536/7163 | 1.22E-09 | 20 | − |
| 0022402 | Cell cycle process | 79/467 | 594/7163 | 2.28E-06 | 20 | − |
| 0007049 | Cell cycle | 80/467 | 637/7163 | 6.80E-06 | 20 | − |
| 0051276 | Chromosome organization | 64/467 | 495/7163 | 5.15E-05 | 20 | − |
| 0050896 | Response to stimulus | 103/467 | 996/7163 | 6.29E-05 | 20 | − |
| 0006974 | Cellular response to DNA damage stimulus | 45/467 | 299/7163 | 8.15E-05 | 20 | − |
| 0048285 | Organelle fission | 52/467 | 390/7163 | 8.16E-05 | 20 | − |
| 0006259 | DNA metabolic process | 59/467 | 448/7163 | 9.67E-05 | 20 | − |
| 0006950 | Response to stress | 73/467 | 655/7163 | 2.20E-04 | 20 | − |
| 0044699 | Single-organism process | 286/467 | 3588/7163 | 2.70E-04 | 20 | − |
| 0009057 | Macromolecule catabolic process | 53/467 | 392/7163 | 4.30E-04 | 20 | − |
| 0009056 | Catabolic process | 79/467 | 710/7163 | 8.20E-04 | 20 | − |
| 0044248 | Cellular catabolic process | 74/467 | 657/7163 | 1.24E-03 | 20 | − |
| 2000113 | Negative regulation of cellular macromolecule biosynthetic process | 42/467 | 302/7163 | 2.04E-03 | 20 | − |
| 0044265 | Cellular macromolecule catabolic process | 49/467 | 370/7163 | 2.36E-03 | 20 | − |
| 0044763 | Single-organism cellular process | 254/467 | 3165/7163 | 4.14E-03 | 20 | − |
| 0044767 | Single-organism developmental process | 38/467 | 268/7163 | 4.48E-03 | 20 | − |
| 0032502 | Developmental process | 38/467 | 271/7163 | 5.87E-03 | 20 | − |
| 0007533 | Mating type switching | 10/467 | 28/7163 | 6.95E-03 | 20 | − |
| 0010526 | Negative regulation of transposition, RNA-mediated | 3/12 | 8/7163 | 2.06E-05 | 5 | + |
| 0031297 | Replication fork processing | 2/12 | 4/7163 | 1.59E-03 | 5 | + |
| 0007005 | Mitochondrion organization | 40/342 | 390/7163 | 2.35E-03 | 20 | + |
| 0017182 | Peptidyl-diphthamide metabolic process | 5/342 | 7/7163 | 4.25E-03 | 20 | + |
| GO-Term ID | Description | Cluster frequency | Background frequency | P-value | Generation | Fitness |
|---|---|---|---|---|---|---|
| 0006974 | Cellular response to DNA damage stimulus | 23/81 | 299/7163 | 3.29E-11 | 5 | − |
| 0006259 | DNA metabolic process | 26/81 | 448/7163 | 4.13E-10 | 5 | − |
| 0006950 | Response to stress | 28/81 | 655/7163 | 7.02E-09 | 5 | − |
| 0050896 | Response to stimulus | 35/81 | 996/7163 | 1.30E-08 | 5 | − |
| 0006310 | DNA recombination | 14/81 | 181/7163 | 4.70E-06 | 5 | − |
| 0051052 | Regulation of DNA metabolic process | 11/81 | 100/7163 | 5.42E-06 | 5 | − |
| 0006260 | DNA replication | 12/81 | 153/7163 | 5.16E-05 | 5 | − |
| 0051053 | Negative regulation of DNA metabolic process | 7/81 | 36/7163 | 5.46E-05 | 5 | − |
| 0065007 | Biological regulation | 38/81 | 1536/7163 | 9.81E-05 | 5 | − |
| 0090304 | Nucleic acid metabolic process | 38/81 | 1573/7163 | 1.70E-04 | 5 | − |
| 0019222 | Regulation of metabolic process | 28/81 | 958/7163 | 3.50E-04 | 5 | − |
| 0006261 | DNA-dependent DNA replication | 10/81 | 128/7163 | 6.80E-04 | 5 | − |
| 0043170 | Macromolecule metabolic process | 54/81 | 2946/7163 | 8.20E-04 | 5 | − |
| 0044260 | Cellular macromolecule metabolic process | 53/81 | 2870/7163 | 9.60E-04 | 5 | − |
| 0007530 | Sex determination | 6/81 | 35/7163 | 1.03E-03 | 5 | − |
| 0022616 | DNA strand elongation | 6/81 | 37/7163 | 1.45E-03 | 5 | − |
| 0007049 | Cell cycle | 21/81 | 637/7163 | 2.71E-03 | 5 | − |
| 0044237 | Cellular metabolic process | 61/81 | 3724/7163 | 3.42E-03 | 5 | − |
| 0006139 | Nucleobase-containing compound metabolic process | 38/81 | 1773/7163 | 4.01E-03 | 5 | − |
| 0071897 | DNA biosynthetic process | 5/81 | 27/7163 | 5.22E-03 | 5 | − |
| 0044238 | Primary metabolic process | 58/81 | 3499/7163 | 7.11E-03 | 5 | − |
| 0071704 | Organic substance metabolic process | 60/81 | 3708/7163 | 8.45E-03 | 5 | − |
| 0065007 | Biological regulation | 163/467 | 1536/7163 | 1.22E-09 | 20 | − |
| 0022402 | Cell cycle process | 79/467 | 594/7163 | 2.28E-06 | 20 | − |
| 0007049 | Cell cycle | 80/467 | 637/7163 | 6.80E-06 | 20 | − |
| 0051276 | Chromosome organization | 64/467 | 495/7163 | 5.15E-05 | 20 | − |
| 0050896 | Response to stimulus | 103/467 | 996/7163 | 6.29E-05 | 20 | − |
| 0006974 | Cellular response to DNA damage stimulus | 45/467 | 299/7163 | 8.15E-05 | 20 | − |
| 0048285 | Organelle fission | 52/467 | 390/7163 | 8.16E-05 | 20 | − |
| 0006259 | DNA metabolic process | 59/467 | 448/7163 | 9.67E-05 | 20 | − |
| 0006950 | Response to stress | 73/467 | 655/7163 | 2.20E-04 | 20 | − |
| 0044699 | Single-organism process | 286/467 | 3588/7163 | 2.70E-04 | 20 | − |
| 0009057 | Macromolecule catabolic process | 53/467 | 392/7163 | 4.30E-04 | 20 | − |
| 0009056 | Catabolic process | 79/467 | 710/7163 | 8.20E-04 | 20 | − |
| 0044248 | Cellular catabolic process | 74/467 | 657/7163 | 1.24E-03 | 20 | − |
| 2000113 | Negative regulation of cellular macromolecule biosynthetic process | 42/467 | 302/7163 | 2.04E-03 | 20 | − |
| 0044265 | Cellular macromolecule catabolic process | 49/467 | 370/7163 | 2.36E-03 | 20 | − |
| 0044763 | Single-organism cellular process | 254/467 | 3165/7163 | 4.14E-03 | 20 | − |
| 0044767 | Single-organism developmental process | 38/467 | 268/7163 | 4.48E-03 | 20 | − |
| 0032502 | Developmental process | 38/467 | 271/7163 | 5.87E-03 | 20 | − |
| 0007533 | Mating type switching | 10/467 | 28/7163 | 6.95E-03 | 20 | − |
| 0010526 | Negative regulation of transposition, RNA-mediated | 3/12 | 8/7163 | 2.06E-05 | 5 | + |
| 0031297 | Replication fork processing | 2/12 | 4/7163 | 1.59E-03 | 5 | + |
| 0007005 | Mitochondrion organization | 40/342 | 390/7163 | 2.35E-03 | 20 | + |
| 0017182 | Peptidyl-diphthamide metabolic process | 5/342 | 7/7163 | 4.25E-03 | 20 | + |
| GO-Term ID | Description | Cluster frequency | Background frequency | P-value | Generation | Fitness |
|---|---|---|---|---|---|---|
| 0006974 | Cellular response to DNA damage stimulus | 23/81 | 299/7163 | 3.29E-11 | 5 | − |
| 0006259 | DNA metabolic process | 26/81 | 448/7163 | 4.13E-10 | 5 | − |
| 0006950 | Response to stress | 28/81 | 655/7163 | 7.02E-09 | 5 | − |
| 0050896 | Response to stimulus | 35/81 | 996/7163 | 1.30E-08 | 5 | − |
| 0006310 | DNA recombination | 14/81 | 181/7163 | 4.70E-06 | 5 | − |
