





Abstract
Mistranslation can follow two events during protein synthesis: production of non-cognate amino acid:transfer RNA (tRNA) pairs by aminoacyl-tRNA synthetases (aaRSs) and inaccurate selection of aminoacyl-tRNAs by the ribosome. Many aaRSs actively edit non-cognate amino acids, but editing mechanisms are not evolutionarily conserved, and their physiological significance remains unclear. To address the connection between aaRSs and mistranslation, the evolutionary divergence of tyrosine editing by phenylalanyl-tRNA synthetase (PheRS) was used as a model. Certain PheRSs are naturally error prone, most notably a Mycoplasma example that displayed a low level of specificity consistent with elevated mistranslation of the proteome. Mycoplasma PheRS was found to lack canonical editing activity, relying instead on discrimination against the non-cognate amino acid by kinetic proofreading. This mechanism of discrimination is inadequate for organisms where translation is more accurate, as Mycoplasma PheRS failed to support Escherichia coli growth. However, minor changes in the defunct editing domain of the Mycoplasma enzyme were sufficient to restore E. coli growth, indicating that translational accuracy is an evolutionarily selectable trait.
INTRODUCTION
Accurate transfer of genetic information is critical for cellular maintenance and integrity, with each stage of gene expression requiring different levels of fidelity (1). DNA replication is an extremely accurate process with an error rate of 1 in 108 (2), whereas messenger RNA transcription and translation are relatively less accurate with misincorporation rates of 10−5 and 10−4, respectively (3–5). Fidelity in translation is dependent on several steps including synthesis of cognate aminoacyl transfer RNAs (aa-tRNAs) by aminoacyl tRNA synthetases (aaRSs), binding of aa-tRNAs by elongation factor Tu (EF-Tu), and accurate selection of aa-tRNAs by the ribosome (6–8). The aaRS family is composed of 23 distinct enzymes, each of which is responsible for ligating a single amino acid to a subset of tRNAs with specific anticodon sequences. Aminoacylation of tRNA by aaRSs is a two-step reaction, consisting of adenosine triphosphate (ATP)-dependent amino acid activation followed by ligation of amino acid to the 3′-end of tRNA forming an aminoacyl ester bond (9). During the activation step, approximately half of the aaRSs display a level of specificity of 3000:1 or greater for cognate versus non-cognate amino acids, which is similar to overall error rates typically observed during protein synthesis (10). For the other aaRSs, the existence of closely related near-cognate substrates precludes a high level of cognate amino acid discrimination, and these enzymes depend on an additional proofreading function called ‘editing’ for quality control (11). Editing is categorized as pre- or post-transfer depending on whether products of the first or second step of the aminoacylation reaction are hydrolyzed, respectively (6,12,13). AaRS editing contributes to quality control as part of a ‘double sieve’ mechanism (14,15). The first sieve is the synthetic active site of the aaRS, which determines the specificity for cognate substrate. The second sieve is an editing or proofreading activity to clear either near-cognate amino acids or mischarged tRNAs.
The aaRSs are an essential family of enzymes, but their corresponding editing activities are not required for cell viability under standard laboratory growth conditions [reviewed in (16)]. Although not lethal, defects in aaRS editing lead to mistranslation that results in stress responses because of the accumulation of misfolded proteins in eukaryotic cells, and increased susceptibility to some antibiotics and induction of the SOS mechanism in bacteria (17–22). Specific mechanisms actively reduce aaRS quality control and elevate misacylation under certain growth conditions, which can be beneficial for cell survival (23–26). The finding that aaRS-dependent mistranslation can be detrimental or advantageous depending on growth conditions suggests that accuracy may be a variable, selectable, trait rather than an absolute requirement for viable translation (27,28). To further investigate the evolution of aaRS quality control mechanisms and their role in regulating the accuracy of translation, we examined the discrimination of near-cognate tyrosine by phenylalanyl-tRNA synthetase (PheRS) from Mycoplasma mobile. Previous studies have shown that PheRS proofreading mechanisms display considerable structural and functional diversity between different organisms and cellular compartments, suggesting they have adapted to divergent cellular requirements for translation quality control (27). M. mobile provides an ideal model system to investigate the evolution of PheRS quality control, as the extensive mistranslation observed in the proteome of this organism was recently proposed to be directly linked to the absence of canonical editing domains in several aaRSs, including PheRS (28). Here, we show that M. mobile PheRS (MmPheRS) has lost post-transfer editing activity against misaminoacylated tRNA, and has acquired an alternative, albeit less efficient, quality control pathway dependent on kinetic proofreading. Sequence alignment-based single amino acid replacements in MmPheRS substantially increased proofreading activity in vivo and in vitro, suggesting that the mechanistic changes observed in the enzyme reflect divergent selection pressures on the translation quality control machinery during evolution. The possible advantages to proteome integrity of regulating translation accuracy via aaRS quality control mechanisms are discussed.
MATERIALS AND METHODS
General methods
Site-directed mutagenesis was performed by polymerase chain reaction using primers obtained from Sigma. Aminoacylation, ATP consumption, deacylation and steady state kinetic assays were performed as described previously (17,29).
Preparation of MmPheRS
GeneIDs for M. mobile pheS encoding the α subunit and pheT encoding the β subunit are MMOB3170 and MMOB5160, respectively. M. mobile gene sequences were codon optimized for expression in Escherichia coli; specifically, TGA codons were substituted with TGG codons, and the modified operon was synthesized (GenScript). The intergenic sequence between pheS and pheT genes was replaced with the sequence from that in E. coli to ensure efficient transcription and translation of the two subunits in E. coli. The MmPheRS-encoding genes were subcloned into pQE31 vector (Qiagen) at SacI and HindIII restriction sites. The resulting plasmid pQE31-His6-MmPheRS was used to transform E. coli BL21(DE3)pLysS cells. Cells were grown to an optical density at 600 nM (OD600) of 0.4 at 37°C, 250 r.p.m., and then grown until an OD600 of 0.7 at 22°C, 250 r.p.m. Isopropyl-β-D-thiogalactoside (IPTG) was then added to a final concentration of 0.5 mM, and cells were grown overnight at 22°C, 250 r.p.m. Cells were harvested; the pellet was resuspended in a buffer containing 25 mM Tris–HCl (pH 8.0), 300 mM NaCl, 10% glycerol and 5 mM imidazole, and flash frozen using liquid N2 before storage at −80°C. The frozen cells were thawed at 37°C for 10 min and sonicated, and supernatant was collected after centrifugation at 50 000 r.p.m. for 1 h. The supernatant was applied to a pre-equilibrated 3 ml TALON® resin metal affinity column (Clontech) followed by washing, and the protein was eluted with Tris–HCl (pH 8.0), 300 mM NaCl, 250 mM imidazole and 10% glycerol. Fractions containing MmPheRS were checked for electrophoretic purity by sodium dodecyl sulphate–polyacrylamide gel electrophoresis, pooled and dialyzed overnight at 4°C into buffer containing 10 mM Tris–HCl (pH 8.0), 200 mM NaCl, 5 mM NaH2PO4 and 10% glycerol, after which the protein was aliquoted and stored at −20°C in the same buffer with 50% glycerol.
Cloning and in vitro transcription of M. mobile tRNAPhe
The gene for M. mobile tRNAPheGAA (GeneID: MMOB9150) was synthesized using synthetic DNA oligomers according to standard procedures (30) and cloned into pUC19 vector using BamHI and HindIII restriction sites to yield pUC19-MmtRNAPhe. This plasmid (200 µg) was digested with BstNI to generate 3′ CCA and used as a template for run-off transcription using T7 RNA polymerase. The tRNA transcript was purified on denaturing 12% polyacrylamide gel and extracted by electrodialysis in 90 mM Tris-borate/2 mM ethylenediaminetetraacetic acid (EDTA) (pH 8.0). The tRNA was phenol and chloroform extracted, ethanol precipitated and resuspended in diethylpyrocarbonate (DEPC)-treated ddH2O. Refolding was carried out by heating the tRNA at 70°C for 2 min, followed by addition of MgCl2 to a concentration of 2 mM and subsequent slow cooling to room temperature.
Tyr-tRNAPhe preparation and hydrolysis assays
Wild-type MmPheRS (1 µM) was used to aminoacylate 5 µM tRNAPhe to produce Tyr-tRNAPhe, which was then purified, and reactions were performed as described previously (17). Reaction mixture contained 1 µM Tyr-tRNAPhe and 100–500 nM wild-type or mutant PheRS. Bovine serum albumin was used as a negative control and 0.1 M NaOH as a positive control.
Steady-state kinetics
Steady-state kinetic assays were carried out at 37°C as previously described (17,29). For ATP-PPi exchange assays, concentrations of substrates were varied from 0.5 to 200 µM for Phe, and 0.1 to 9 mM for Tyr. Enzymes were added to a final concentration of 100–150 nM. For steady-state aminoacylation assays, concentrations of substrates were varied from 0.5 to 100 µM for Phe, 20 to 400 µM for Tyr and 0.5 to 40 µM for tRNAPhe. For cognate and non-cognate charging reactions, final PheRS concentrations used were 100 and 500 nM, respectively. Kinetic parameters were determined as averages from three independent reactions, and their standard errors are shown.
Single-turnover kinetics
Experiments were performed at 22°C on a quench flow RQF-3 KinTek instrument, using the constant quench option. The buffer for PheRS contained 100 mM Na-Hepes (pH 7.2), 30 mM KCl, 10 mM MgCl2 and 10 mM dithiothreitol (DTT), whereas the quench solution contained 3 M sodium acetate (pH 4.5). The PheRS-aminoacyl-AMP complex is not stable following purification, so the complex was formed in situ in the first syringe as described previously (31). The sample was then mixed rapidly with tRNAPhe in the second syringe and quenched at various time intervals. In syringe A, enzyme-aminoacyl-adenylate complex was formed in situ by incubating 6–8 µM PheRS, 40 µM [14C]-Phe (215 cpm/pmol) or 80 µM [14C]-Tyr (360 cpm/pmol), 4 mM ATP and 2 units/mL of inorganic pyrophosphatase. Syringe B contained 2–4 µM tRNAPhe. Reaction aliquots of 36 µl (18 µl in each syringe) were quenched and precipitated in 5% trichloroacetic acid. The radiolabeled aa-tRNA products were quantified by scintillation counting. Amount of aa-tRNA formed was plotted versus time and fitted to the single exponential equation, y = C + A*[1 − exp( − ktrans × t)], where C is the y intercept, A is the amplitude, ktrans is the rate of aminoacyl transfer and t is time in seconds. Phe and Tyr transfer rate constants were determined by single-turnover kinetics using 6–8 µM PheRS and 3–4 µM MmtRNAPhe or EctRNAPhe. For EcPheRS, the αA294G βG318W editing-defective variant was used to prevent significant hydrolysis of Tyr-tRNAPhe during the assay. Data represent averages of three independent experiments and the corresponding standard errors.
E. coli NP37 pheSts complementation
The E. coli NP37 strain [pheSts, (32)] was co-transformed with plasmids pQE31-MmPheRS (ampr) and pREP4 (kanr, Qiagen) to ensure tight transcriptional regulation of plasmid-encoded PheRS. The transformants were plated on Luria broth (LB) supplemented with 100 µg/ml ampicillin and 50 µg/ml kanamycin and incubated at 30°C. Individual colonies were then streaked and grown on the same media as aforementioned at both 30°C and 42°C for 24–48 h. To ensure the colonies appearing at 42°C were not revertants, the presence of the pheSts mutation in E. coli NP37 was confirmed by sequencing.
RESULTS
MmPheRS lacks canonical quality control activities
Previous analyses of components of the M. mobile proteome showed mistranslation of up to 0.7% of Phe codons, as Tyr and preliminary sequence analyses suggested this was, in part, because of PheRS-specific editing defects (28). In E. coli PheRS (EcPheRS), the highly conserved residues βR244, βH265, βG318, βE334, βT354 and βA356 are all involved in editing (33). βR244, which interacts with the tRNA backbone primarily at C75, is replaced by Lys in most Mycoplasma species. Although MmPheRS retains the well-conserved His residue in the signature motif QPxHxFD, other key residues are not well conserved, including the second signature motif, GVMGGxxS/T, which aligns poorly because of extensive sequence changes, possibly indicative of changes in the editing activity of the M. mobile enzyme (Figure 1). For the cognate amino acid Phe, some differences were observed between MmPheRS and EcPheRS, with the M. mobile enzyme having a 2-fold higher kcat/KM in ATP-PPi exchange and the E. coli enzyme a 9-fold higher kcat/KM for aminoacylation [Tables 1 and 2; (35)]. For non-cognate Tyr, MmPheRS showed negligible post-transfer editing of Tyr-tRNAPhe compared with either wild-type EcPheRS or the αA294G βG318W variant, which has ∼80-fold reduced activity compared with wild-type [Figure 2A; (33)]. The absence of post-transfer editing rendered MmPheRS considerably more error prone than its E. coli counterpart, as shown by its ability to synthesize and accumulate Tyr-tRNAPhe (Figure 2B). The level of mischarging by MmPheRS was comparable with that of the αA294G βG318W editing-deficient E. coli variant and is in contrast to WT EcPheRS, which does not produce any detectable mischarged Tyr-tRNAPhe. These results indicate that MmPheRS lacks post-transfer editing activity and is inherently error prone.

Editing site sequence alignments for bacterial PheRSs. Sequences are from the first (top) and second (bottom) signature motifs using E. coli numbering. See main text for details. Inset is the structure of the Thermus thermophilus editing site in complex with Tyr, with E. coli numbering in parentheses (34).
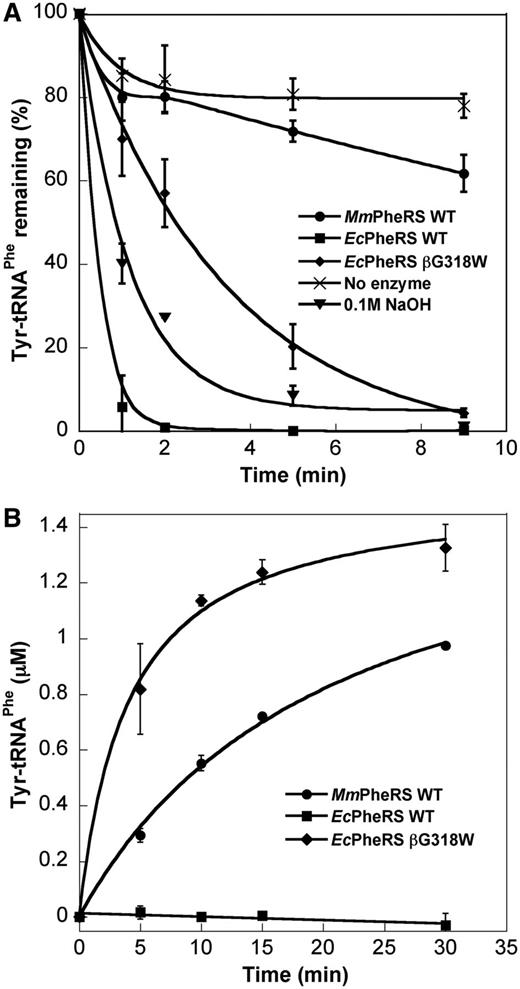
Aminoacylation and editing activities of MmPheRS. (A) Post-transfer editing of preformed Tyr-tRNAPhe by MmPheRS. WT EcPheRS and 0.1 M NaOH serve as positive controls and editing-deficient αA294G βG318W E. coli PheRS as a negative control. (B) Tyrosylation of MmtRNAPhe by MmPheRS, EcPheRS and the αA294G βG318W E. coli PheRS editing-deficient mutant. Data represent averages of three independent experiments and the corresponding standard errors.
Steady-state kinetic constants for tRNA aminoacylation by MmPheRS
| Amino acid | kcat (s−1) | KM (µM) | kcat/KM (s−1 µM−1) | Specificity (kcat/KM) Phe/(kcat/KM) Tyr |
|---|---|---|---|---|
| Phe | 0.10 ± 0.03 | 3.3 ± 0.5 | 0.040 ± 0.003 | |
| Tyr | 0.0100 ± 0.0005 | 130 ± 40 | 9 × 10−5 ± 0.3 × 10−5 | 440 |
| Amino acid | kcat (s−1) | KM (µM) | kcat/KM (s−1 µM−1) | Specificity (kcat/KM) Phe/(kcat/KM) Tyr |
|---|---|---|---|---|
| Phe | 0.10 ± 0.03 | 3.3 ± 0.5 | 0.040 ± 0.003 | |
| Tyr | 0.0100 ± 0.0005 | 130 ± 40 | 9 × 10−5 ± 0.3 × 10−5 | 440 |
Steady-state kinetic constants for tRNA aminoacylation by MmPheRS
| Amino acid | kcat (s−1) | KM (µM) | kcat/KM (s−1 µM−1) | Specificity (kcat/KM) Phe/(kcat/KM) Tyr |
|---|---|---|---|---|
| Phe | 0.10 ± 0.03 | 3.3 ± 0.5 | 0.040 ± 0.003 | |
| Tyr | 0.0100 ± 0.0005 | 130 ± 40 | 9 × 10−5 ± 0.3 × 10−5 | 440 |
| Amino acid | kcat (s−1) | KM (µM) | kcat/KM (s−1 µM−1) | Specificity (kcat/KM) Phe/(kcat/KM) Tyr |
|---|---|---|---|---|
| Phe | 0.10 ± 0.03 | 3.3 ± 0.5 | 0.040 ± 0.003 | |
| Tyr | 0.0100 ± 0.0005 | 130 ± 40 | 9 × 10−5 ± 0.3 × 10−5 | 440 |
Steady-state kinetic constants for ATP-[32P]PPi exchange by wild-type and MmPheRS variants
| Phe | Tyr | ||||||
|---|---|---|---|---|---|---|---|
| MmPheRS | kcat (s−1) | KM (µM) | kcat/KM (s−1 µM−1) | kcat (s−1) | KM (µM) | kcat/KM (s−1 µM−1) | Specificitya (Phe/Tyr) |
| WT | 10.0 ± 0.2 | 10 ± 1 | 1.0 ± 0.2 | 8.0 ± 0.4 | 3900 ± 540 | 0.0021 ± 0.0003 | ∼500 |
| (T305A | 24 ± 4 | 11 ± 1 | 2.3 ± 0.5 | 3.4 ± 0.8 | 2060 ± 40 | 0.0017 ± 0.0004 | ∼1350 |
| βQ306A | 18 ± 3 | 10 ± 4 | 2.1 ± 0.6 | 5 ± 1 | 3850 ± 1590 | 0.0014 ± 0.0002 | ∼1500 |
| (T305A/Q306A | 15 ± 4 | 9.0 ± 0.1 | 1.7 ± 0.4 | ND | ND | 0.0006 ± 0.000b | ∼3000 |
| Phe | Tyr | ||||||
|---|---|---|---|---|---|---|---|
| MmPheRS | kcat (s−1) | KM (µM) | kcat/KM (s−1 µM−1) | kcat (s−1) | KM (µM) | kcat/KM (s−1 µM−1) | Specificitya (Phe/Tyr) |
| WT | 10.0 ± 0.2 | 10 ± 1 | 1.0 ± 0.2 | 8.0 ± 0.4 | 3900 ± 540 | 0.0021 ± 0.0003 | ∼500 |
| (T305A | 24 ± 4 | 11 ± 1 | 2.3 ± 0.5 | 3.4 ± 0.8 | 2060 ± 40 | 0.0017 ± 0.0004 | ∼1350 |
| βQ306A | 18 ± 3 | 10 ± 4 | 2.1 ± 0.6 | 5 ± 1 | 3850 ± 1590 | 0.0014 ± 0.0002 | ∼1500 |
| (T305A/Q306A | 15 ± 4 | 9.0 ± 0.1 | 1.7 ± 0.4 | ND | ND | 0.0006 ± 0.000b | ∼3000 |
aSpecificity (Phe/Tyr) = (kcat/KM)Phe/(kcat/KM)Tyr.
bkcat/KM was estimated using sub-saturating Tyr concentrations from the slope of the equation, V = kcat [E][S]/KM.
ND, not determined.
Steady-state kinetic constants for ATP-[32P]PPi exchange by wild-type and MmPheRS variants
| Phe | Tyr | ||||||
|---|---|---|---|---|---|---|---|
| MmPheRS | kcat (s−1) | KM (µM) | kcat/KM (s−1 µM−1) | kcat (s−1) | KM (µM) | kcat/KM (s−1 µM−1) | Specificitya (Phe/Tyr) |
| WT | 10.0 ± 0.2 | 10 ± 1 | 1.0 ± 0.2 | 8.0 ± 0.4 | 3900 ± 540 | 0.0021 ± 0.0003 | ∼500 |
| (T305A | 24 ± 4 | 11 ± 1 | 2.3 ± 0.5 | 3.4 ± 0.8 | 2060 ± 40 | 0.0017 ± 0.0004 | ∼1350 |
| βQ306A | 18 ± 3 | 10 ± 4 | 2.1 ± 0.6 | 5 ± 1 | 3850 ± 1590 | 0.0014 ± 0.0002 | ∼1500 |
| (T305A/Q306A | 15 ± 4 | 9.0 ± 0.1 | 1.7 ± 0.4 | ND | ND | 0.0006 ± 0.000b | ∼3000 |
| Phe | Tyr | ||||||
|---|---|---|---|---|---|---|---|
| MmPheRS | kcat (s−1) | KM (µM) | kcat/KM (s−1 µM−1) | kcat (s−1) | KM (µM) | kcat/KM (s−1 µM−1) | Specificitya (Phe/Tyr) |
| WT | 10.0 ± 0.2 | 10 ± 1 | 1.0 ± 0.2 | 8.0 ± 0.4 | 3900 ± 540 | 0.0021 ± 0.0003 | ∼500 |
| (T305A | 24 ± 4 | 11 ± 1 | 2.3 ± 0.5 | 3.4 ± 0.8 | 2060 ± 40 | 0.0017 ± 0.0004 | ∼1350 |
| βQ306A | 18 ± 3 | 10 ± 4 | 2.1 ± 0.6 | 5 ± 1 | 3850 ± 1590 | 0.0014 ± 0.0002 | ∼1500 |
| (T305A/Q306A | 15 ± 4 | 9.0 ± 0.1 | 1.7 ± 0.4 | ND | ND | 0.0006 ± 0.000b | ∼3000 |
aSpecificity (Phe/Tyr) = (kcat/KM)Phe/(kcat/KM)Tyr.
bkcat/KM was estimated using sub-saturating Tyr concentrations from the slope of the equation, V = kcat [E][S]/KM.
ND, not determined.
In the absence of post-transfer editing activity, it is possible that MmPheRS relies instead for quality control on highly stringent Phe recognition and Tyr discrimination. This mechanism is also used by yeast mitochondrial PheRS to compensate for its inability to edit Tyr-tRNAPhe (27). Steady-state kinetic analyses of MmPheRS revealed a Phe:Tyr specificity ratio during aminoacylation of 440:1 (Table 1). Examination of amino acid specificity during ATP-dependent activation indicated that the MmPheRS active site displays specificity for Phe over Tyr of 480:1 (Table 2). The specificity for Phe over Tyr of MmPheRS is 14-fold lower than that of EcPheRS, demonstrating a substantial reduction in discrimination against the near-cognate amino acid.
Pre-transfer editing provides a potential additional quality control mechanism to prevent usage of near-cognate Tyr by PheRS. To investigate possible pre-transfer editing by MmPheRS, ATP consumption assays were performed using either cognate Phe or near-cognate Tyr, in the presence or absence of tRNAPhe. The level of ATP consumption in the presence of Tyr was similar to that in the presence of Phe. ATP consumption increased on addition of tRNA in the presence of Tyr, but not Phe, suggesting weak tRNA-dependent editing activity (Figure 3A). To distinguish between cis-post-transfer editing and tRNA-dependent pre-transfer editing, we used a non-chargeable substrate, 2′-deoxy EctRNAPhe (17). In the presence of Tyr and 2′-deoxy EctRNAPhe, the ATP consumption rate was similar to that found in the presence of Tyr and chargeable tRNA (Figure 3B). These data indicate that MmPheRS exhibits tRNA-dependent pre-transfer hydrolysis activity, which provides a modest, ∼2-fold, discrimination between Phe and Tyr (Figure 3A). The relatively low substrate specificity of amino acid activation, weak pre-transfer-editing and absence of post-transfer editing together show that MmPheRS lacks all of the canonical quality control mechanisms normally used by aaRS enzymes to prevent non-cognate aa-tRNA synthesis.
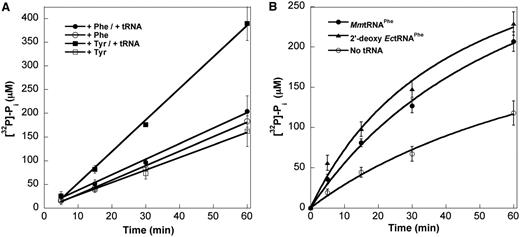
ATP hydrolysis by MmPheRS. (A) ATP hydrolysis activity in the presence of cognate Phe (1 mM) or noncognate Tyr (5 mM), with or without MmtRNAPhe (10 µM). (B) tRNA-dependent ATP hydrolysis in the presence of non-cognate Tyr (5 mM) with MmtRNAPhe (5 µM), 2′-deoxy EctRNAPhe (5 µM) or no tRNA. ATP hydrolysis was monitored in the absence of enzyme during each experiment and subtracted from data points. Data represent averages of three independent experiments and the corresponding standard errors.
Kinetic proofreading during amino acid transfer by MmPheRS
Steady-state amino acid activation rates (kcat) for MmPheRS were similar with both Phe and Tyr, whereas the overall rate of tRNA charging with Tyr was ∼10-fold lower than that with Phe. This finding suggests MmPheRS may kinetically discriminate against near-cognate Tyr at a step subsequent to activation, either aminoacyl transfer to tRNA or release of aminoacyl-tRNA. Previous studies showed that aminoacyl-tRNA release is not the rate-determining step during aminoacylation by EcPheRS (36–38), and this was also found to be the case for MmPheRS (Figure 4). Using αA294G and αA294S EcPheRS variants, which can accommodate unnatural Phe analogs such as p-chlorophenylalanine (p-Cl Phe), aminoacyl transfer rate constants were previously measured for cognate Phe and p-Cl Phe (38). p-Cl Phe transferred to tRNAPhe at a faster rate than Phe, indicating that near- or non-cognate aminoacyl-AMP intermediates form less stable complexes with EcPheRS than their cognate counterparts. Using rapid quench assays under single-turnover conditions, ktrans was measured for Tyr and Phe. An EcPheRS editing-deficient mutant (αA294G βG318W) was used to prevent hydrolysis of mischarged Tyr-tRNAPhe during the timescale of the reaction. The observed ktrans for Tyr was ∼2-fold higher (30 ± 2 s−1) than that for Phe (16 ± 3 s−1), consistent with previous studies (38). In contrast, for MmPheRS, the rate of Phe transfer (16 ± 2 s−1) was 5-fold faster than the rate of Tyr transfer (3 ± 0.4 s−1). These data reveal that MmPheRS, unlike its E. coli counterpart, uses a distinct mechanism to discriminate cognate from near-cognate substrates based on kinetic proofreading of aminoacyl-adenylates during the transfer step of the aminoacylation reaction.
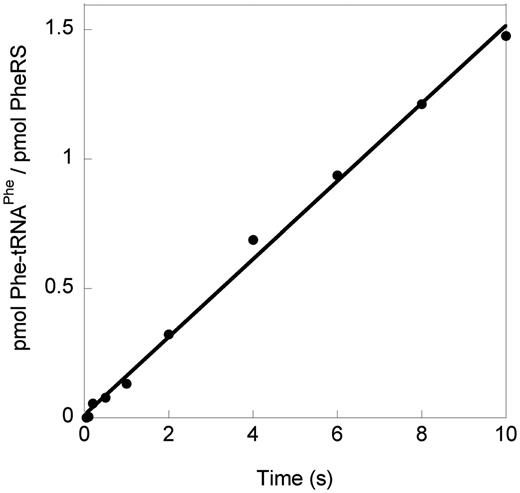
Transient kinetics of MmPheRS. Time course of aminoacylation in pre-steady-state burst conditions (0.5 μM PheRS and 20 μM tRNAPhe). The absence of a burst of product formation indicates that product release is not rate limiting, in contrast to aaRSs where product release is rate limiting (31).
Restoring canonical quality control in MmPheRS
In vitro studies (see earlier) showed that MmPheRS lacks canonical quality control mechanisms and suggest that the enzyme may not be able to support growth of organisms, such as E. coli, where translation is predicted to be more accurate than in M. mobile. The in vivo activity of MmPheRS in a comparatively accurate cellular context was investigated using the E. coli strain NP37, which contains a temperature-sensitive (ts) chromosomal pheS allele that restricts growth at 42°C. Wild-type EcPheRS displayed robust complementation, while MmPheRS was unable to rescue growth of E. coli NP37 at 42°C (Figure 5A). E. coli tRNAPhe is highly homologous to M. mobile tRNAPhe, and both EcPheRS and MmPheRS can cross-aminoacylate either tRNA (Supplementary Figure S1), suggesting the lack of complementation by MmPheRS does not result from a tRNA recognition defect. Cross-species tRNA recognition was further supported by the ability of PheRS M. mobile α-subunits and E. coli β-subunits produced together in vivo to rescue growth (Supplementary Figure S2). To investigate whether the absence of complementation in vivo stemmed from the relatively poor quality control observed in vitro with MmPheRS, sequence alignment-based variants of MmPheRS were constructed in an attempt to restore proofreading activity. Replacements were confined to the second editing motif of the β-subunit (G315VMGGxxS/T, E. coli numbering), which is considerably diverged in M. mobile when compared with the canonical, conserved, bacterial signature sequence (Figure 1). In the second editing motif of MmPheRS, all three Gly residues have been replaced by relatively bulkier residues (βY302, βT305 and βQ306), which may occlude Tyr from the editing active site. The impact of these changes on the quality control activities of MmPheRS was investigated by making Ala and Gly substitutions of βY302, βT305 and βQ306. βY302A/G, βT305A/G and βQ306A/G MmPheRS-encoding genes were then screened for their in vitro activities and ability to rescue the ts phenotype of E. coli NP37 (Table 3). Only genes encoding βT305A and βQ306A MmPheRS rescued growth at 42°C, as did the βT305A/Q306A variant, albeit poorly (Figure 5A).
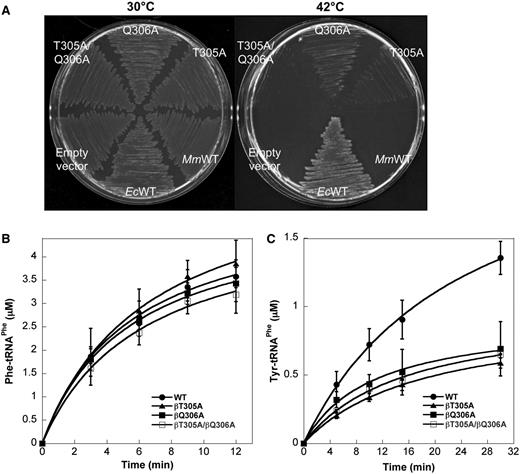
Activity of MmPheRS quality control variants. (A) Rescue of growth phenotype of E. coli NP37 (pheSts). E. coli NP37 was transformed with plasmids producing WT MmPheRS, MmPheRS variants βT305A, βQ306A and βT305A/Q306A and WT EcPheRS as positive and empty vector as negative controls, respectively. (B) Phenylalanylation and (C) tyrosylation by WT MmPheRS and variants βT305A, βQ306A and βT305A/Q306A. Data represent averages of three independent experiments and the corresponding standard errors.
Properties of MmPheRS quality control variants
| MmPheRS | Phe-tRNAPhe (Relative)a | Tyr-tRNAPhe (Relative)b | Viabilityc |
|---|---|---|---|
| WT | 1 | 1 | No |
| βP301G | 0.2 | 0.1 | No |
| βY302G | 1 | 2 | No |
| βT305G | 1 | 2.5 | No |
| βQ306G | 1 | 1 | No |
| βT305G/βQ306G | NDd | ND | No |
| βY302G/βT305G/βQ306G | ND | ND | No |
| βT305A | 1 | 0.4 | Yes |
| βQ306A | 1 | 0.4 | Yes |
| βT305A/βQ306A | 1 | 0.4 | Yes |
| MmPheRS | Phe-tRNAPhe (Relative)a | Tyr-tRNAPhe (Relative)b | Viabilityc |
|---|---|---|---|
| WT | 1 | 1 | No |
| βP301G | 0.2 | 0.1 | No |
| βY302G | 1 | 2 | No |
| βT305G | 1 | 2.5 | No |
| βQ306G | 1 | 1 | No |
| βT305G/βQ306G | NDd | ND | No |
| βY302G/βT305G/βQ306G | ND | ND | No |
| βT305A | 1 | 0.4 | Yes |
| βQ306A | 1 | 0.4 | Yes |
| βT305A/βQ306A | 1 | 0.4 | Yes |
aSteady-state aminoacylation level after 12 min compared with WT.
bSteady-state aminoacylation level after 15 (Gly substitutions) or 30 min (Ala substitutions) compared to WT.
cAbility to rescue growth of E. coli NP37 at 42°C.
dND, no detectable activity.
Properties of MmPheRS quality control variants
| MmPheRS | Phe-tRNAPhe (Relative)a | Tyr-tRNAPhe (Relative)b | Viabilityc |
|---|---|---|---|
| WT | 1 | 1 | No |
| βP301G | 0.2 | 0.1 | No |
| βY302G | 1 | 2 | No |
| βT305G | 1 | 2.5 | No |
| βQ306G | 1 | 1 | No |
| βT305G/βQ306G | NDd | ND | No |
| βY302G/βT305G/βQ306G | ND | ND | No |
| βT305A | 1 | 0.4 | Yes |
| βQ306A | 1 | 0.4 | Yes |
| βT305A/βQ306A | 1 | 0.4 | Yes |
| MmPheRS | Phe-tRNAPhe (Relative)a | Tyr-tRNAPhe (Relative)b | Viabilityc |
|---|---|---|---|
| WT | 1 | 1 | No |
| βP301G | 0.2 | 0.1 | No |
| βY302G | 1 | 2 | No |
| βT305G | 1 | 2.5 | No |
| βQ306G | 1 | 1 | No |
| βT305G/βQ306G | NDd | ND | No |
| βY302G/βT305G/βQ306G | ND | ND | No |
| βT305A | 1 | 0.4 | Yes |
| βQ306A | 1 | 0.4 | Yes |
| βT305A/βQ306A | 1 | 0.4 | Yes |
aSteady-state aminoacylation level after 12 min compared with WT.
bSteady-state aminoacylation level after 15 (Gly substitutions) or 30 min (Ala substitutions) compared to WT.
cAbility to rescue growth of E. coli NP37 at 42°C.
dND, no detectable activity.
The ability of the sequence alignment-based MmPheRS variants to synthesize Phe-tRNAPhe and Tyr-tRNAPhe was examined in vitro. βT305A, βQ306A and βT305A/Q306A MmPheRS all showed normal levels of Phe-tRNAPhe and reduced Tyr-tRNAPhe synthesis compared with wild-type, indicative of elevated quality control and consistent with the complementation activities of the corresponding genes (Table 3; Figures 5B and 5C). Trans-editing assays showed that the Ala substitutions at T305A and Q306A had not restored post-transfer editing activity, nor did ATP consumption assays show an increased rate of AMP production, indicating that none of the three MmPheRS variants had acquired editing function. Amino acid activation kinetics showed that the βT305A and βQ306A replacements all increased specificity for Phe over Tyr by 3- to 6- fold compared with wild-type, consistent with the observed reduction in Tyr-tRNAPhe synthesis (Table 2). These data indicate that the sequence alignment-based replacements in the second editing motif support E. coli growth by partially restoring a canonical quality control activity normally absent from MmPheRS. Despite all the MmPheRS variants being at residues in the defunct editing domain, increased quality control resulted from improved amino acid discrimination at the active site in the α-subunit rather than restoration of editing activity in the β-subunit. This suggests communication between the editing and synthetic active sites, analogous to the recent observation that fusion of a CP1 editing domain to M. mobile leucyl-tRNA synthetase (LeuRS) leads to enhancement of amino acid discrimination during the activation step in addition to conferring post-transfer editing activity (39).
DISCUSSION
Structural and functional divergence of PheRS
The proteome of the obligate intracellular pathogen M. mobile contains significant amino acid ambiguities, with misincorporation rates at Leu and Phe codons estimated to be as high as 1 in 200 (28). The absence of the canonical CP1 domain, and the accompanying loss of editing activity, in M. mobile LeuRS suggested that mistranslation of Leu codons resulted from elevated levels of mischarged tRNALeu. Our data suggests that the same is true for mistranslation of Phe codons in M. mobile, which likely results from elevated levels of mischarged tRNAPhe. Detailed in vitro analyses showed that the relatively low accuracy of MmPheRS aminoacylation results from changes at all steps in the reaction rather than loss of a single quality control step. Previous studies have shown that PheRS displays considerable divergence in quality control; some organisms, such as E. coli, retain multiple quality control steps while others, such as yeast, depend on single steps to maintain cellular viability (27,29). In addition, even when only a single quality control mechanism is retained, as in both yeast PheRSs, this is still not essential for cellular viability (27). These findings suggest that, under appropriate growth conditions, PheRS could evolve to dispense entirely with canonical quality control and still maintain cellular viability. This was confirmed by the in vitro and in vivo data presented here for MmPheRS. In addition to losing canonical quality control pathways, MmPheRS has acquired the ability to kinetically discriminate against non-cognate intermediates during the aminoacyl transfer step. Kinetic proofreading provides a mechanism to increase fidelity at lower time and energy costs compared with recycling reactions such as post-transfer editing (40,41), but its capacity to maintain adequately fast and accurate aa-tRNA synthesis during translation has been questioned (14). Analysis of MmPheRS aminoacylation shows that kinetic proofreading alone can support viable translation, but only in cellular systems with relatively low requirements for translational accuracy.
The functional divergence of MmPheRS may also have been facilitated by the expected effect on the proteome of the high AT content of Mycoplasma genomes (42,43). Bioinformatic analyses have shown that organisms containing AT-rich genomes possess a greater bias to AT-rich codons encoding the amino acids Phe, Tyr, Met, Ile, Asn and Lys rather than GC-rich codons coding for Gly, Ala, Arg and Pro (44). Comparison of the sequences of MmPheRS and EcPheRS shows a strong tendency toward the amino acid substitutions predicted from changes in nucleotide bias (Figure 6). This trend away from certain amino acids would be expected to favor substitutions at various key synthetic and editing site residues (Figure 1). Therefore, nucleotide bias appears to have acted as a non-specific factor facilitating a shift in the amino acid composition and functional properties of MmPheRS.
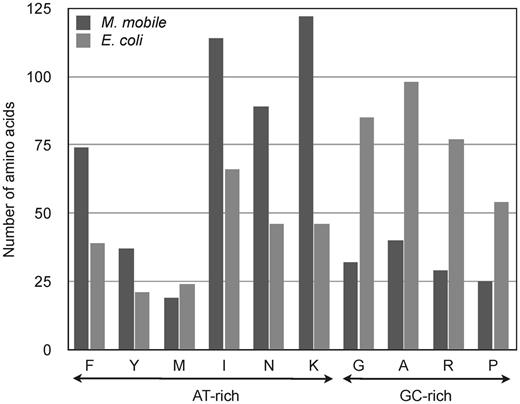
Amino acid composition of PheRS. Dark gray bars represent MmPheRS and light gray bars represent E. coli PheRS.
Relaxation of aaRS quality control confines mistranslation to structurally related amino acid pairs
The Mycoplasma proteome contains a substantially higher level of ambiguous decoding than routinely observed in other bacteria such as E. coli (28). Translational ambiguity can arise via different mechanisms. For instance, ribosomal ambiguity (ram) mutations cause miscoding by increasing the affinity of ribosomes for non-cognate aa-tRNA in a non-specific codon-independent manner (45). Another general mechanism of mistranslation involves mismethionylation of several tRNA species by MetRS in bacteria, as well as mammalian cells, which is believed to protect against oxidative stress (24,25). In contrast to these general mechanisms that lead to mistranslation, variability in the M. mobile proteome is confined to specific amino acid substitutions. In particular, decoding of Leu and Phe codons is highly erroneous with rates of ∼1 in 200 versus an error rate of ≤1 in 3000 for translation of other codons. Data presented here, together with recent studies on MmLeuRS (25), suggest that functional divergence of aaRSs allows for error modulation by restricting mistranslation to a subset of codons rather than causing a global defect. Reducing the stringency of the quality control steps used by M. mobile aaRSs targets translation errors to near-cognate amino acids, ensuring that any changes in the composition of the proteome following mistranslation are structurally and functionally conservative. While modulating aaRS-quality control has less impact on the overall integrity of the proteome than non-specific mistranslation, the selective advantage of reducing the accuracy of translation is unclear. One potential advantage, as proposed by Li et al. (28), is that mistranslation may protect M. mobile from host defenses by increasing antigen diversity. This is analogous to some mechanisms proposed to promote the diversity of peptides presented by major histocompatibility complex molecules (46). Our studies now show that modulating aaRS amino acid specificity may provide a potential mechanism to adapt to different stresses, and further studies of the divergence of quality control mechanisms may provide new insights into the physiology of the corresponding organisms.
FUNDING
National Science Foundation (NSF) [MCB-1052344 to M.I.]. Funding for open access charge: NSF [MCB-1052344].
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Drs Tammy Bullwinkle, Assaf Katz and Jenni Shepherd for critical reading of the manuscript.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments