





ABSTRACT
Membrane proteins are important for many processes in the cell and used as main drug targets. The increasing number of high-resolution structures available makes for the first time a characterization of local structural and functional motifs in α-helical transmembrane proteins possible. MeMotif (http://projects.biotec.tu-dresden.de/memotif) is a database and wiki which collects more than 2000 known and novel computationally predicted linear motifs in α-helical transmembrane proteins. Motifs are fully described in terms of several structural and functional features and editable. Motifs contained in MeMotif can be used in different biological applications, from the identification of biochemically important functional residues which are candidates for mutagenesis experiments to the improvement of tools for transmembrane protein modeling.
INTRODUCTION
Membrane proteins are important for many cellular processes, ranging from transport of ions and molecules across the membrane to signal transduction. Given the low number of high-resolution structures of membrane proteins in the Protein Data Bank (PDB) (1), compared with the number of globular proteins (2,3), detailed analysis of their structural features is widely needed for understanding their function, for annotation of large scale genome sequencing data, improve topology predictions and drug design.
A strategy for protein function assignment consists in detecting local sequence or structure patterns, associated with a particular function, which can be common to proteins with different folds. Protein annotation effort benefits immensely from knowledge of functional signatures in primary, secondary and tertiary structure. Motif databases that derive motifs or family signatures at sequence level (from multiple sequence alignments of homologous proteins) are: Pfam (4), PROSITE (5), SMART (6), ProDom (7), PRINTS (8), BLOCKS (9), InterPro (10) and others.
While protein domains or family signatures, as those contained in Pfam or PROSITE, can be identified from alignments of evolutionary related sequences, the identification of short sequence motifs, shared between different folds and functional classes is much harder.
Sequence patterns derived from structure-based approaches, and known to be associated with a particular function or fold nucleation site, offer an attractive way to annotate proteins because of their applicability to both sequences and structures with unknown function. Many databases of structural motifs have been developed for proteins in general, but very few motif databases focus on motifs in transmembrane proteins. Databases of fully annotated structural motifs for proteins in general include, among others, the I-sites library (11) and the MSDmotif database (12), which classify three-dimensional (3D) structural motifs, through properties of hydrogen bonding and ϕ/ψ angles.
In this work we have developed MeMotif, a database of linear motifs in α-helical transmembrane proteins. This is the first database, to our knowledge, that focuses on structural motifs in α-helical transmembrane proteins, fully described also in terms of functional features and editable, by means of a wiki-based web interface, by experts in different fields.
There are some specialized databases that collect members of certain membrane protein families (13,14), but detailed and complete structure-based classification schemes, such as SCOP (15) and CATH (16) for globular proteins, do not entirely exist, as well as comprehensive classifications of membrane proteins in unambiguous functional classes. All currently known high-resolution membrane protein structures have been organized into the PDBTM database (17), which assigns membrane-spanning regions by means of the TMDET algorithm (18) and TOPDB database (19), which contains experimentally derived topologies of transmembrane proteins. Sequence-based, non-hierarchical classifications of membrane protein domains are available in Pfam and other similar databases. Recently, Simon and co-workers developed the TOPODOM database (20), a collection of domains and sequence motifs from known motif databases, located consistently on the same side of the membrane in α-helical transmembrane proteins. Another database that focuses on membrane protein features is the TMFuncion database (21), a collection of experimentally observed functional residues in membrane proteins.
Membrane protein structures are often too complex to fit completely into a simple topology model (3). Membrane proteins are rich in non-standard structural features, which usually have a functional role. These are: helix distortions or kinks in the middle of transmembrane helices, which are thought to facilitate conformational changes for many receptors; reentrant loops dipping into the lipid bilayer, which function as selectivity filters in many channels; elements at the membrane–water interface, such as helix caps or short helices running parallel to the membrane plane which might be needed for structural reasons or channel gating mechanisms; helix–helix contact patterns, important for protein folding and protein–lipid interaction motifs, which have an impact on correct insertion, folding and functionality of the protein. Sequence motifs associated with such features could be used to predict such features from sequence alone and used to constrain or enrich topology prediction models (22).
A library of structural and functional motifs specific to membrane proteins can help classifying their different functions or shed light on the biochemical role of hot-spot amino acids, as well as helping in transmembrane protein modeling.
MeMotif describes the specific functional and structural role of known and novel motifs, which are computationally predicted and available for experimental validation, together with their statistical evaluation, references to the scientific literature and cross-links to other databases such as Pfam, PROSITE, GO (23), UniProt (24), SCOPPI (25), Entrez Gene (26) and GoGene (27). In contrast to databases which derive motifs or family signatures at sequence level, MeMotif derives sequence patterns from structural clusters of fragments from known transmembrane 3D structures. MeMotif covers different functional motifs, such as ligand and lipid binding sites, disease-linked mutations, protein–protein interaction sites, thanks to a comprehensive automated functional annotation process and expert knowledge-based manual curation. Structural motifs in α-helical transmembrane proteins, such as reentrant regions, helix kinks, interface helices and helix–helix contacts, are also annotated.
MeMotif has different fields of applicability: First, it allows the identification of hot-spot amino acids important for the protein's function or stability and that can be targeted for inhibition. Second, motifs can be used to improve functional annotation from structure or characterize the function of sequences of unknown structure. Third, motifs can be useful as constraints to add to 2D or 3D models of transmembrane proteins.
MATERIALS AND METHODS
The structure fragment clustering
In this section we briefly describe the procedure of structure fragment clustering for deriving the sequence-structure motifs contained in MeMotif. The procedure consists of six steps.
Step 1—data collection
Non-redundnant (NR 90%) and high-resolution (RMSD <3.5 Å) protein structures are collected from the PDBTM database (17). The filtered dataset contained 168 non-redundant α-helical transmembrane protein chains from 97 different PDB structures.
Step 2—fragment generation and structural characterization
Protein structures were fragmented and fragments of different sizes, ranging from 3 to 14 amino acids, were labeled according to their location annotation in the PDBTM and TOPDB databases. The region types to which each fragment could be assigned are: Cytoplasm and Extracellular (corresponding to the two sides of the membrane), Helix core (corresponding to the membrane-embedded part), Reentrant (corresponding to reentrant loops) and Interface (corresponding to protein's segments at the membrane-water interface region). For each fragment, a backbone torsion angle profile was derived from the corresponding PDB file and the associated hydrogen bonding pattern, if it existed, calculated by means of the Chimera algorithm.
Step 3—clustering
Hierarchical clustering of fragments of the same length and region was performed by implementing two different distance measures: one based on similar hydrogen bond patterns and the other one based on similar backbone torsion angle profiles.
Step 4—sequence codification
Sequence motifs in PROSITE language were derived for each structural class, if possible, by using the Pratt program. If no significant motif could be associated with a cluster, a further sequence-based clustering step was performed to filter significant sequence patterns.
Step 5—functional annotation
Functional annotation of fragments was performed by using multiple sources of information. Fragment clusters were annotated with enriched GO categories, whose significance was assessed by means of P-values from the hyper-geometric distribution. Each fragment in a cluster was associated with a UniProtKB/Swiss-Prot (24) Feature (FT) field annotation, when this annotation existed. The SCOPPI database, which classifies all protein domain-domain interactions contained in the PDB, was used to annotate fragments that are part of the interaction interface of protein complexes. The whole PDB database was screened for fragments belonging to ligand binding pockets or binding sites. Furthermore, residues in fragments that have been experimentally mutated, and their function reported in literature, were also annotated by means of an automated text-mining approach (28). Finally, for each cluster-derived sequence pattern, its total or partial overlap with a PROSITE pattern was checked and reported.
Step 6—filtering of statistically significant novel motifs
In total, 4842 motifs were derived and further filtered by statistical significance and specificty to membrane proteins, resulting in 2228 motifs. The statistical significance of a motif was assessed by means of the P-value of the occurrence distribution, derived by randomly permuting the transmembrane protein dataset from Swiss-Prot. In order to assess the specificity of a given motif for transmembrane proteins, in contrast to globular proteins, a false positive rate number was calculated, defined as the number of hits of the motif in all globular proteins in the Swiss-Prot database divided by the total number of hits (both in globular and in transmembrane proteins) in the Swiss-Prot database. Beta-barrel proteins were excluded from this analysis. Novelty of motifs was checked by comparing all regular expressions from our motifs directly against PROSITE patterns, by means of a regular expression-based comparison algorithm.
The MeMotif web server
The collected sequence/structure motifs are freely available to the scientific community in a web-interface based on a open source, java-based, wiki software, called JAMWiki. Concerning the layout, wiki syntax and functionality, JAMWiki is based on MediaWiki (the current software for Wikipedia). Users can edit existing topics or create new topics. Not all users are allowed to have all roles: most users are allowed to edit documents or to create new ones but only the administrator of the database is allowed to dynamically create the basic content of each page.
RESULTS
The MeMotif database
The data relative to each motif are stored in a MySQL database. The database is already freely available at http://projects.biotec.tu-dresden.de/memotif/ and contains 2228 entries corresponding to statistically significant structure/sequence motifs in transmembrane proteins. 213 motifs in MeMotif are novel (according to our comparison with motifs in the PROSITE database) and are fully annotated (or in the process of being annotated) from the database developers and the experts in the field. Motifs cover different topologies, functions and locations with respect to the lipid bilayer planes. About 61% of the motifs are helical motifs, belonging to several categories of α-helices with differences in the regularity of the helix, e.g. regular helices, 310-helices, pi-bulges and kinks. About 33% of the motifs are found at the membrane-water interface region, such as helix caps, loops and short parallel helices. Only about 5% of the motifs are structured loops located on the cytoplasmic or extracellular side of the membrane. About 1% of the motifs form reentrant loops.
The vast majority of motifs (94%) appear across evolutionary unrelated families, indicating that the conformation types are not merely a result of sequence homology or evolutionary relationships among proteins but highlight the modularity of functional design in membrane proteins. Almost all the family-specific motifs are found in ligand/cofactor binding sites, suggesting that they are specific for the protein's function. About 30% of the total motifs are found at protein–protein interaction interfaces of transmembrane complexes; 12% are found to bind specific kinds of lipids such as cholesterol or cardiolipins, which are known to modulate protein function via specific protein–lipid interactions. Finally, about 15% of the motifs are found to participate in helix-helix contacts.
Some examples of functional motifs in MeMotif, directly involved in the biochemical activity of the protein, are: First, the A-R-Y-[AI]-D-W-[LM]-[FV]-T-T-P motif in bacteriorhodopsins, which contains the crucial residues involved in the proton translocation pathway, such as the first protonation site Asp 85, and Thr 89, Trp 86, Tyr 83 and Arg 82 (PDB ID: 1c8s, Halobacterium salinarum) which, together with water molecules and other polar side chains, form a 3D hydrogen-bonded network and have been identified by mutagenesis to participate in proton release to the surface (29). This motif is very similar to the bacteriorhodopsin family signature in the PROSITE database (PROSITE ID: PS00950), except that it is shorter with respect to the PROSITE pattern, it contains a highly conserved Ala in the first position of the motif and a highly conserved hydrophobic residue (Leu or Met) in the seventh position.
Second, the selectivity filter motif G-[AGT]-x-[FIM]-N-P-A-[ST]-[FI]-[AG] in water-glycerol transporters contains crucial residues which contribute with a favorable electrostatic environment to the passage of water molecules across cell membranes (30). Third, the selectivity filter motif V-[ST]-[ILM]-[AT]-T-V in potassium channels contains residues whose electro-negative carbonyl oxygen atoms align toward the center of the filter pore and allow the selective passage of potassium ions (31).
Compared with other 3D structural and protein motif databases that solely archive data, MeMotif permits anyone knowledgeable with respect to a particular protein or a motif to add information regarding its function and relate the information directly to the sequence-structure motif. Mistakes are easily corrected by users and adding textual content to the web site is simple, taking advantage of the Wikipedia interface, which is familiar to millions of users. When appropriate or necessary, an entry may be protected from being edited except by a selected group of scientists.
Typical entry of a fully annotated motif
A typical entry of a fully annotated motif is shown in Figure 1. The motif is named according to its function or its special structural features. Each page includes the 3D molecule of one of the PDB structures containing the motif, visualized in Jmol (http://www.jmol.org/). A schematic representation of the membrane planes is also shown. The structural motif in the protein in visualized in red and different transmembrane regions are shown in different colors, as explained in the legend below the 3D image of the molecule.
The page starts with a text that summarizes, in a descriptive way, the main features of the motif, its putative function and contains references to PubMed articles. This is the manually curated part of each motif entry. For example, in Figure 1, the cross-family reentrant motif L-[AT]-G-[FI]-[AILV]-x-[IPV]-[IL] is described as a pattern found in different Pfam families, and located in the reentrant regions of different transport proteins. This structural motif, which is a combination of small and hydrophobic amino acids is typical of reentrant regions and functions as selectivity filter for many ion/water/glycerol/drug transporters.
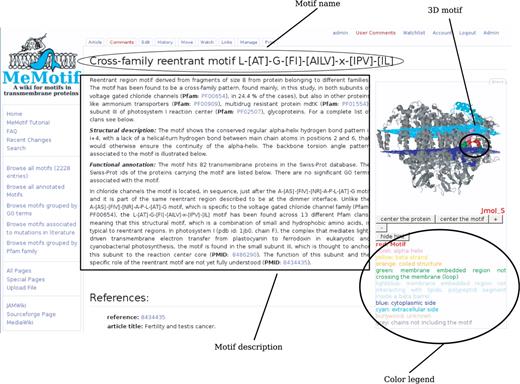
Typical entry of a fully annotated motif. Entry of a fully annotated motif in MeMotif with emphasis on the manually curated description.
The description is followed by a sequence web logo of the motif and a table of contents. The table of contents points to different sections in the page. The Motif info section shortly summarizes some basic information about the motif such its specificity for membrane proteins (false positive rate), the number of hits in the Swiss-Prot database, its length and its location with respect to the lipid bilayer.
The Structural details section describes in detail structural features associated with the motif, such as its positions in known PDB structures, its secondary structure content, its associated hydrogen bond pattern and backbone torsion angle profile, by means of schematic representations.
The Functional annotation section provides functional annotation of the motif, such as list of enriched GO terms, annotation in the Swiss-Prot database, specificity to protein–protein interaction interfaces or ligand binding sites, Pfam families which contain the motif, links to similar patterns in the PROSITE database and to point mutations automatically extracted from literature.
See the MeMotif tutorial for more details about the meaning of different fields in a motif entry.
Search options
Different search functionalities are implemented in MeMotif by clicking on the left panel the link Search. A search by keyword allows the user to search for motifs corresponding to: a given topic, e.g. ‘helix-helix interaction’; transmembrane region, e.g. ‘interface’; enriched GO_term, e.g. ‘photosynthesis’; PubMed id; motif-associated regular expression, e.g. L-x-[AGSV]-A-x-P-x(2)-G. Search results are displayed in the following order: fully annotated motifs appear first ordered by increasing false positive rate.
Another feature in MeMotif is the search of motifs in a protein sequence in fasta format by using the Sequence search box field. The result of the search is a list of motifs that match in the protein sequence. For more detailed explanations about the different search options see the MeMotif tutorial.
First example of usage—characterize hot-spot residues in cytochrome b
Let us assume that a structural biologist, working on the protein cytochrome b wants to know which residues or motifs are crucial for the protein's stability and function, in order to design ad hoc mutagenesis experiments. Cytochrome b is the main subunit of the third complex in the electron transport chain. It couples electron transfer to the generation of the proton gradient driving the ATP synthesis. In Figure 2, the result page of the search by proteinname:“cytochrome b” is shown. All the entries containing the word ‘cytochrome b’ (2174 in total) appear on the screen. Fully annotated motifs appear first, as they are the ones that provide the most reliable information about the specific motif function for that family. Among the motifs associated with cytochrome b we find the T-A-F-[LMV]-G-Y-x(0,2)-V-x(1,3)-G motif (Figure 2a), a regular helix followed by the irregular 310 helix pattern, according to the hydrogen bond diagram. The motif is part of the Q0-inhibitor binding site and found from the automated protein–ligand analysis to be involved in non-covalent interactions with stigmatellin, which binds in the Q0 site (32). The mutation analysis from literature indicates that the mutation of phenylalanine 129 to leucine in CYB_YEAST [UniProt_id: P00163 (33)] confers the protein substantial resistance to the inhibitor with minor effects on the bc1 complex activity. Phenylalanine 129 participates in van der Waals interactions with stigmatellin and, if replaced by leucine, leads to an unfavorable increase in binding free energy (33). The importance of this residue for the protein function is further highlighted from our automated analysis of residues associated to Swiss-Prot features: phenylalanine 129 in yeast cytochrome b is annotated to be associated to a loss o of binding affinity for ubiquinone and ubiquinol when mutated to leucine or serine. Another motifs associated with cytochrome b is the membrane-embedded heme packing motif L-[IMV]-x-Q-I-[LV]-T-G-[IL] (Figure 2b). It has been reported in the scientific literature that in the Rhodobacter sphaeroides cytochrome b, mutation of glycine 48, which is part of this motif, to aspartate or valine results in the loss of photosynthetic growth of the organism (34). The motif seems to be important for heme packing as a mutation to Ala is tolerated in this position, suggesting that a small residue is important for packing of the heme in the helix core (34). Other two important motifs for cytochrome b are: the irregular helix motif R-F-F-[AS]-[FL]-H-[FY] (Figure 3b), which contains an histidine residue involved in heme binding and the loop motif P-[DN]-x-L-G-[DH]-P-[DE] (Figure 2d) on the extracellular side of the membrane (intramembrane space in mitochondria). This last motif is part of the interaction interface between cytochrome b and cytochrome c1, suggesting its importance for the assembly of the complex. The role of the two proline residues is probably structural, as they confer rigidity to the loop.
The retrieved motifs for cytochrome b, and their detailed structural and functional characterization help understanding which residues are crucial for the complex stability and/or function and worth to investigate experimentally.

Results from the search: proteinmane: ‘cytochrome b’. Result page from the search by means of the keyword cytochrome b. Four significant matching motifs are highlighted on the cytochrome b protein structure (PDB ID: 2a06). Motifs are highlighted in violet on the PDB structure.
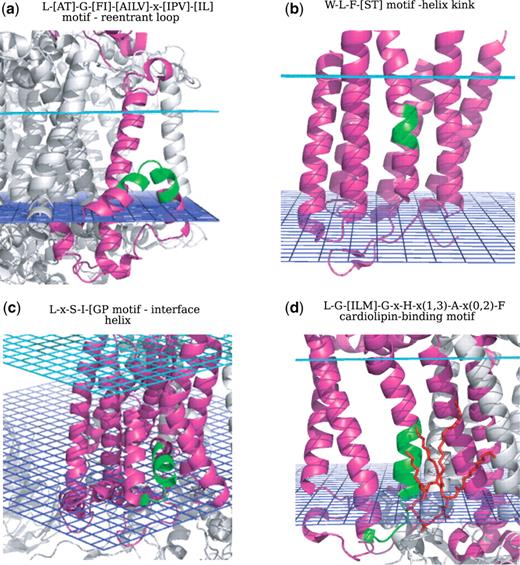
Structural motifs. (a) Reentrant loop; (b) Helix kink; (c) Interface helix; (d) Cardiolipin-binding motif. The motifs are highlighted in green on the corresponding PDB structures and the stigmatellin molecule is shown in red on (d).
Second example of usage—search for specific structural motifs
Let us assume that the user would like to retrieve all the structural motifs associated with reentrant regions in α-helical transmembrane proteins. This is done by typing region:reentrant in the search field by keyword. Thirty-seven significant and overlapping motifs are retrieved and showed in the result page. Among them, we find some reentrant loops specific to protein families such as: the well-known NPA motif, whose extended signature is G-[AGT]-x-[FIM]-N-P-A-x-[ST]-[FI]-[AG], that plays a functional role as selectivity filter in water/glycerol transporters; the V-[ST]-[ILM]-[AT]-T-V motif, a selectivity filter in most of the potassium channel family members and a novel reentrant region-associated motif (not documented in any known motif database), the A-[AS]-[FIV]-[NR]-A-P-L-[AT]-G motif at the dimer interface of voltage-gated chloride channels. An example of a reentrant loop associated with proteins of different folds is the L-[AT]-G-[FI]-[AILV]-x-[IPV]-[IL] motif, found across different protein families involved in transport functions (see Figure 3a).
Searching for sequence motifs associated with helix kinks can be done by typing, for example, content:kink in the search field by keyword. The retrieved helix kinks motif contain proline residues or aromatic amino acids, such as tryptophane and tyrosine, that break the regular hydrogen bond pattern of α-helices and induce the helix to bend. This feature can be observed in the hydrogen bond diagrams associated with each motif. Examples of kinks specific to some protein families are: the proline-kink motif in bacteriorhodopsins L-W-x-[AG]-Y-P-[IV]-[LV]-W, part of the retinal binding pocket and the proline-kink motif G-H-P-x-V-Y-[FY] in cytochrome c and quinol oxidases, associated with a copper binding site through the histidine and tyrosine residues. An example of a kink motif found across different protein families is: the W-L-F-[ST] motif (see Figure 3b), which builds a kink in the middle of transmembrane helices in G protein-coupled receptors and in other kinds of receptors, due to the presence of the bulky amino acids tryptophane and phenylalanine.
In the same way the user can search for sequence motifs associated with parallel helices, by typing content: “interface helix”. Most of the retrieved interface helix-associated motifs are convergently evolved motifs, such as the L-x-S-I-[GP] motif (see Figure 3c) or motifs specific to the function of a certain protein family, such as the the G-L-Y-Y-G-S-Y motif, a small parallel helix-turn found in cytochrome b, and part of the heme binding pocket.
Third example of usage—cardiolipin-binding motifs
Anionic phospholipids, such as phosphatidyl glycerol (LHG) and cardiolipin (CDL) play essential roles in a variety of processes carried out by membrane proteins such as cytochrome c oxidase and photosynthetic reaction center (35). By typing in the search field by keyword msdchem: CDL OR LHG (three-letter codes according to the PDB standard), all possible motifs associated with cardiolipin binding are retrieved. Among the search results we find the L-G-[ILM]-G-x-H-x(1,3)-A-x(0,2)-F motif, a cardiolipin-binding motif in the photosynthetic reaction center (see Figure 3d), an integral membrane protein complex that uses light energy to pump electrons across the cytoplasmic membrane of photosynthetic bacteria. This motif has an experimental evidence: in X-ray crystallographic studies (35), it was found that the head group of the cardiolipin comes into close contact with conserved residues from all three unique chains of the protein. Several possible interactions were observed between the lipid headgroup and the surrounding protein, in particular histidine 145 and lysine 144 in Rhodobacter sphaeroides, as confirmed from our automated protein–ligand interaction analysis. According to the crystal structure, the tail region interacts over a large surface area within the transmembrane region of the protein, suggesting that the strength of lipid/protein interactions is contributed by both ionic interactions with the cardiolipin headgroups and the van der Waals' interactions in the tail region. It is intriguing that residues that interact with cardiolipin form a conserved structure-sequence motif among the subunit M in the photosynthetic reaction center family. These residues may contribute a conserved site for binding of cardiolipin in bacterial reaction centers, confirming the hypothesis already suggested in (35), that cardiolipin is a key component of energy-transducing membranes and important for the maintenance of optimal functional activity of many integral membrane proteins, by controlling or enhancing the processes catalyzed from these proteins, such as electron transfer (35). This motif was not annotated in any known motif database.
DISCUSSION
In this work we have introduced MeMotif, a database of linear motifs in α-helical transmembrane proteins, described in detail with respect to structural features and putative functional association. MeMotif is based on the results of a structure fragment clustering based on known transmembrane protein structures. We have shown the utility of the database for many biological applications such as (i) identification of hot-spot residues which are candidates for mutagenesis experiments, (ii) identification of motifs that, even if not directly involved in the core biochemical activity of the protein, modulate the protein's function and (iii) classification of structural motifs, such as reentrant regions, interface helices and helix-kinks which can be used for transmembrane modeling purposes. The motifs contained in MeMotif do not necessarily correspond to family or fold signatures but can be found across different protein families, underlying the modularity of functional design.
In general, structure-based methods for motif retrieval are not meant to replace sequence-based methods. One limitation of structural methods is that the use of static crystal structures for motif derivation misses those motifs associated to flexible regions or those patterns for which not enough structure information is available. On the other hand, structure-based methods can recover those short patterns which are difficult to identify by sequence alignment-based methods (36).
Furthermore, the short motifs contained in MeMotif can complement the information of family signature databases such as Pfam, as they shed light on the structural properties, sometimes essential to understand the function of specific residues, which are not detectable by sequence analysis.
An analysis of enrichment or certain amino acids in MeMotif with respect to their number of occurrences in transmembrane protein structures (see Supplement 1) shows that some residues such as Asp, Glu, and Lys, which are functionally very important, are significantly under-represented in MeMotif. In contrast, residues such as Leu, Ile, and Val, are over-represented. This is likely due to the under-representation of certain protein families in the PDB database, which leads to the incapability of deriving significant sequence-structure clusters. As soon as the number of high-resolution structures in the PDB will increase, the survey of other family-specific functional motif will become possible.
All the motifs in the database have been automatically annotated with respect to several structural and functional features and so far 50 of them have been manually assessed, through an accurate check in the scientific literature and qualitatively described from the biological point of view. We hope in the future that expert structural biologists from the scientific community will contribute to the annotation process by making corrections or by adding annotations to help understanding the role of a motif in a particular protein family or across different families. This would enhance the usefulness of MeMotif for the scientific community. We hope that MeMotif will have the capacity to leverage the resources of many diverse experts in the fields rather than just the curators of the database site and be a platform for cooperative work and discussions in membrane protein functional and structural studies. The database will be periodically updated, as new transmembrane protein structures will be available. New motifs can be easily added, their novelty and statistical significance checked and linked to similar motifs in the database. With the growth of structural data, MeMotif will be able to cover the increasing number of membrane protein functional classes and structural folds and help improving systematic membrane protein classification into distinct unambiguous classes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors thank Boris Vassilev for contributing valuable data. They further thank Loic Royer and Julia Winter for the design of the MeMotif web logo.
FUNDING
SeaLife project (grant IST-2006-027269). Funding for open access charge: Bundesministerium fur Bildung und Forschung (BMBF) program format.
Conflict of interest statement. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
Comments