| 0051052 | Regulation of DNA metabolic process | 11/81 | 100/7163 | 5.42E-06 | 5 | − |
| 0006260 | DNA replication | 12/81 | 153/7163 | 5.16E-05 | 5 | − |
| 0051053 | Negative regulation of DNA metabolic process | 7/81 | 36/7163 | 5.46E-05 | 5 | − |
| 0065007 | Biological regulation | 38/81 | 1536/7163 | 9.81E-05 | 5 | − |
| 0090304 | Nucleic acid metabolic process | 38/81 | 1573/7163 | 1.70E-04 | 5 | − |
| 0019222 | Regulation of metabolic process | 28/81 | 958/7163 | 3.50E-04 | 5 | − |
| 0006261 | DNA-dependent DNA replication | 10/81 | 128/7163 | 6.80E-04 | 5 | − |
| 0043170 | Macromolecule metabolic process | 54/81 | 2946/7163 | 8.20E-04 | 5 | − |
| 0044260 | Cellular macromolecule metabolic process | 53/81 | 2870/7163 | 9.60E-04 | 5 | − |
| 0007530 | Sex determination | 6/81 | 35/7163 | 1.03E-03 | 5 | − |
| 0022616 | DNA strand elongation | 6/81 | 37/7163 | 1.45E-03 | 5 | − |
| 0007049 | Cell cycle | 21/81 | 637/7163 | 2.71E-03 | 5 | − |
| 0044237 | Cellular metabolic process | 61/81 | 3724/7163 | 3.42E-03 | 5 | − |
| 0006139 | Nucleobase-containing compound metabolic process | 38/81 | 1773/7163 | 4.01E-03 | 5 | − |
| 0071897 | DNA biosynthetic process | 5/81 | 27/7163 | 5.22E-03 | 5 | − |
| 0044238 | Primary metabolic process | 58/81 | 3499/7163 | 7.11E-03 | 5 | − |
| 0071704 | Organic substance metabolic process | 60/81 | 3708/7163 | 8.45E-03 | 5 | − |
| 0065007 | Biological regulation | 163/467 | 1536/7163 | 1.22E-09 | 20 | − |
| 0022402 | Cell cycle process | 79/467 | 594/7163 | 2.28E-06 | 20 | − |
| 0007049 | Cell cycle | 80/467 | 637/7163 | 6.80E-06 | 20 | − |
| 0051276 | Chromosome organization | 64/467 | 495/7163 | 5.15E-05 | 20 | − |
| 0050896 | Response to stimulus | 103/467 | 996/7163 | 6.29E-05 | 20 | − |
| 0006974 | Cellular response to DNA damage stimulus | 45/467 | 299/7163 | 8.15E-05 | 20 | − |
| 0048285 | Organelle fission | 52/467 | 390/7163 | 8.16E-05 | 20 | − |
| 0006259 | DNA metabolic process | 59/467 | 448/7163 | 9.67E-05 | 20 | − |
| 0006950 | Response to stress | 73/467 | 655/7163 | 2.20E-04 | 20 | − |
| 0044699 | Single-organism process | 286/467 | 3588/7163 | 2.70E-04 | 20 | − |
| 0009057 | Macromolecule catabolic process | 53/467 | 392/7163 | 4.30E-04 | 20 | − |
| 0009056 | Catabolic process | 79/467 | 710/7163 | 8.20E-04 | 20 | − |
| 0044248 | Cellular catabolic process | 74/467 | 657/7163 | 1.24E-03 | 20 | − |
| 2000113 | Negative regulation of cellular macromolecule biosynthetic process | 42/467 | 302/7163 | 2.04E-03 | 20 | − |
| 0044265 | Cellular macromolecule catabolic process | 49/467 | 370/7163 | 2.36E-03 | 20 | − |
| 0044763 | Single-organism cellular process | 254/467 | 3165/7163 | 4.14E-03 | 20 | − |
| 0044767 | Single-organism developmental process | 38/467 | 268/7163 | 4.48E-03 | 20 | − |
| 0032502 | Developmental process | 38/467 | 271/7163 | 5.87E-03 | 20 | − |
| 0007533 | Mating type switching | 10/467 | 28/7163 | 6.95E-03 | 20 | − |
| 0010526 | Negative regulation of transposition, RNA-mediated | 3/12 | 8/7163 | 2.06E-05 | 5 | + |
| 0031297 | Replication fork processing | 2/12 | 4/7163 | 1.59E-03 | 5 | + |
| 0007005 | Mitochondrion organization | 40/342 | 390/7163 | 2.35E-03 | 20 | + |
| 0017182 | Peptidyl-diphthamide metabolic process | 5/342 | 7/7163 | 4.25E-03 | 20 | + |
To assess the contribution of individual sirtuins to NAM-induced fitness defects, we compared our results with available genetic interaction data (49) and found that our data set significantly overlaps with negative genetic interactions involving HST3 and HST4, but not SIR2, HST1 or HST2 (Figure 2E and data not shown). Our experiments also identified a limited number of genes whose mutation improved fitness in response to NAM (12 and 342 genes at 5 and 20 generations, respectively). In particular, deletion of genes that are genetically and biochemically linked to the H3K56ac cellular pathway (RTT101, RTT107 and MMS1) (43,58) improved fitness in the presence of NAM. These three genes are negative regulators of retrotransposition (59), and promote the response to damaged DNA replication forks, explaining their associated GO terms in Table 2 (60,61).
To validate these results, we tested the influence of NAM on the growth of individual mutant strains presenting high Z-scores in our fitness assays. This was done by evaluating the optical density (OD630) of cultures of the corresponding haploid deletion mutants after 48 h of growth in NAM-containing medium (Figure 2G–I). Results from these experiments are in line with our fitness assays, as mutations in DDR genes caused NAM sensitivity. Importantly, deletion of the gene encoding the H3K56 acetyltransferase Rtt109 rescued the NAM sensitivity of several DDR mutants, e.g. pol32Δ, yku70Δ, mrc1Δ, rad59Δ, slx4Δ and pph3Δ (Figures 2I, 3A, 4B). These data confirm that various DDR pathways respond to H3K56ac-dependent DNA damage induction caused by NAM.

The NAM sensitivity of slx4Δ cells requires H3K56ac, Rtt107 and Rtt101. (A) slx4Δ mutants are sensitive to NAM-induced H3K56 constitutive acetylation. Cells were grown in 96 well plates and OD readings were acquired as described in Materials and Methods. (B–C) Mutation of Rtt101 and Rtt107 cause growth inhibition and DNA damage in NAM. (B) Cells were treated as in A. (C) Exponentially growing yeast cultures were exposed for 8 h to 20 mM NAM and samples were collected for microscopy analysis of Rad52-YFP foci. Results are represented as the ratio of cells with Rad52-YFP foci after and before NAM treatment. The numbers below the graph indicate the fraction of cells containing foci in NAM-treated and untreated cells. At least 300 cells were examined per time point, and the experiment was performed in triplicate. (D–E) RTT107 is part of the H3K56ac genetic pathway. Cells were serially diluted, spotted on the indicated medium and incubated at the indicated temperature (D) or 30°C (E). (F) Evaluation of the NAM sensitivity of mutants of genes encoding nucleases interacting with Slx4. Cells were treated as in A. MMS: Methyl methanesulfonate, NT: Non-treated, *: P-value < 0.05 as calculated with an unpaired one-tailed Student's t-test. SC-URA: synthetic medium lacking uracil. 5-FOA: 5-Fluoroorotic Acid-containing medium.
