





ABSTRACT
Over the last years, the publicly available knowledge on interactions between small molecules and proteins has been steadily increasing. To create a network of interactions, STITCH aims to integrate the data dispersed over the literature and various databases of biological pathways, drug–target relationships and binding affinities. In STITCH 2, the number of relevant interactions is increased by incorporation of BindingDB, PharmGKB and the Comparative Toxicogenomics Database. The resulting network can be explored interactively or used as the basis for large-scale analyses. To facilitate links to other chemical databases, we adopt InChIKeys that allow identification of chemicals with a short, checksum-like string. STITCH 2.0 connects proteins from 630 organisms to over 74 000 different chemicals, including 2200 drugs. STITCH can be accessed at http://stitch.embl.de/.
INTRODUCTION
The effects of small molecules on organisms have long been the focus of biochemistry and pharmacology. Over the last years there has been a considerable increase in the number of high-throughput screens that have been performed using chemical libraries (1–3). At the same time, the molecular targets of individual chemicals are being studied in ever greater detail (4,5). There also is a great interest in chemical biology approaches, using small molecules to perturb cellular functions (6). For the design and interpretation of these studies, the context of the chemicals and proteins needs to be considered. For example, in the case of high-content screening for specific cellular effects, it is important to know whether the active chemicals already have known activities that can explain the observed effects, or whether novel mechanisms of actions might be present. Therefore, we have developed a ‘search tool for interactions of chemicals’ (STITCH) both as a large-scale, downloadable database of interaction data and as an interactive web tool for the exploration of interaction networks (Figure 1). Since its first release (7), STITCH is being accessed by over one hundred scientists each week and has been used as a source of protein–chemical associations e.g. by Prathipati et al. (8), who used the STITCH network to automatically extract the targets of anti-tuberculosis compounds in Mycobacterium tuberculosis.
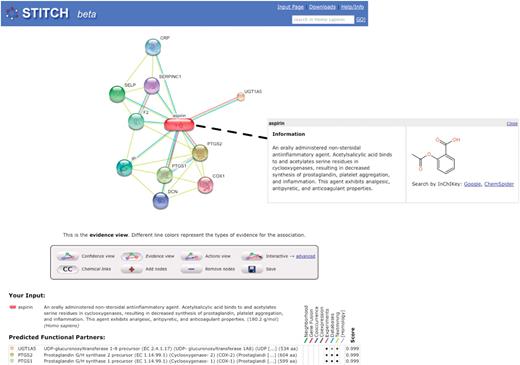
Interaction network around aspirin. Human proteins predicted to interact with aspirin according to different sources of evidence are shown. Edges are colored according to the source of evidence (magenta: experimental information, cyan: manually curated databases, yellow: text-mining). Clicking on the node ‘aspirin’ will display a pop-up showing the structure and description.
Here, we present the second version of STITCH. In addition to the sources of protein–chemical interactions included in the previous version—PDSP Ki Database (9), Protein Data Bank (PDB) (10), KEGG (11), Reactome (12), NCI-Nature Pathway Interaction Database (http://pid.nci.nih.gov), DrugBank (13) and MATADOR (14)—we now further include interactions imported from GLIDA (15), PharmGKB (16,17), Comparative Toxicogenomics Database (CTD) (18) and BindingDB (19). These added databases mainly provide information on interactions between human proteins and drugs or drug-like molecules.
The imported sources of information are scored separately and then combined with information from text-mining (7). Databases which contain manually annotated interactions receive high scores, while interactions based on experimental information are scored by the confidence or relevance of the reported interaction. The number of high-confidence (score ≥ 0.7) human chemical–protein interactions increased from 51 000 to 85 000. For these high-confidence interactions, the number of interacting human proteins increases from 5300 to 7400 (as STITCH is locus-based, only one gene product is counted per gene).
INCREASING THE NUMBER OF SPECIFIED ACTIONS
The STITCH network is created by mapping interactions from the sources mentioned above and from text-mining onto a consolidated set of chemicals that has been derived from PubChem, assigning a confidence score for each interaction (7). The newly-derived protein–chemical and chemical–chemical associations are then complemented with protein–protein interactions from the STRING database (20). In the previous version of STITCH (7), we began to import ‘actions’ derived from natural language processing (NLP), pathway and interaction databases. These actions specify the nature of the interaction independent of the source of interaction information. For example, a ‘binding’ action could be derived from a binding affinity database and an ‘inhibition’ action could be imported from NLP. We have greatly extended the set of available actions by further importing action types from GLIDA (15), PharmGKB (16,17), CTD (18), BindingDB (19) and a manually annotated set of interactions. This set of interactions has been curated from DrugBank (13) records, results from NLP analysis of PubMed abstracts, Medical Subject Headings (MeSH) pharmacological actions, Anatomical Therapeutic Chemical classification (ATC) entries and a review paper on drugs and their targets (21). An action has been assigned to 81% of the high-confidence human chemical–protein interactions. The number of available edges with a high-confidence action annotation increased from 44 000 to 65 000 human chemical–protein interactions.
HANDLING OF CHEMICAL STRUCTURES
As described previously (7), STITCH creates a consolidated set of chemicals from PubChem (22) by merging stereo isomers and salt forms of the same molecule into one compound. This is done to ensure that all information about the same biologically active entity is merged. While this works very well for drugs that can be supplied in different formulations (e.g. different salt forms), it also has limitations, especially regarding carbohydrates. It is our long-term goal to associate interactions both with the individual isomer and the merged structure. For now, we have taken the step to explicitly display all the different compounds that have been deemed biologically equivalent (Figure 2).
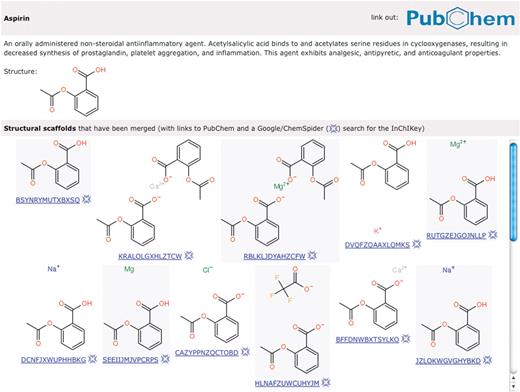
Different structural scaffolds corresponding to aspirin. For the drug aspirin, a link to PubChem and a short description is shown. Different salts of aspirin that will have the same bioactivity have been consolidated and merged with the main, uncharged form. Below each chemical structure, the first part of the InChIKey is shown, corresponding to an encoded (hashed) description of the structure. This short string can be used to search for more information about the compound on the Internet.
Recently, the International Union of Pure and Applied Chemistry (IUPAC) has standardized an open format for chemical structures, namely the IUPAC International Chemical Identifier (InChI). In addition to the existing capability to search chemical structures using SMILES string, we have now also implemented a search for InChIs. We use the tool Open Babel to convert InChIs to SMILES strings, which are in turn searched against our chemical database by using hashed fingerprints as implemented in the open-source Chemical Development Kit (23). Furthermore, we have implemented a search for InChIKeys, which are short strings that represent an encoded (hashed) form of the chemical structure. InChIKeys consist of two parts, the first of which is based on the chemical connectivity, whereas the second part contains information about stereochemistry, tautomers and other structural variations. As STITCH currently considers structures with the same connectivity to be equivalent (thus merging stereo isomers), only the first part of the InChIKey is queried against our chemical database. We also use this part of the InChIKey to provide links to Google and ChemSpider.
USER INTERFACE IMPROVEMENTS
Many proteins, especially drug targets, have a large number of high-scoring interactions with small molecules in the STITCH network. In this case, a network centered on such a protein will only show chemicals unless very many interaction partners are requested to be shown (Figure 3a). In order to allow the user to see more of the context of the query protein, we now offer the option to show a network in which proteins and chemicals each make up more than a third of the nodes (Figure 3b). When this option is selected, only a limited number of the highest-scoring chemicals are displayed. Further chemicals are omitted in favor of proteins (or vice versa) and their number is shown to the user. If the network consists of only chemicals, but no proteins are available at the current settings (e.g. due to a minimum score limit), then the option to show more context is not shown.
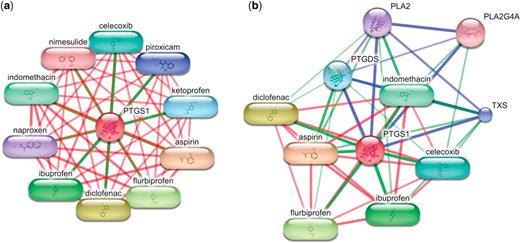
Interactions of prostaglandin-endoperoxide synthase 1 (PTGS1). (a) The highest-scoring interaction partners of PTGS1 are non-steroidal anti-inflammatory drugs (NSAIDs). As the confidence scores for these interactions are very high, no interacting proteins are shown. (b) The user may ask STITCH to display more of the interaction context and to let at least one-third of the interaction partners be proteins. In this case, STITCH is skipping 19 high-scoring chemicals in order to include four interacting proteins. In both networks, the color of the edge corresponds to the type of connected nodes (e.g. green: chemical–protein interaction) and the width of the edge correlates with the confidence score.
Previously, STITCH required the user to select an organism when searching for interactions with a chemical. Now, this is not required anymore. When no organism is selected, the organism with the highest-scoring interaction partners is selected. In case of multiple organisms with equal scores, human and several model organisms are preferentially selected. (Human is one of the highest-ranking species for 60% of the chemicals with protein–chemical interactions.) For example, the binding between the antipsychotic agent fluspiperone and the 5–hydroxytryptamine (serotonin) receptor 7 has only been studied in mouse and rat. Consequently, a user searching for this compound would be directed to the protein–chemical interaction network in mouse. It is also possible to restrict the search to different levels of the NCBI taxonomy (24), e.g. bacteria, fungi or rodents.
While central repositories of gene annotations exist, no such information is available in a centralized manner for chemicals. To be able to display text annotation for chemicals, we have imported information from the following databases: DrugBank (13), National Cancer Institute (NCI) thesaurus (25), MeSH descriptors and qualifiers. Using STITCH's dictionary of chemical synonyms we mapped compounds from these databases to STITCH identifiers. In case where descriptions are available for different forms of the same compound (e.g. different salt forms, which have been merged in STITCH), we have automatically assigned the description of the main compound. Any remaining inconsistencies were manually resolved. For each chemical we have assigned the text annotation from only one source, prioritizing sources as follows: NCI (descriptions), DrugBank (descriptions), DrugBank (pharmacology), DrugBank (drug category), MeSH (pharmacological action), NCI (tags) and MeSH (scope note). Descriptions are available for 33 352 chemicals, covering 33% of the chemicals with interactions.
USE CASES
The STITCH homepage offers several short tutorials to introduce the different query options (e.g. searching for a single identifier or multiple chemical structures). A search for ‘aspirin’ on the homepage will lead to the interaction network shown in Figure 1. Here, the main interactors of the drug are shown in human (which is selected automatically as described above). The known main targets, PTGS1 and PTGS2, are connected by very high scores. While most interaction partners are backed up by evidence from manually curated databases and are therefore very reliable, one interaction is derived only from text-mining: COX1 is actually a false positive arising from an ambiguous synonym.
Taken together, STITCH 2 offers an enlarged set of protein–chemical interactions, extended inter-database operability, increased query options and an improved user interface. STITCH can be accessed at http://stitch.embl.de/. Users can explore the interaction network interactively or download the complete set of interactions. In addition, we provide an application programming interface (API) to let scripts resolve identifiers and retrieve interaction networks either as an image or in standard network formats (20).
FUNDING
Klaus Tschira Foundation (to M.K. and A.B.). Novo Nordisk Foundation Center for Protein Research (partial). Funding for open access charge: European Molecular Biology Laboratory.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
Comments