





ABSTRACT
Peptidome is a public repository that archives and freely distributes tandem mass spectrometry peptide and protein identification data generated by the scientific community. Data from all stages of a mass spectrometry experiment are captured, including original mass spectra files, experimental metadata and conclusion-level results. The submission process is facilitated through acceptance of data in commonly used open formats, and all submissions undergo syntactic validation and curation in an effort to uphold data integrity and quality. Peptidome is not restricted to specific organisms, instruments or experiment types; data from any tandem mass spectrometry experiment from any species are accepted. In addition to data storage, web-based interfaces are available to help users query, browse and explore individual peptides, proteins or entire Samples and Studies. Results are integrated and linked with other NCBI resources to ensure dissemination of the information beyond the mass spectroscopy proteomics community. Peptidome is freely accessible at http://www.ncbi.nlm.nih.gov/peptidome.
INTRODUCTION
With the abundance of DNA sequence information currently available, researchers are now looking to comprehensively identify and characterize the proteomic products of the genetic blueprint. The most widespread and high-throughput methodology being used to address this goal is mass spectrometry (MS). Using MS to perform large-scale experiments generates substantial amounts of peptide and protein mass spectra. The informatics issues involved in dealing with these volumes of data can be overwhelming even within an individual laboratory. This matter, together with a lack of open standard formats, has contributed towards the general shortage of publicly available MS-based proteomic data. However, there is increasing recognition within the community, granting bodies and publishing agencies (1) that published proteomic data can, and should, be fully accessible. Open access policies such as these allow the community to review and comprehensively re-examine the data upon which experimental conclusions are based. From the researcher's; perspective, the long-term archiving of proteomic data at a centralized repository not only increases the data usability and visibility but also decreases the risk of data loss. In addition, the availability of large collections of MS data can benefit the field in a wider sense, for example, by enabling informaticians to develop better algorithms or construct spectral libraries.
Several databases already exist to store and disseminate proteomic data including PRIDE (2), PeptideAtlas (3), Tranche (4), The GPM (5), and Human Proteinpedia (6). Peptidome (7) aims to complement these resources; the major goals of the Peptidome project are to:
build and maintain a robust database in which to efficiently store tandem MS data at a level of detail appropriate for both MS professionals and the wider biological community,
develop simple deposit procedures that minimize the burden of submission while supporting well-annotated data deposits from the research community,
exchange data with established MS repositories,
offer user-friendly tools that enable users to query, locate, analyze and review the data of interest.
ORGANIZATION OF THE DATABASE
The information captured in the online database is designed to represent the final results based upon the submitter's; interpretation of the raw MS/MS spectra. This is intended to provide a quick overview for mass spectrometrists and accessibility for other non-specialists. The original submitted files are the true record, and will always be available for download. A graphical sketch of the database schema is shown in Figure 1.
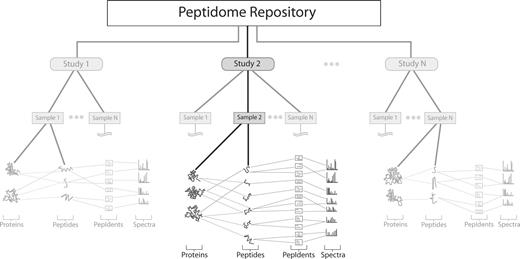
A schematic overview of Peptidome. The main logical entity is a ‘Study’ which contains a set of related ‘Samples’. Each Sample contains a list of both identified ‘proteins’ and ‘peptides’. Proteins contain links to their member peptides, and peptides know which proteins they are members of. Furthermore, peptides are linked to individual ‘spectra’ through peptide identifications (pepIdents), thus allowing more than one interpretation of a spectrum, and providing storage for post-translational modifications and identification scores.
The two major components of Peptidome are Studies and Samples. A Study is a collection of related Samples and provides a focal point for, and description of, the whole experiment. A Sample contains all the data related to the biological material, which may be derived from one or more MS instrument runs. Each Sample record contains a list of identified proteins and a list of identified peptides. Each protein points to a sublist of peptides by which it was identified, and each peptide is linked to the protein(s) that it is a member of. Each peptide contains a set of peptide identifications (pepIdents). These pepIdents denote any post-translational modifications as well as any identification scores for the match between a given spectrum and a peptide. Note that this allows a spectrum to have more than one peptide associated with it. A Sample also contains descriptive information about the biological material, protocols used to generate the data, instrumentation and informatics parameters.
Both Studies and Samples are accessioned objects; each record is assigned a unique and stable Peptidome accession number that may be cited in a manuscript describing the data. The accession consists of a number and a letter prefix indicating whether the record is a Peptidome Study (PSExxx) or Peptidome Sample (PSMxxxx).
SUBMISSION PROCEDURES
Our goal is to make data submission procedures as straightforward as possible, while encouraging a high level of experimental annotation. To minimize the burden of data submission, Peptidome accepts native file formats from which required information is extracted.
There are four components that are required for a complete submission:
A metadata file that describes the overall experiment, each associated biological sample, the instruments and protocols used to generate the data and the relationship of the corresponding data files. This information is provided by completing a metadata spreadsheet; templates and example spreadsheets are provided within the Peptidome submission guidelines from a link on the Petidome homepage.
Raw data files contain the original MS and MS/MS information from the instrument. All spectra should be submitted, irrespective of whether they are identified or not. Peptidome currently accepts any of the standard XML formats (mzData, mzXML or mzML) that contain both the MS and MS/MS data from a single fraction. In addition, text formats (.mgf, .pkl, .sqt, .dta) are also accepted, but not preferred. Proprietary binary data directly from the manufacturers (e.g. .raw or .wiff) are not accepted.
Output files from any peptide identification program. These files contain matches of the MS/MS scans to the peptides. Currently, Peptidome supports DAT files from Mascot, ASN.1 or XML formatted files from OMSSA, and any search engine output files that have been converted to PepXML format (e.g. X!Tandem or Sequest). If manual identification was used to interpret the spectra, or the search engine output formats are not supported by Peptidome, the submitted results table must include references to spectrum files and additional information that is usually extracted from the search engine output files, e.g. charge state and identification scores.
Results tables that describe the submitter's; view of the final, processed results according to whatever criteria they use to determine acceptability. Results tables list the proteins discovered in each Sample in the Study. For each protein, the peptides should be listed, and for each peptide, the matching spectrum files should be listed. Any natural or artificial post-translational modifications can also be specified. If the matching spectrum file list is omitted, then every spectrum matched to that peptide/protein in the Peptide identification output files is assumed to be correct. Similarly, if the Results table contains only proteins, then all associated peptides and spectra will be gleaned from the Peptide identification output files.
There are many different methods for MS quantification, with new ones being invented all the time. Therefore, for peptide and protein quantification, each quantification value with a single number per protein, peptide and/or spectrum per Sample is denoted, where the units (if applicable) and methodology used to quantify the sample are required in the Metadata annotation. More complete information about the quantification method used may be submitted in Ssupplementary Data.
When all submission files are assembled, they can be transferred to Peptidome via FTP. A curator will collect the files and manually curate and validate them before depositing in the database. Work on more automated methods for depositing records is currently in progress.
Some journals require accession numbers for MS proteomic data before acceptance of a paper for publication. Thus, ideally, data should be deposited in Peptidome before a manuscript describing the data is sent to a journal for review. Authors can cite the Peptidome accession number(s) in their manuscripts. The submission may remain private until a manuscript describing the data is published. A reviewer URL can be generated and disclosed to journal editors; this URL grants anonymous, confidential access to private data during the manuscript review period.
BUILDING THE DATABASE
All submissions undergo both syntactic validation and manual review by a curator before being uploaded to the Peptidome database, thus enforcing good quality data deposits. When any issues, such as mangled formats or missing components, are identified, a curator will work with the submitter until the problems are resolved. Early exemplar submissions in Peptidome were gathered from selected data in PeptideAtlas (3), with the metadata manually enriched from publications associated with those experiments.
Submissions are processed using custom software to upload the metadata and results into the database. The spectra files are not loaded into the database, instead they are converted to a custom format, based upon HDF5 (available from http://www.hdfgroup.org/HDF5/), that is both more compact and allows faster retrieval of individual spectra than the original XML format.
Proteins are linked with the corresponding entries in NCBI Entrez in a best effort fashion. This flexibility allows submitters to reference novel proteins that are not yet in the mainstream databases, and to use custom protein databases. Peptidome protein links are updated in an ongoing process, in order that they remain up-to-date.
RETRIEVING PEPTIDOME DATA
The data in Peptidome may be browsed by Studies or Samples, and the individual Samples may be examined from the identified proteins and peptides down to the individual spectra as shown in Figure 2.

Metadata for all public Studies and Samples, and the associated proteins for each Sample, are loaded into NCBI's; powerful Entrez search and linking system (9). This facilitates cross-linkage with other NCBI resources like Entrez Protein, PubMed and Taxonomy. The Entrez interface also allows users to search Peptidome using simple free text or complex fielded queries.
Additionally, all original data, including spectra files, output results and Supplementary Data are available for bulk download via anonymous FTP at ftp://ftp.ncbi.nih.gov/pub/peptidome/.
CONCLUSIONS AND FUTURE DEVELOPMENT
Peptidome was recently established at NCBI with the goal to enhance proteomic research by providing a high-quality mass spectra repository. We are now accepting submissions and invite researchers to deposit their tandem MS data sets with us so that we can disseminate them to the wider community.
Currently, the Peptidome team is working to expand and improve existing indexing, linking, searching, exploring and retrieving functionalities. Additional advanced data mining and visualization tools for the convenience of proteomic researchers are under design and development.
ACKNOWLEDGEMENTS
The authors acknowledge and thank Ron Edgar for all his input into establishing the Peptidome resource. Also, advice was provided by Lewis Geer, Salvatore Sechi and Sandy Markey and the rest of the Laboratory of Neurotoxicology, NIMH, NIH. This project was initiated as part of the NIH Building Blocks, Biological Pathways and Networks Roadmap.
FUNDING
Funding for open access charge: Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Conflicts of interest statement. None declared.

{kind=link}
{kind=link}
Comments