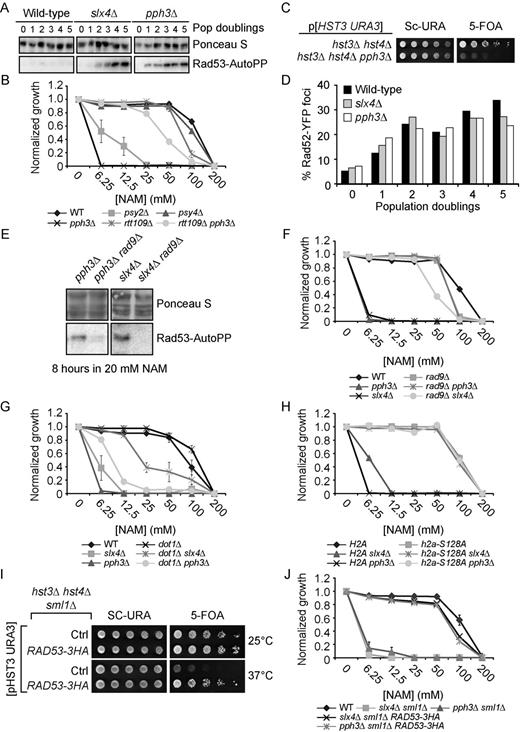
DNA damage-induced signaling inhibits cell growth in NAM. (A) slx4Δ and pph3Δ mutants strongly activate Rad53 in response to NAM. Exponentially growing cells were incubated in YPD with 20 mM NAM and samples were taken for Rad53 in situ autophosphorylation assays. A population doubling is defined as the doubling time of wild-type cells in NAM. The ‘0 population doubling’ sample was taken immediately prior NAM exposure and therefore represents an untreated control. (B) NAM inhibits growth of pph3Δ and psy2Δ but not psy4Δ mutants in an H3K56ac-dependent manner. Cells were grown in 96 well plates and OD readings were acquired as described in Materials and Methods. (C) Deletion of PPH3 in hst3Δ hst4Δ cells causes synthetic lethality. Cells were serially diluted, spotted on the indicated medium and incubated at 30°C. (D) slx4Δ and pph3Δ mutations do not increase the frequency of NAM-induced Rad52-YFP foci. Samples were taken at indicated population doublings and processed for fluorescence microscopy analysis. Population doublings are defined as in A. (E–F) RAD9 deletion inhibits NAM-induced Rad53 activation and growth defects in slx4Δ and pph3Δ mutants. Cells were incubated for 8 h in YPD + 20 mM NAM at 30°C, and samples were processed for Rad53 autophosphorylation assays. (G–H) Abolishing H3K79me and H2A S128 phosphorylation suppress NAM-induced growth defects in slx4Δ and pph3Δ mutants. Cells were treated as in B. (I) The RAD53–3HA hypomorphic allele rescues the thermosensitivity of hst3Δ hst4Δ mutants. Cells were serially diluted, spotted on the indicated medium and incubated at the indicated temperature. (J) Reducing Rad53 activity rescues the NAM-induced growth defects of slx4Δ and pph3Δ mutants. Cells were treated as in B.
As a complementary approach to our fitness assays, mRNA profiling was performed to document transcriptional changes caused by NAM. To permit comparison between transcriptional and phenotypic responses, cells were grown under the same conditions as for the fitness assays, i.e. using the same S. cerevisiae diploid strain (BY4743), NAM concentration (20 mM) and time points (5 and 20 generations). We also included a short time point (1 h) to allow detection of early changes in mRNA expression patterns. We identified 213, 430 and 306 genes that were differentially expressed in cells exposed to NAM for 1 h, and for 5 and 20 generations, respectively (absolute fold change ≥ 2.0; Supplementary Table S3). A majority of the identified genes (91–95%) were influenced by NAM at every time point analyzed, with a core set of 133 induced genes. Results from GO term analysis are only presented for genes whose expression is significantly modulated after 5 generations, since analyses performed with genes modulated at other time points yielded similar results (data not shown). Many significantly enriched GO terms were clustered into subsets of processes known to be regulated by Hst1 (and to a lesser extent Sir2) at the transcriptional level, and are expected to respond to NAM-induced inhibition of these sirtuins, e.g. sexual reproduction (sporulation) (62,63) and metabolism (‘de novo’ NAD biosynthetic process, sulfur amino acid, carboxylic acid, energy reserve metabolic processes) (17,18) (Table 3). GSEA also revealed that transcripts repressed in NAM-treated cells were significantly enriched in ribosome biogenesis, translation and tRNA modification (Supplementary Figure S1, Supplementary Table S4). Genes involved in general transcription regulation including mediators and RNA elongation complexes were also repressed. Transcripts upregulated by NAM were significantly correlated with those found to be activated by NAM treatment in a previous study, thereby validating our methodology (18). Consistent with the GO term analysis presented in Table 3, GSEA indicated that genes involved in sporulation were activated by NAM. Finally, we found that genes bound by the transcription factor Sum1, which acts together with Hst1 to repress middle sporulation-specific genes (16), were significantly upregulated by NAM.
GO term analysis of genes whose expression is modulated by NAM at 5 generations
| GO-Term ID | Description | Cluster frequency | Background frequency | P-value |
|---|---|---|---|---|
| 0048646 | Anatomical structure formation involved in morphogenesis | 53/430 | 142/7164 | 1.85E-26 |
| 0043935 | Sexual sporulation resulting in formation of a cellular spore | 49/430 | 120/7164 | 1.97E-26 |
| 0070726 | Cell wall assembly | 34/430 | 54/7164 | 6.59E-26 |
| 0048856 | Anatomical structure development | 56/430 | 165/7164 | 1.33E-25 |
| 0043934 | Sporulation | 51/430 | 138/7164 | 3.95E-25 |
| 0048869 | Cellular developmental process | 58/430 | 199/7164 | 1.13E-22 |
| 0051704 | Multi-organism process | 62/430 | 260/7164 | 2.96E-19 |
| 0022414 | Reproductive process | 63/430 | 276/7164 | 1.60E-18 |
| 0044767 | Single-organism developmental process | 60/430 | 270/7164 | 6.74E-17 |
| 0032502 | Developmental process | 60/430 | 273/7164 | 1.21E-16 |
| 0045229 | External encapsulating structure organization | 39/430 | 154/7164 | 2.87E-12 |
| 0000003 | Reproduction | 73/430 | 464/7164 | 3.70E-12 |
| 0071554 | Cell wall organization or biogenesis | 39/430 | 201/7164 | 2.56E-08 |
| 0006112 | Energy reserve metabolic process | 12/430 | 34/7164 | 2.30E-04 |
| 0034627 | ‘de novo’ NAD biosynthetic process | 5/430 | 5/7164 | 5.80E-04 |
| 0019752 | Carboxylic acid metabolic process | 44/430 | 349/7164 | 1.25E-03 |
| 0044281 | Small molecule metabolic process | 71/430 | 684/7164 | 1.48E-03 |
| 0006082 | Organic acid metabolic process | 44/430 | 363/7164 | 3.59E-03 |
| 0000096 | Sulfur amino acid metabolic process | 11/430 | 36/7164 | 3.72E-03 |
| GO-Term ID | Description | Cluster frequency | Background frequency | P-value |
|---|---|---|---|---|
| 0048646 | Anatomical structure formation involved in morphogenesis | 53/430 | 142/7164 | 1.85E-26 |
| 0043935 | Sexual sporulation resulting in formation of a cellular spore | 49/430 | 120/7164 | 1.97E-26 |
| 0070726 | Cell wall assembly | 34/430 | 54/7164 | 6.59E-26 |
| 0048856 | Anatomical structure development | 56/430 | 165/7164 | 1.33E-25 |
| 0043934 | Sporulation | 51/430 | 138/7164 | 3.95E-25 |
| 0048869 | Cellular developmental process | 58/430 | 199/7164 | 1.13E-22 |
| 0051704 | Multi-organism process | 62/430 | 260/7164 | 2.96E-19 |
| 0022414 | Reproductive process | 63/430 | 276/7164 | 1.60E-18 |
| 0044767 | Single-organism developmental process | 60/430 | 270/7164 | 6.74E-17 |
| 0032502 | Developmental process | 60/430 | 273/7164 | 1.21E-16 |
| 0045229 | External encapsulating structure organization | 39/430 | 154/7164 | 2.87E-12 |
| 0000003 | Reproduction | 73/430 | 464/7164 | 3.70E-12 |
| 0071554 | Cell wall organization or biogenesis | 39/430 | 201/7164 | 2.56E-08 |
| 0006112 | Energy reserve metabolic process | 12/430 | 34/7164 | 2.30E-04 |
| 0034627 | ‘de novo’ NAD biosynthetic process | 5/430 | 5/7164 | 5.80E-04 |
| 0019752 | Carboxylic acid metabolic process | 44/430 | 349/7164 | 1.25E-03 |
| 0044281 | Small molecule metabolic process | 71/430 | 684/7164 | 1.48E-03 |
| 0006082 | Organic acid metabolic process | 44/430 | 363/7164 | 3.59E-03 |
| 0000096 | Sulfur amino acid metabolic process | 11/430 | 36/7164 | 3.72E-03 |
| GO-Term ID | Description | Cluster frequency | Background frequency | P-value |
|---|---|---|---|---|
| 0048646 | Anatomical structure formation involved in morphogenesis | 53/430 | 142/7164 | 1.85E-26 |
| 0043935 | Sexual sporulation resulting in formation of a cellular spore | 49/430 | 120/7164 | 1.97E-26 |
| 0070726 | Cell wall assembly | 34/430 | 54/7164 | 6.59E-26 |
| 0048856 | Anatomical structure development | 56/430 | 165/7164 | 1.33E-25 |
| 0043934 | Sporulation | 51/430 | 138/7164 | 3.95E-25 |
| 0048869 | Cellular developmental process | 58/430 | 199/7164 | 1.13E-22 |
| 0051704 | Multi-organism process | 62/430 | 260/7164 | 2.96E-19 |
| 0022414 | Reproductive process | 63/430 | 276/7164 | 1.60E-18 |
| 0044767 | Single-organism developmental process | 60/430 | 270/7164 | 6.74E-17 |
| 0032502 | Developmental process | 60/430 | 273/7164 | 1.21E-16 |
| 0045229 | External encapsulating structure organization | 39/430 | 154/7164 | 2.87E-12 |
| 0000003 | Reproduction | 73/430 | 464/7164 | 3.70E-12 |
| 0071554 | Cell wall organization or biogenesis | 39/430 | 201/7164 | 2.56E-08 |
| 0006112 | Energy reserve metabolic process | 12/430 | 34/7164 | 2.30E-04 |
| 0034627 | ‘de novo’ NAD biosynthetic process | 5/430 | 5/7164 | 5.80E-04 |
| 0019752 | Carboxylic acid metabolic process | 44/430 | 349/7164 | 1.25E-03 |
| 0044281 | Small molecule metabolic process | 71/430 | 684/7164 | 1.48E-03 |
| 0006082 | Organic acid metabolic process | 44/430 | 363/7164 | 3.59E-03 |
| 0000096 | Sulfur amino acid metabolic process | 11/430 | 36/7164 | 3.72E-03 |
| GO-Term ID | Description | Cluster frequency | Background frequency | P-value |
|---|---|---|---|---|
| 0048646 | Anatomical structure formation involved in morphogenesis | 53/430 | 142/7164 | 1.85E-26 |
| 0043935 | Sexual sporulation resulting in formation of a cellular spore | 49/430 | 120/7164 | 1.97E-26 |
| 0070726 | Cell wall assembly | 34/430 | 54/7164 | 6.59E-26 |
| 0048856 | Anatomical structure development | 56/430 | 165/7164 | 1.33E-25 |
| 0043934 | Sporulation | 51/430 | 138/7164 | 3.95E-25 |
| 0048869 | Cellular developmental process | 58/430 | 199/7164 | 1.13E-22 |
| 0051704 | Multi-organism process | 62/430 | 260/7164 | 2.96E-19 |
| 0022414 | Reproductive process | 63/430 | 276/7164 | 1.60E-18 |
| 0044767 | Single-organism developmental process | 60/430 | 270/7164 | 6.74E-17 |
| 0032502 | Developmental process | 60/430 | 273/7164 | 1.21E-16 |
| 0045229 | External encapsulating structure organization | 39/430 | 154/7164 | 2.87E-12 |
| 0000003 | Reproduction | 73/430 | 464/7164 | 3.70E-12 |
| 0071554 | Cell wall organization or biogenesis | 39/430 | 201/7164 | 2.56E-08 |
| 0006112 | Energy reserve metabolic process | 12/430 | 34/7164 | 2.30E-04 |
| 0034627 | ‘de novo’ NAD biosynthetic process | 5/430 | 5/7164 | 5.80E-04 |
| 0019752 | Carboxylic acid metabolic process | 44/430 | 349/7164 | 1.25E-03 |
| 0044281 | Small molecule metabolic process | 71/430 | 684/7164 | 1.48E-03 |
| 0006082 | Organic acid metabolic process | 44/430 | 363/7164 | 3.59E-03 |
| 0000096 | Sulfur amino acid metabolic process | 11/430 | 36/7164 | 3.72E-03 |
The above data suggest that transcriptional changes are unlikely to significantly contribute to DNA damage-induced inhibition of cell proliferation caused by NAM, which depends largely on H3K56 hyperacetylation (Figure 1D). No GO term associated with the DDR was identified in our analyses of mRNA profiling data. Nevertheless, keyword searches identified three DDR genes in our lists of NAM-modulated genes: HUG1 encoding a protein involved in the Mec1p-mediated checkpoint pathway (64); SRL4 of unknown function but whose deletion suppresses the lethality of rad53 mutations (65); and HIM1 encoding a poorly characterized protein involved in DNA repair (66). Northern blot analysis of these three genes confirmed their induction in response to NAM (data not shown). However, mutant cells lacking these genes are not sensitive to NAM (Figure 2J), suggesting that their NAM-induced expression likely reflects non-specific responses to DNA damage.
Slx4 is essential for growth in the presence of NAM-induced sirtuin inhibition
Slx4 acts as a scaffold by forming complexes with several important DDR proteins (67). slx4Δ homozygote diploid cells displayed strong NAM-induced fitness defects in our assays (Supplementary Table S1), and the corresponding haploid deletion strain grew poorly in medium containing NAM (Figure 3A). Deletion of RTT109 restored growth of slx4Δ cells in NAM, suggesting that Slx4 promotes cell survival in response to H3K56 hyperacetylation (Figure 3A). Consistently, we confirmed that deletion of SLX4 in hst3Δ hst4Δ mutants was synthetically lethal (Figure 3D), as previously reported (22).
We sought to exploit the NAM sensitivity of slx4Δ mutants as a means of probing the function of this gene in the H3K56ac pathway. Slx4 interacts with several DDR proteins including Rtt107 (67–69), Dpb11 (67), Mus81-Mms4 (70), Rad1-Rad10 (71) and Slx1 (72). In agreement with fitness data (Supplementary Table S1), rtt107Δ cells presented improved cell growth compared to wild-type in NAM (Figure 3B) and mutation of RTT107 abrogated NAM-induced growth defects in slx4Δ (Figure 3B). One model to explain this intriguing observation would be that Rtt107 contributes to induction of DNA lesions in an H3K56ac-dependent manner, which would require Slx4 for their resolution. In such a case, the absence of Rtt107-dependent NAM-induced DNA lesions would circumvent any eventual requirement for the Slx4-Rtt107 complex. Consistent with this, the fraction of cells presenting Rad52-YFP foci was increased after 8 h in NAM to a significantly greater extent in wild-type cells (Figure 3C, ∼5-fold) as compared to rtt107Δ and rtt109Δ mutants (Figure 3C, ∼1 to 1.5-fold). Rtt107 physically interacts with the Rtt101-Mms1-Mms22 complex, which is genetically linked to H3K56ac (43,58,73–74). Interestingly, deletion of RTT101 also diminished the frequency of NAM-induced Rad52-YFP foci as compared to WT cells (Figure 3C), suggesting that Rtt107 and Rtt101 promote the formation of DNA lesions in conjunction with NAM-induced H3K56ac.
The genetic relationship between RTT107 and H3K56ac is controversial as recently published data suggest, in direct contradiction with our results, that deletion of RTT107 does not suppress the phenotypes of hst3Δ hst4Δ cells (75), and that slx4Δ hst3Δ hst4Δ cells are viable. We sought to address these discrepancies by further examining the genetic relationships between H3K56ac, RTT109, HST3, HST4 and RTT107. We found that rtt109Δ is epistatic to rtt107Δ in response to MMS, a genotoxic drug causing replication-blocking DNA lesions, and that mutation of RTT107 clearly rescues the temperature sensitivity of hst3Δ hst4Δ mutants (Figure 3D–E). Furthermore, our results indicate that deletion of RTT107 abolishes the synthetic lethality caused by hst3Δ hst4Δ slx4Δ mutations (Figure 3D). Taken together, our results suggest that cells with constitutive H3K56ac require Slx4 to process Rtt107-induced DNA lesions.
Dampening DDR activity promotes growth in response to NAM-induced H3K56 hyperacetylation
We next sought to identify functions of Slx4 that are important for the processing or tolerance of H3K56ac-dependent DNA damage. Mus81/Mms4, Rad1/Rad10 and Slx1 form distinct structure-specific endonuclease complexes that physically interact with Slx4 and are involved in resolving branched DNA structures (72,76–77). We found that the haploid deletion strains of each of these genes displayed only modest NAM sensitivity in comparison to slx4Δ (Figure 3F). This suggests that loss of function of any among these Slx4-containing complexes cannot explain the NAM sensitivity of slx4Δ mutants, although we cannot exclude that simultaneously compromising the activity of all these complexes may sensitize slx4Δ cells to NAM.
Alternatively, we hypothesized that other functions of Slx4 could account for the extreme NAM sensitivity of slx4Δ mutants. Slx4 acts with Rtt107 to dampen DDR signaling by competing with Rad9 for binding to phosphorylated H2A, thus limiting Rad53 activation in response to DNA damage (68). Consistently, NAM engendered strong Rad53 activation in slx4Δ mutants compared to WT cells (Figure 4A). Interestingly, 3 genes encoding subunits of the yeast PP4 phosphatase complex presented high Z-scores in our fitness assays: PPH3, PSY2 and PSY4 (78,79) (Figure 2A–B, Supplementary Table S1). This complex dephosphorylates Rad53 and H2A S128, well-known targets of Mec1 kinase, thereby counteracting DNA damage-induced signaling (80,81). Consistent with a model in which constitutive H3K56ac causes lethality in the absence of Pph3, haploid psy2Δ and pph3Δ mutants, but not psy4Δ, displayed RTT109-dependent growth defects in NAM-containing medium, while deletion of PPH3 in hst3Δ hst4Δ cells caused synthetic lethality (Figure 4B–C). Psy4 is required for dephosphorylation of H2A S128, but not Rad53 (80,81), indicating that activated Rad53 is likely to be a critical PP4 substrate in the context of NAM exposure. Consistently, Rad53 was strongly activated in NAM-treated pph3Δ cells (Figure 4A). NAM did not cause a higher frequency of Rad52-YFP foci in slx4Δ or pph3Δ cells as compared to WT (Figure 4D), arguing against the notion that DNA lesions are induced at higher frequency in response to NAM in these mutants.
We and others showed that spontaneous activation of Rad53 in hst3Δ hst4Δ cells depends in large part on the Rad9 adaptor protein (22,23). Strikingly, mutation of RAD9 abrogated NAM-induced Rad53 activation in slx4Δ and pph3Δ mutants (Figure 4E) and rescued growth of these mutants in NAM-containing medium (Figure 4F). Rad9 recruitment to damaged chromatin depends on its interaction with trimethylated histone H3 lysine 79 (H3K79me3) and with phosphorylated serine 128 of histone H2A (H2A-P) via its tudor and BRCT domains, respectively (82,83). Dot1 is the methyltransferase responsible for H3K79me3 in yeast (84). We found that introducing a histone mutation abolishing H2A-P (H2A S128A) or deleting DOT1 in slx4Δ and pph3Δ mutants improved their growth in NAM (Figure 4G–H). However, for unknown reasons suppression of NAM sensitivity in pph3Δ by dot1Δ was not as striking as in slx4Δ mutants, although the sensitivity of both mutants was similarly suppressed by H2A S128A. Expression of a hypomorphic RAD53-HA allele (85) also rescued both thermosensitivity of hst3Δ hst4Δ mutants and growth defects of pph3Δ and slx4Δ mutants in NAM (Figure 4I–J). Overall, these results indicate that Rad9-mediated Rad53 activation inhibits cell growth in response to NAM-induced H3K56 hyperacetylation.
Hyperactive DDR signaling compromises completion of DNA replication in response to NAM
Rad53 delays S phase completion by inhibiting activation of late origins of DNA replication in response to DNA damage (86). Using flow cytometry-based DNA content analysis, we found that compared to WT cells slx4Δ and pph3Δ mutants accumulated in S phase during NAM exposure (Figure 5A). This phenotype was abolished by rtt109Δ or rad9Δ mutations, indicating that defects in S phase progression were caused by H3K56 hyperacetylation and Rad9-dependent DNA damage signaling (Figure 5A). Rad53-mediated phosphorylation of several residues in Dbf4 and Sld3 prevents late origin firing in response to DNA damage (87). Expression of non-phosphorylatable dbf4–4A and sld3-A alleles in yeast does not influence Rad53 activation (87), but bypasses the inhibition of late replication origins in response to DNA lesions. Interestingly, we found that the dbf4–4A sld3-A mutations partially rescue the growth defects of slx4Δ and pph3Δ cells in NAM (Figure 5B), and that dbf4–4A sld3-A pph3Δ and dbf4–4A sld3-A slx4Δ mutants display reduced accumulation in S phase in response to NAM in comparison to slx4Δ and pph3Δ cells (Figure 5C). We note that cells of the W303 genetic background accumulate in late S phase in NAM, in contrast to BY4741 cells which accumulate in mid S (compare Figure 5C and A). Although the reasons for this remain unclear, we excluded the possibility that the rad5-G535R mutation present in certain W303 backgrounds explains these differences, as cells harboring this mutation respond to NAM in a manner indistinguishable from W303 RAD5 cells (Supplementary Figure S2). Overall, these data are consistent with a model in which Rad53-mediated phosphorylation of Dbf4 and Sld3 contributes to inhibition of cell cycle progression in cells with constitutive H3K56ac.
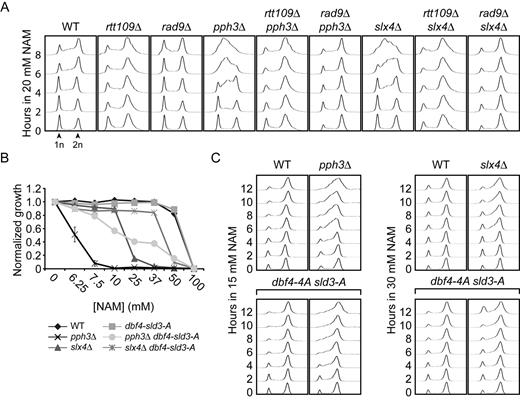
NAM inhibits the completion of DNA replication in slx4Δ and pph3Δ mutants. (A) NAM-induced S-phase arrest in slx4Δ and pph3Δ mutants depends on Rad9 and Rtt109. Exponentially growing cells were incubated in YPD with 20 mM NAM and samples were taken at the indicated time points for DNA content analysis by flow cytometry. (B–C) Rad53-dependent inhibition of the activation of late DNA replication origins contributes to growth defects of slx4Δ and pph3Δ in NAM. (B) Cells were grown in 96 well plates and OD readings were acquired as described in Materials and Methods. (C) Cells were treated as in A. 1n, 2n: DNA content.
Rad53 activity has been shown to inhibit the mutagenic translesion synthesis (TLS) pathway of DNA damage tolerance in response to MMS (85,88). Because of this, mutations that compromise Rad9-dependent Rad53 activation, i.e. dot1Δ or H3K79R, increase cellular resistance to MMS in a TLS-dependent manner (85,88). Our previous data suggested that the MMS sensitivity of hst3Δ hst4Δ cells may result in part from Rad53-dependent inhibition of Pol Zeta, a TLS polymerase involved in the bypass of MMS-damaged DNA bases such as 3-methyladenine (23,89–90). We sought to verify whether Rad53-mediated inhibition of TLS contributes to cell cycle progression defects of NAM-treated slx4Δ and pph3Δ mutants. REV3 encodes the catalytic subunit of Pol Zeta, and has been shown to mediate TLS-induced spontaneous mutagenesis in cells lacking Slx4 (71). We found that deletion of REV3 had no effect on NAM sensitivity of slx4Δ dot1Δ or pph3Δ dot1Δ mutants (Figure 6A and Supplementary Figure S3A–B). This suggests either that DNA lesions caused by NAM are not bypassed by Pol Zeta, or that relief of Rad53-mediated TLS inhibition does not contribute to the suppressive effect of dot1Δ on NAM-associated phenotypes in slx4Δ and pph3Δ mutants. To distinguish these possibilities, we tested whether ubiquitination (ub) of the DNA replication processivity clamp PCNA was involved in the suppressive effect of dot1Δ and rad9Δ on NAM-induced phenotypes. PCNA mono-ub on lysine 164 by the Rad6/Rad18 ub ligase complex favors TLS in response to damaged DNA bases, whereas subsequent poly-ub of this residue by Mms2-Rad5-Ubc13 promotes error-free homologous recombination-mediated template switching (91). Interestingly, mutation of PCNA lysine 164 to a non-ubiquitylable arginine residue diminished the extent of rad9Δ-dependent rescue of the NAM sensitivity of slx4Δ and pph3Δ mutants (Figure 6B). Moreover, deletion of RAD18 abrogated the rescue of NAM sensitivity conferred to slx4Δ and pph3Δ mutants by the dot1Δ and rad9Δ mutations, respectively (Figure 6C–D). This effect depends on TLS-promoting PCNA mono-ub rather than subsequent Mms2-Rad5-Ubc13-mediated poly-ub since deletion of MMS2 had no impact on the NAM sensitivity of slx4Δ dot1Δ and pph3Δ rad9Δ mutants (Figure 6C–D). However, we note that even though our data suggest that relief of Rad53-mediated TLS inhibition may contribute to the rescue of the phenotypes of slx4Δ and pph3Δ mutants in the context of NAM exposure, the identity of eventual TLS polymerase (s) involved in the bypass of NAM-induced DNA lesions remain unknown.
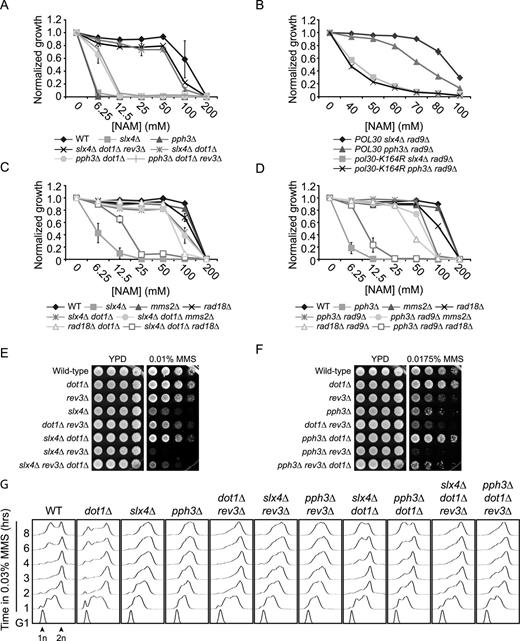
The translesion synthesis pathway of DNA damage tolerance promotes growth in response to NAM and MMS in slx4Δ and pph3Δ mutants. (A) dot1Δ-mediated suppression of the NAM sensitivity of slx4Δ and pph3Δ mutants does not depend on the Rev3 subunit of translesion DNA polymerase Zeta. Cells were grown in 96 well plates and OD reading were assessed as described in Materials and Methods. (B) rad9Δ-mediated suppression of the NAM sensitivity of slx4Δ and pph3Δ mutants depends on PCNA K164. Cells were treated as in A. (C–D) dot1Δ- and rad9Δ-mediated rescue of the NAM sensitivity of slx4Δ and pph3Δ mutants require RAD18 but not MMS2. Cells were treated as in A. (E–F) dot1Δ-mediated rescue of the MMS sensitivity of slx4Δ and pph3Δ depends on the Rev3 subunit of translesion DNA polymerase Zeta. Cells were serially diluted, spotted on the indicated medium and incubated at 30°C. (G) Translesion synthesis promotes completion of the cell cycle, but not S phase progression, in dot1Δ, dot1Δ slx4Δ and dot1Δ pph3Δ mutants treated with MMS. Cells were synchronized in G1 with alpha factor and released in YPD containing 0.03% MMS. Samples were taken at indicated time and processed for DNA content analysis by flow cytometry. MMS: methyl methanesulfonate. 1n, 2n: DNA content.
To further investigate the role of TLS in slx4Δ and pph3Δ cells, we decided to use MMS as a model mutagen, which produces 3-methyladenines that are bypassed in large part by Pol Zeta. As was the case with NAM, DOT1 deletion partially suppressed MMS sensitivity of both slx4Δ and pph3Δ mutants (Figure 6E–F). This suppression was reversed in large part by REV3 deletion, consistent with the notion that TLS could be involved in this phenomenon (Figure 6E–F). However, we found that DOT1 slx4Δ rev3Δ and pph3Δ rev3Δ cells are also extremely sensitive to MMS, and that dot1Δ slightly suppressed the severe MMS sensitivity of these mutants. We conclude that even though dot1Δ-mediated suppression of the phenotypes of slx4Δ and pph3Δ mutants requires Rev3 in large part, other cellular pathways also appear to contribute.
Deletion of SLX4 or PPH3 causes delays in S phase completion in response to MMS (71,92). To investigate the impact of Rad53-dependent inhibition of TLS on this phenomenon, we synchronized cells in G1 using alpha factor and released them toward S-phase in MMS-containing medium. Cell cycle progression was monitored by flow cytometry at regular intervals. As expected, slx4Δ and pph3Δ mutants exhibited delays in passage through S in MMS compared to WT cells (Figure 6G). In contrast, deletion of DOT1 in these mutants permitted completion of DNA replication with kinetics similar to WT, and furthermore allowed cells to progress toward the next G1 and S-phase. The rev3Δ mutation did not enhance the replication defects observed in pph3Δ and slx4Δ single mutants, and also did not dramatically compromise S phase progression in dot1Δ, dot1Δ slx4Δ and dot1Δ pph3Δ. Instead, these latter strains were found to accumulate in late S-G2/M in the absence of Rev3. The above data (i) are in agreement with the notion that cells lacking Dot1 rely on Rev3-mediated TLS for growth in the presence of MMS (Figure 6E–F) (88), and (ii) further suggest that Rev3 acts to bypass MMS-induced DNA lesions mostly in late S-G2/M under these conditions, thus operating in a manner that is uncoupled from the bulk of DNA replication, as described (93). Our overall results therefore suggest that Rad53-mediated inhibition of TLS contributes at least in part to the phenotypes of slx4Δ and pph3Δ mutants in response to DNA damage, but does not explain the striking S-phase progression defects of these mutants when challenged with MMS. These data also highlight the fact that the genetic requirements for dot1Δ-mediated suppression of slx4Δ and pph3Δ differ in cells treated with NAM versus MMS.
Reactive oxygen species generate DNA damage in NAM-treated cells
We next sought to identify the source of DNA lesions observed in NAM-treated cells (Figures 1C and 4D). ROS are generated as by-products of ATP production during oxidative phosphorylation. ROS induce highly-mutagenic replication-blocking DNA lesions that can be bypassed by TLS polymerases (94,95). Interestingly, data from our NAM fitness assays significantly overlaps with that of similar assays performed in the presence of paraquat, a chemical known to induce ROS (51,96) (Figure 7A–B). Moreover, addition of the antioxidants N-acetylcysteine (NAC) or glutathione (GSH) improved growth of slx4Δ and pph3Δ mutants in NAM (Figure 7C–D), and NAC reduced both the frequency of NAM-induced Rad52-YFP foci in WT cells and activation of Rad53 in slx4Δ and pph3Δ mutants following NAM exposure (Figure 7E–F). Overall, these data are consistent with the notion that cellular ROS may contribute at least in part to NAM-induced DDR and growth defects.
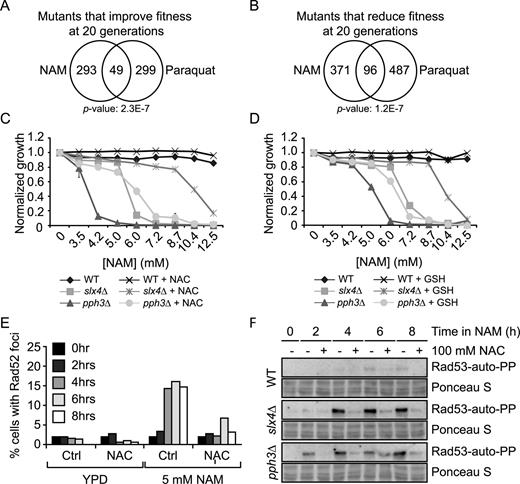
Reactive oxygen species generate DNA damage in NAM-treated cells. (A–B) Growth in NAM and paraquat share similar genetic requirements. Fitness assays data sets were compared and Venn diagrams were generated as described in Materials and Methods. (C–D) N-acetylcysteine (NAC) and glutathione (GSH) partially suppress NAM-induced growth defects in slx4Δ and pph3Δ mutants. OD630 was measured after 48 h in YPD containing NAM at 30°C in 96 well plates, with or without 100 mM NAC or GSH. (E) NAC suppresses NAM-induced formation of Rad52-YFP foci. Exponentially growing cells were incubated in YPD at 30°C with or without 5 mM NAM and/or 100 mM NAC. Samples were taken at indicated times and processed for fluorescence microscopy. (F) NAC suppresses the NAM-induced Rad53 activation in slx4Δ and pph3Δ mutants. Cells were treated as in (E) and samples were taken at indicated time points for Rad53 in situ autophosphorylation assays. NAC: N-acetylcysteine, GSH: Glutathione.
As initial explanation for the above-described observations, we hypothesized that NAM exposure could lead to increased ROS production, thereby causing growth defects and DDR induction. This model predicts that mutations causing ROS sensitivity should also sensitize cells to NAM, and vice-versa. Contrary to this, growth of slx4Δ and pph3Δ mutants was only mildly hampered by H2O2 (Figure 8A). In addition, deletion of RTT109 in slx4Δ and pph3Δ cells caused synthetic growth defects in response to H2O2, but rescued their NAM sensitivity. To more directly assess whether intracellular ROS levels were influenced by NAM, we stained control and NAM-treated cells with dihydrorhodamine 123 (DHR123), a reagent that fluoresces by reacting with ROS, and analyzed cell fluorescence by flow cytometry (Figure 8B). We also assessed DHR123 fluorescence in cells exposed to 2.5 mM H2O2, a concentration that inhibits cell proliferation to a similar extent as 20 mM NAM (data not shown). NAM exposure did not increase DHR123 fluorescence compared to untreated control cells, whereas H2O2 caused strong increase in fluorescence signal, as expected (Figure 8B). Overall, our data do not support the concept that NAM exposure per se directly causes elevated intracellular ROS.
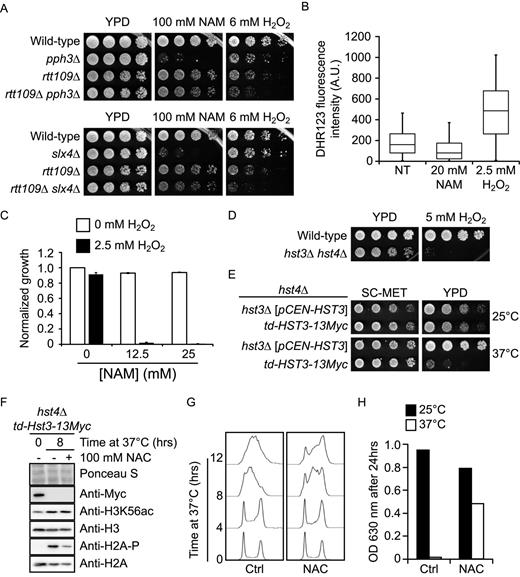
NAM-induced H3K56 hyperacetylation sensitizes cells to reactive oxygen species. (A) Growth in NAM and H2O2 require different genetic pathways. Cells were serially diluted, spotted on the indicated media, and incubated at 30°C. (B) NAM does not elevate intracellular ROS. Exponentially growing cells were incubated in synthetic medium at 25°C with or without 20 mM NAM or 2.5 mM H2O2 for 8 h. Cells were stained with dihydrorhodamine 123 and cellular fluorescence was analyzed by flow cytometry. The distribution of fluorescence signals is represented in a box and whiskers plot in which the whiskers show the 5th and 95th percentiles. (C) NAM and H2O2 cause synergistic growth defects. Cells were grown in YPD at 30°C with indicated concentrations of NAM or H2O2. OD630 measurements were taken after 48 h and values were normalized relative to the untreated control. (D) hst3Δ hst4Δ cells are hypersensitive to H2O2. Cells were treated as in A, but were incubated at 25°C. (E) hst4Δ cells expressing a temperature sensitive-degron of Hst3 under the control of a methionine-repressible promoter grow poorly at 37°C. td: temperature-sensitive degron, including methionine-repressible promoter. SC-MET: synthetic medium lacking methionine. Cells were treated as in A, and incubated at the indicated temperature. (F) NAC reduces H2A serine 128 phosphorylation upon growth of td-HST3 at 37°C. Cells were grown to the exponential phase in synthetic medium lacking methionine. The medium was then changed to YPD and cells were incubated at 37°C. Samples were taken after 8 h and analyzed by immunoblotting. (G) NAC promotes cell cycle progression in cells lacking Hst3 and Hst4. Cells were treated as in F, and DNA content was analyzed by flow cytometry at the indicated time after changing the medium to YPD from SC-MET. (H) NAC improves the growth of cells lacking Hst3 and Hst4 activity at the restrictive temperature. td-Hst3–13Myc cells were grown overnight in SC-MET. An identical number of cells were then diluted in YPD containing either 0 or 100 mM NAC. Cells were then incubated at 25°C or 37°C, and OD readings at 630 nm were taken after 24 h. NT: non-treated, A.U.: Arbitrary units.
As an alternative explanation, we reasoned that NAM-induced H3K56 hyperacetylation could sensitize cells to DNA damage caused by normal ROS levels, thereby compromising cell proliferation. Consistent with this, we found that combined exposure to NAM and ROS-inducing agents led to strong synergistic growth defects (Figure 8C), and that hst3Δ hst4Δ mutants are exquisitely sensitive to H2O2 (Figure 8D). These results strongly suggest that H3K56 hyperacetylation sensitizes cells to ROS. To further validate this idea, we exploited an hst4Δ strain expressing a temperature-inducible degron allele of HST3 (td-Hst3) under the control of a methionine-repressible promoter. This strain exhibits strong temperature sensitivity in YPD, but not in synthetic medium lacking methionine (SC-MET; Figure 8E). Our results show that cells expressing td-Hst3 arrest in S phase when grown in YPD at the restrictive temperature of 37°C and accumulate elevated H3K56ac and H2A-P, a marker of DNA damage (Figure 8F). We found that addition of NAC to the growth medium noticeably improved growth at 37°C, alleviated S phase accumulation, and reduced H2A-P in cells expressing td-Hst3 grown at 37°C (Figure 8F–H). Overall, these data support a model in which endogenously-produced ROS contribute to growth defects caused by NAM-induced constitutive H3K56ac in yeast.
DISCUSSION
In the present study, we probed the role of the sirtuin family of histone deacetylases in promoting cell proliferation in Saccharomyces cerevisiae. Our results show that NAM-induced sirtuin inhibition promotes the induction of DNA lesions that impede cell growth, and that such DNA damage results largely from inhibition of Hst3 and Hst4 and consequent constitutive H3K56ac. This is consistent with known phenotypes of hst3Δ hst4Δ cells, which grow poorly and present spontaneous HR foci and activation of DNA damage-induced kinases (22,23). Mutations in several genes known to cause synthetic lethality when combined with hst3Δ hst4Δ, e.g. SRS2, SLX4 and DUN1, reduced cell fitness in NAM, thereby supporting our interpretation of the data (22). Our screens provide a comprehensive assessment of genetics networks responding to misregulated H3K56ac, and as such represent an important resource toward elucidation of the biological functions of this histone modification. Even though H3K56ac plays a dominant role, we note that compromising H4K16ac (via a H4K16A mutation) also partially suppressed NAM-induced growth inhibition. This is in agreement with our previous results indicating that lack of H4K16ac partially suppresses the temperature and genotoxin-sensitivity of hst3Δ hst4Δ cells, although the molecular mechanisms involved in such suppression remain unclear (23). We speculate that localized increase in H4K16ac in specific regions of the genome in response to NAM, e.g. at subtelomeric regions or other silent loci known to be deacetylated by the SIR complex, may cause DNA damage via unknown mechanisms. Further experiments will be required to understand the basis of such putative H4K16ac-induced DNA lesions, and to verify whether DNA damage specifically occurs at silent genomic loci in response to NAM.
Our data support a model in which cells with constitutive H3K56ac require Slx4- and Pph3-containing complexes to counteract Rad9-dependent activation of Rad53 in response to spontaneous DNA damage. Such a model is consistent with our published results indicating that histone mutations known to cripple Rad9 binding to chromatin, e.g. H3K79R, partially suppress the phenotypes of hst3Δ hst4Δ mutants (23). Interestingly, while formation of the Rtt107-Slx4 complex is necessary to limit Rad53 activity in response to replicative stress (68), deletion of either gene caused opposite effects in NAM. We propose that Slx4-Rtt107 complexes permit cells to tolerate NAM-induced DNA lesions, whereas complexes containing Rtt101 and/or Rtt107 generate DNA damage in an H3K56ac-dependent manner. We note that the biological significance of the Rtt101-Rtt107 complex and its links to H3K56ac is poorly characterized. Rtt101 is part of a ubiquitin ligase complex which promotes chromatin assembly behind DNA replication forks by ubiquitinating newly synthesized histones before their deposition into chromatin, thereby promoting the flow of new histones from the chaperone Asf1 to the CAF1 and Rtt106 chromatin assembly factors (97). However, Rtt107 is not required for such ubiquitination events to occur (97), suggesting that abnormal chromatin assembly is unlikely to explain Rtt107-dependent DNA damage in NAM-treated cells. On the other hand, Rtt109 and Rtt101 have been shown to promote Rtt107 recruitment to chromatin in response to replicative stress induced by MMS (74), and recently published data indicate that the Slx4-Rtt107 complex is recruited specifically behind stalled DNA replication forks during genotoxic stress (68,98). Since H3K56ac is normally only present behind DNA replication forks, we hypothesize that this histone modification may serve to restrict recruitment of the Slx4-Rtt107 or Rtt107-Rtt101 complexes behind stalled replisomes. We further speculate that constitutive H3K56ac may cause inappropriate localization of these complexes during DNA replication, leading to the formation of DNA lesions. Additional studies will be required to investigate the validity of such models.
NAM-induced activation of Rad53 was found to impede S phase progression, and lack of PPH3 and SLX4 strongly enhanced this effect. Our genetic data suggest that these phenotypes are attributable in part to Rad53-mediated inhibition of late replication origin firing. Our data also indicate that while Rad53-mediated inhibition of TLS (85,88) does not contribute significantly to S phase progression defects in slx4Δ and pph3Δ cells, such inhibition does prevent these mutants from initiating subsequent cell cycles after MMS exposure. This is consistent with the known ability of the TLS DNA damage tolerance pathway to act in G2 (93), and clarify the molecular mechanisms that impede cell cycle progression in slx4Δ and pph3Δ mutants. Our results and those of others (71) also show that deletion of REV3 in pph3Δ and slx4Δ cells cause synergistic sensitivity to MMS, raising the possibility that alternative pathways of DNA damage tolerance, e.g. homologous recombination, may be compromised in these mutants. In agreement with this, recently published data indicate that Slx4- and Pph3-mediated dampening of Rad53 activity promotes the functions of the Mus81-Mms4 structure-specific endonuclease, thereby permitting resolution of homologous recombination structures in response to MMS (99). Our finding that reduction of Rad9-dependent Rad53 activation via DOT1 deletion partially rescues the MMS sensitivity of slx4Δ rev3Δ and pph3Δ rev3Δ mutants also supports this notion.
Several observations suggest that NAM-induced constitutive H3K56ac promotes the induction of DNA lesions that differ from those bypassed by Pol Zeta, a TLS polymerase known to act on methylated DNA bases resulting from MMS exposure: (i) hst3Δ hst4Δ mutations cause increased rates of spontaneous CAN1 mutation in a Pol Zeta-independent manner (100), (ii) Rev3 is required for Dot1-mediated rescue of the MMS, but not temperature sensitivity of hst3Δ hst4Δ mutants (23) and (iii) suppression of NAM sensitivity in slx4Δ and pph3Δ cells by dot1Δ or rad9Δ is unaffected by REV3 deletion. Nevertheless, our results indicate that TLS-promoting PCNA mono-ub, but not PCNA poly-ub (which favors template switching), mitigates NAM-induced growth defects in slx4Δ dot1Δ and pph3Δ rad9Δ mutants. This in turn suggest that Rad53-mediated inhibition of TLS DNA polymerases other than Pol Zeta may contribute to growth defects caused by constitutive H3K56ac. Although the precise nature of the DNA lesions causing the phenotypes of cells with constitutive H3K56ac remains elusive, our data suggest that such lesions result at least in part from endogenous ROS. ROS-induced DNA adducts can be bypassed by mutagenic TLS polymerases (101), and can cause severe replicative stress (102). Indeed, we found that H3K56 hyperacetylation strongly sensitizes cells to H2O2, and that treatment with antioxidants mitigates DDR induction in both NAM-treated WT cells and in hst3Δ hst4Δ mutants. Overall, our results suggest a model in which sirtuin-mediated deacetylation of marks associated with newly synthesized histones is critical toward preventing growth inhibition due to endogenous genotoxins.
Sirtuins are the focus of intense investigation because of their conserved roles in modulating aging in several model systems (103). In addition, misregulated sirtuin expression is observed in several tumor types, and pharmacological inhibition of these enzymes may hold promise for cancer treatment (104). Our published results also demonstrate that pan-inhibition of sirtuins by nicotinamide prevents growth in several species of pathogenic fungi (24). Further understanding the biology of sirtuins therefore harbours important clinical ramifications. However the therapeutically-relevant targets of these enzymes, and their molecular mechanism of action, are far from being completely characterized. Toward enhancing our knowledge in this regard, results presented herein provide novel insight into the cellular and genetic networks responding to sirtuin inhibition and outline their role in modulating important aspects of the DNA damage response.
We thank Dr Dindial Ramotar (Université de Montréal, Canada), Dr John Diffley (Francis Crick Institute, Cancer Research UK), Dr Jef D. Boeke (New York University Langone, USA), Dr Alain Verreault (Université de Montreal, Canada) and Dr Peter Burgers (Washington University, St. Louis, USA) for yeast strains, antibodies and plasmids. We also thank the McGill University and Genome Quebec Innovation Center for the expression profiling experiment, Edlie St-Hilaire and Eirndeep Boparai for their technical support and Dr Elliot Drobetsky for critical reading of the manuscript.
FUNDING
Canadian Institutes of Health Research [MOP 123438]; Cole Foundation; Canadian Foundation for Innovation (to H.W.); Mérieux Institute to (M.R.); Canadian Cancer Society Research Institute [20380]; infrastructure funding from the National Human Genome Research Institute (to G.G. and C.N.). HW is the recipient of a Fonds de la recherche du Québec-Santé Junior 1 scholarship. AS is the recipient of a PhD scholarship from the Fonds de la recherche du Québec-Santé and from the Canadian Institutes for Health Research. Funding for open access charge: Canadian Institutes of Health Research [MOP 123438 to H.W.].
Conflict of interest statement. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments