





ABSTRACT
Advances in high-throughput next-generation sequencing technology have reshaped the transcriptomic research landscape. However, exploration of these massive data remains a daunting challenge. In this study, we describe a novel database, deepBase, which we have developed to facilitate the comprehensive annotation and discovery of small RNAs from transcriptomic data. The current release of deepBase contains deep sequencing data from 185 small RNA libraries from diverse tissues and cell lines of seven organisms: human, mouse, chicken, Ciona intestinalis, Drosophila melanogaster, Caenhorhabditis elegans and Arabidopsis thaliana. By analyzing ∼14.6 million unique reads that perfectly mapped to more than 284 million genomic loci, we annotated and identified ∼380 000 unique ncRNA-associated small RNAs (nasRNAs), ∼1.5 million unique promoter-associated small RNAs (pasRNAs), ∼4.0 million unique exon-associated small RNAs (easRNAs) and ∼6 million unique repeat-associated small RNAs (rasRNAs). Furthermore, 2038 miRNA and 1889 snoRNA candidates were predicted by miRDeep and snoSeeker. All of the mapped reads can be grouped into about 1.2 million RNA clusters. For the purpose of comparative analysis, deepBase provides an integrative, interactive and versatile display. A convenient search option, related publications and other useful information are also provided for further investigation. deepBase is available at: http://deepbase.sysu.edu.cn/.
INTRODUCTION
Next-generation ‘deep-sequencing’ technologies have enabled the detection and profiling of both known and novel small noncoding RNAs (ncRNAs) at unprecedented sensitivity and depth (1–3). Most studies to date have used 454 and Solexa technologies to discover new and different ncRNA classes in a multitude of species, including human (4–6), mouse (5,7,8), chicken (9,10), Ciona intestinalis (11), Drosophila melanogaster (12–15), Caenorhabditis elegans (16–18) and Arabidopsis thaliana (19–22). However, the analysis of these massive and heterogeneous deep sequencing data sets poses several challenges, including effective data mapping, annotation and visualization; efficient data storage and retrieval; integration and interpretation of data from multiple technological platforms, tissues and cell lines; and customizing the analysis so that a variety of biological questions can be addressed. Although the above-mentioned studies have targeted some of these individual steps in a specific genome, an integrated database that can meet all these basic needs for deep sequencing data is not yet available for animal and plant genomes.
Recent studies have shown that many small RNAs derived from annotated genomic elements, such as long ncRNAs, transcription start sites (TSSs) and transposable elements (TEs), can modulate diverse biological functions (6,23–29), raising the possibility that a large group of small RNAs originating from annotated genomic elements may still be hiding in eukaryotic genomes (6,23–29). However, in the past, sequence reads mapped to non-miRNA or non-piRNA gene families have been routinely discarded and not analyzed further. Intriguingly, a large number of highly abundant small RNAs derived from known ncRNAs often span the entire RNA locus, indicating that we not only can recapitulate known ncRNAs but also can identify novel ncRNAs by grouping these nearby small RNAs into clusters.
In this study, we describe the newly developed deepBase database for the comprehensive annotation and mining of deep sequencing data from 185 small RNA libraries from diverse tissues and cell lines of seven organisms (Figure 1). deepBase contains millions of small RNAs derived from known ncRNAs, protein-coding genes and repeat elements, as well as a massive number of unannotated small RNAs. In addition, we report about 1.2 million RNA clusters that include multiple classes of infrastructural ncRNAs (e.g. tRNAs, rRNAs, snRNA and snoRNAs), miRNAs, piRNA precursors and repeat-associated siRNA precursors, as well as numerous novel ncRNAs, some of which can be predicted as novel miRNAs and snoRNAs by miRDeep (5) and our snoSeeker programme (30). Finally, deepBase provides an integrative, interactive and versatile web graphical interface to display these data and facilitate transcriptomic research and the discovery of novel ncRNAs.
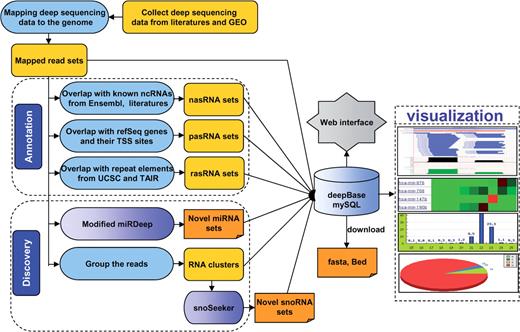
The basic framework of deepBase. All results generated by deepBase are deposited in relational databases and displayed in the visual browser and web page. The web-interface programmes and browser can be accessed by a wide range of research biologists to analyze and visualize data over the internet.
MATERIALS AND METHODS
One hundred and eighty-five small-RNA libraries from diverse tissues and cell lines from seven organisms were compiled from 34 related studies (Supplementary Data) and downloaded from the NCBI GEO website (31). Known ncRNAs were downloaded from Ensembl (32) or UCSC bioinformatics website (33), or were obtained from the literature. All known miRNAs were downloaded from miRBase [release 13.0, (34)]. Human and Arabidopsis snoRNAs were downloaded from snoRNABase (35) and the Plant snoRNA Database (36), respectively. All known tRNAs were downloaded from the Genomic tRNA Database (37). All refSeq genes and repeat elements (38,39) for animal genomes were downloaded from the UCSC bioinformatics website (33). Human (UCSC hg18), mouse (NCBI Build 37), chicken (Gallus gallus, v2.1), C. intestinalis (JGI v2.0), D. melanogaster (BDGP Release 5) and C. elegans (WS190) genome sequences were downloaded from the UCSC bioinformatics website (33). Arabidopsis (TAIR8) genome sequences, repeat elements and protein-coding genes were downloaded from the TAIR website (40).
All deep sequencing data downloaded from the NCBI GEO database is in SOFT format, and some raw data included 3′ adapters or barcodes. If the raw data included 3′ adapters or barcodes, we clipped the reads using our in-house Perl scripts. Upon removal of adapters, the sequences shorter than 15 nt were discarded. The low-complexity reads were also discarded (41). All unique reads without adapters in each library were mapped to the seven genomes using Bowtie (version 0.9.9.3) (42) with options: −f −k 200 −v 0, and only perfect matches over their entire length were set aside. Specifying the parameters (−k 200 –v 0) instructs Bowtie to report up to 200 perfect hits for each read (42). Together with all mapped reads in each library, we found a total of ∼14.6 million unique reads that perfectly mapped to more than 284 million genomic loci. Finally, up to 50 perfect hits to each genome were considered per query read in the subsequent analysis. Considering mismatch is not allowed between the genome and the small RNA reads in deepBase, current deepBase does not contains isomiRs with at least one mismatch to the genome (4). These mismatches are usually generated by adding the untemplated nucleotide to the 3′-terminal of miRNAs (4) or RNA editing (4,43,44). The large amount of data that is generated and that needs to be analyzed in such a large-scale screen requires appropriate computational means for storage and processing. For this task, a MySQL database was created to store the mapped reads.
We define an RNA cluster as a group of small RNAs in which each small RNA is ≤70 nt from its nearest neighbour and whose cluster length is ≥45 nt. These parameters were determined based on our statistics for the distribution of the distance between two nearest neighbouring reads that mapped to known ncRNAs (Supplementary Data). Our analysis revealed that more than 92% of the known ncRNA precursors can be grouped into clusters (Supplementary Data). RNAfold (45) was applied to predict the RNA secondary structures of ncRNAs and RNA clusters.
Novel miRNA and snoRNA candidates were predicted from deep sequencing data by a modified miRDeep (5) and snoSeeker (30), respectively. RNA cluster sequences, extended by an additional 100 nt in both the 5′- and 3′-directions for each of the species, were extracted as the snoSeeker input data set. We applied the snoSeeker programme (30) to these RNA clusters with the following options: guide C/D ≥ 37.5 bits, orphan C/D ≥ 26.5 bits, guide H/ACA ≥ 40 bits and orphan H/ACA ≥ 27.0 bits. The novel snoRNA candidates that significantly overlapped with exons, repeat elements or other known ncRNAs were discarded. Novel miRNA candidates were predicted from deep sequencing data by a modification of the miRDeep programme (5) with default option scores. To improve search speed of miRDeep, we introduced the following modifications: (i) the sequence reads were mapped to the genome using Bowtie (42), rather than BLAST (41), and (ii) the sequences were extracted from the huge genomes using our fetchSeq programme (the programme are available from the authors upon request), which was written in C language.
Relative expression analysis was sought to determine the expression preferences of individual miRNA and ncRNA across all small RNA libraries. The number of reads matching a particular ncRNA was calculated. Each ncRNA count from each library was normalized to the total read number for that library. The normalized count of a particular ncRNA in a particular library was divided by the sum of normalized count for that ncRNA across all libraries. Those normalized counts were transformed to 100 percentiles, and each bar in heatmap represents the normalized level. Except the miRNAs, the heatmap reflects a rough measure of ncRNA total expression because most of the reads mapped to the other ncRNA species might be the degenerated products.
deepBase DATABASE
Annotation and identification of about 380 000 nasRNAs from millions of deep sequencing reads
Recent studies have shown that many small RNAs generated from long ncRNAs by specific biogenesis pathways can modulate and silence gene expression, indicating that further investigation of these small RNA data sets is worthwhile for discovering novel functional small RNAs (23,24,46). Moreover, miRNA-offset RNAs (moRs) generated from 60-nt pre-miRNAs have been identified in C. intestinalis, suggesting an intrinsic property of the miRNA processing machinery (11). In this study, all mapped sequences were intersected against all types of long ncRNAs, including miRNA precursors (miRBase v13), snoRNAs, tRNAs, rRNAs, snRNAs, scRNAs, Mt_tRNAs and misc_RNAs. A total of ∼58 800 unique reads and ∼380 000 unique ncRNA-associated small RNAs (nasRNAs) originated from 2013 miRNA precursors and the other 9719 known long ncRNAs (Table 1), respectively. All reads overlapping these RNA genes were stored in the MySQL database for searching and browsing in deepBase.
Statistics in deepBase
| Human | Mouse | Chicken | C. intestinalis | D. melanogaster | C. elegans | Arabidopsis | |
| small RNA library | 9 | 63 | 4 | 4 | 31 | 25 | 49 |
| Unique read | 1 456 537 | 1 490 531 | 137 801 | 340 879 | 2 522 289 | 3 156 821 | 5 478 930 |
| Locus number | 22 437 894 | 215 546 228 | 782 488 | 3 590 208 | 19 760 563 | 7 402 057 | 14 613 634 |
| nasRNA | 49 703 | 99 657 | 10 370 | 5448 | 63 565 | 137 904 | 12 507 |
| pasRNA | 62 791 | 105 413 | 5633 | 46 411 | 142 645 | 459 139 | 697 750 |
| easRNA | 160 347 | 354 524 | 6666 | 1687 | 751 728 | 1 990 763 | 674 086 |
| rasRNA | 616 070 | 658 476 | 8099 | 34 300 | 1 409 439 | 293 658 | 2 907 928 |
| RNA cluster | 151 245 | 538 138 | 8801 | 62 583 | 77 113 | 215 226 | 114 235 |
| Predicted miRNA | 705 | 588 | 275 | / | 134 | 336 | / |
| Predicted snoRNA | 378 | 603 | 124 | 263 | 145 | 197 | 179 |
| Human | Mouse | Chicken | C. intestinalis | D. melanogaster | C. elegans | Arabidopsis | |
| small RNA library | 9 | 63 | 4 | 4 | 31 | 25 | 49 |
| Unique read | 1 456 537 | 1 490 531 | 137 801 | 340 879 | 2 522 289 | 3 156 821 | 5 478 930 |
| Locus number | 22 437 894 | 215 546 228 | 782 488 | 3 590 208 | 19 760 563 | 7 402 057 | 14 613 634 |
| nasRNA | 49 703 | 99 657 | 10 370 | 5448 | 63 565 | 137 904 | 12 507 |
| pasRNA | 62 791 | 105 413 | 5633 | 46 411 | 142 645 | 459 139 | 697 750 |
| easRNA | 160 347 | 354 524 | 6666 | 1687 | 751 728 | 1 990 763 | 674 086 |
| rasRNA | 616 070 | 658 476 | 8099 | 34 300 | 1 409 439 | 293 658 | 2 907 928 |
| RNA cluster | 151 245 | 538 138 | 8801 | 62 583 | 77 113 | 215 226 | 114 235 |
| Predicted miRNA | 705 | 588 | 275 | / | 134 | 336 | / |
| Predicted snoRNA | 378 | 603 | 124 | 263 | 145 | 197 | 179 |
Statistics indicating the numbers of small RNA library, unique read mapped to one or more loci, locus number, ncRNA-associated small RNAs (nasRNAs), promoter-associated small RNAs (pasRNAs), exon-associated small RNAs (easRNAs), repeat-associated small RNAs (rasRNAs), RNA cluster, predicted miRNAs and snoRNAs for the seven organisms, including human, mouse, chicken, C. intestinalis, D. melanogaster, C. elegans and Arabidopsis. Arabidopsis miRNA data are not present in the table because miRDeep (5) cannot effectively predict plant miRNAs. C. intestinalis miRNAs have been predicted previously by miRDeep (11).
Statistics in deepBase
| Human | Mouse | Chicken | C. intestinalis | D. melanogaster | C. elegans | Arabidopsis | |
| small RNA library | 9 | 63 | 4 | 4 | 31 | 25 | 49 |
| Unique read | 1 456 537 | 1 490 531 | 137 801 | 340 879 | 2 522 289 | 3 156 821 | 5 478 930 |
| Locus number | 22 437 894 | 215 546 228 | 782 488 | 3 590 208 | 19 760 563 | 7 402 057 | 14 613 634 |
| nasRNA | 49 703 | 99 657 | 10 370 | 5448 | 63 565 | 137 904 | 12 507 |
| pasRNA | 62 791 | 105 413 | 5633 | 46 411 | 142 645 | 459 139 | 697 750 |
| easRNA | 160 347 | 354 524 | 6666 | 1687 | 751 728 | 1 990 763 | 674 086 |
| rasRNA | 616 070 | 658 476 | 8099 | 34 300 | 1 409 439 | 293 658 | 2 907 928 |
| RNA cluster | 151 245 | 538 138 | 8801 | 62 583 | 77 113 | 215 226 | 114 235 |
| Predicted miRNA | 705 | 588 | 275 | / | 134 | 336 | / |
| Predicted snoRNA | 378 | 603 | 124 | 263 | 145 | 197 | 179 |
| Human | Mouse | Chicken | C. intestinalis | D. melanogaster | C. elegans | Arabidopsis | |
| small RNA library | 9 | 63 | 4 | 4 | 31 | 25 | 49 |
| Unique read | 1 456 537 | 1 490 531 | 137 801 | 340 879 | 2 522 289 | 3 156 821 | 5 478 930 |
| Locus number | 22 437 894 | 215 546 228 | 782 488 | 3 590 208 | 19 760 563 | 7 402 057 | 14 613 634 |
| nasRNA | 49 703 | 99 657 | 10 370 | 5448 | 63 565 | 137 904 | 12 507 |
| pasRNA | 62 791 | 105 413 | 5633 | 46 411 | 142 645 | 459 139 | 697 750 |
| easRNA | 160 347 | 354 524 | 6666 | 1687 | 751 728 | 1 990 763 | 674 086 |
| rasRNA | 616 070 | 658 476 | 8099 | 34 300 | 1 409 439 | 293 658 | 2 907 928 |
| RNA cluster | 151 245 | 538 138 | 8801 | 62 583 | 77 113 | 215 226 | 114 235 |
| Predicted miRNA | 705 | 588 | 275 | / | 134 | 336 | / |
| Predicted snoRNA | 378 | 603 | 124 | 263 | 145 | 197 | 179 |
Statistics indicating the numbers of small RNA library, unique read mapped to one or more loci, locus number, ncRNA-associated small RNAs (nasRNAs), promoter-associated small RNAs (pasRNAs), exon-associated small RNAs (easRNAs), repeat-associated small RNAs (rasRNAs), RNA cluster, predicted miRNAs and snoRNAs for the seven organisms, including human, mouse, chicken, C. intestinalis, D. melanogaster, C. elegans and Arabidopsis. Arabidopsis miRNA data are not present in the table because miRDeep (5) cannot effectively predict plant miRNAs. C. intestinalis miRNAs have been predicted previously by miRDeep (11).
Annotation and identification of abundant pasRNAs and easRNAs
A new class of transcripts were recently reported to originate near the expected TSSs upstream of protein-coding sequences (6,25–27,29). The existence of these promoter-associated small RNAs (pasRNAs) challenges our simplistic models of how the DNA sequences known as ‘promoters’ define TSSs (28). Moreover, many endogenous small interfering RNAs (endo-siRNAs) derived from protein-coding regions modulate gene expression and silencing (47,48). Thus, a genome-wide investigation of all of these small RNAs remains desirable due to the light it could shed on their biogenesis and function. In this study, all mapped reads were also intersected against the known refSeq genes and the upstream 350 nucleotides and downstream 150 nucleotides of TSSs. Those mapped reads overlapping TSSs were designated as pasRNAs (49,50). We divided the small RNAs overlapping with exons into sense and antisense exon-associated small RNAs (easRNAs) according to their strand. A total of ∼1.5 million unique pasRNAs and ∼4.0 million unique easRNAs were identified from TSSs and protein-coding sequences, producing the most comprehensive database of pasRNAs to date (Table 1).
Annotation and identification of abundant rasRNAs
A major system that controls the activity of TEs in flies and vertebrates is mediated by Piwi-interacting RNAs (piRNAs), 24–30 nucleotide RNAs that are bound by Piwi-class effectors (51–53). Previously, these piRNAs were grouped together based on their genomic location as repeat-associated small interfering RNAs (rasiRNAs) (54–61). Recent studies have also shown that many small interfering RNAs (siRNAs) from TEs play important roles in plants, fungi, Drosophila and vertebrates (54–61). To annotate and identify these repeat-associated small RNAs (rasRNAs), all mapped reads were also intersected with RepeatMasker annotations (38,39). These mapped small RNAs-overlapping repeats were divided into sense and antisense rasRNAs. A total of ∼3.0 million unique sense and ∼3.0 million unique antisense rasRNAs were identified from repeat elements, producing the most comprehensive database for rasRNAs to date (Table 1).
RNA clusters and novel ncRNA discovery
When we finished the annotation and identification of nasRNAs, we found that a large number of highly abundant ncRNA-associated small RNAs often span part of and even the entire RNA locus. Thus, an analysis of genomic clustering can be used to identify novel ncRNAs, hunt for hidden transcripts and determine whether small RNAs and clusters are differentially expressed in the sampled tissues. To cluster these small RNAs, we grouped all the mapped reads into about 1.2 million RNA clusters according to their distance (details in ‘Materials and Methods’ section). These clusters ranged in size from 45 nt to thousands of nt. All RNA clusters were intersected with known ncRNAs, and 1684 and 8364 RNA clusters were found to overlap known miRNAs and ncRNAs, respectively (Supplementary Data). Moreover, we found that 285 530 RNA clusters overlapped with the evolutionarily conserved elements generated by the PhastCons programme (62) in five organisms (Supplementary Data). These data suggest the possibility that a large group of novel ncRNAs, and perhaps even a novel class of ncRNAs, may still be hiding in eukaryotic genomes. To test the hypothesis, we applied a modified miRDeep (5) and our snoSeeker programmes (30) to the deep sequencing data and these RNA clusters (details in ‘Materials and Methods’ section). We identified 1161 novel miRNA and 857 novel snoRNA candidates, in addition to 877 known miRNAs and 1032 known snoRNAs.
WEB INTERFACE
deepBase provides a variety of interfaces and graphical visualization to facilitate analysis of the massive and heterogeneous small RNA data sets from different tissues, cell lines and technology platforms. We have also developed a new visualization tool, deepView genome browser, to provide a quick overview of a particular region in the genome and for visually correlating various types of features (Figure 2, Supplementary Data). The deepView browser in deepBase provides an integrated view of mapped reads, known and predicted ncRNAs, protein-coding genes and RNA clusters and their expression peaks (Figure 2, Supplementary Data). Clicking a prediction or gene of interest launches a multiple-alignment trace viewer that displays all traces of genes or links to external resources such as NCBI, UCSC, miRBase and TAIR to obtain more comprehensive information. The libView browsers provide the graphical comparisons of multiple libraries for the distribution of length and 5′-terminal nucleotide of small RNAs (Supplementary Data). We also provide the nasView graphical browser to facilitate the comparisons of multiple small RNA libraries of ncRNAs, including miRNAs, snoRNAs, tRNAs, rRNAs, snRNAs, scRNAs, Mt_tRNAs and misc_RNAs (Supplementary Data). The expression profiles for ncRNAs are also provided to test for differential expression pattern among different tissues and cell lines (Supplementary Data). For small RNAs derived from diverse RNAs, RNA clusters and predicted ncRNAs, the database provides the sequence, genomic location, RNA secondary structures, references and annotations.
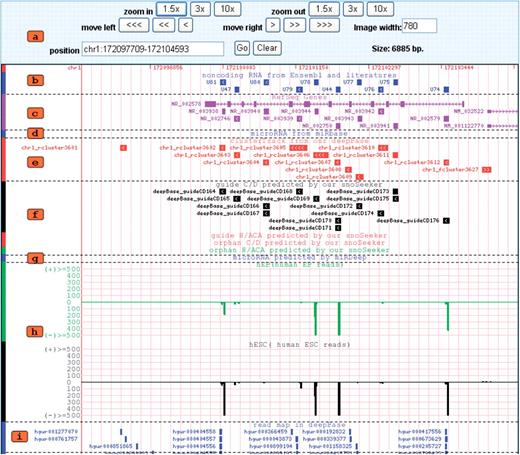
Snapshot of the deepView browser. (a) The controls directly underneath position the browser over a specific region in the genome. (b) RNA genes from Ensembl or the literature. (c) refSeq Gene. (d) microRNA gene from miRBase v13. (e) RNA clusters generated by this study. (f) The predicted snoRNAs from deep sequencing data using snoSeeker. (g) The predicted miRNA genes from deep sequencing data using miRDeep. (h) Strand-specific cluster expression peak (mapped small RNA density) generated for diverse tissues and cell lines. (i) Reads mapped to the genome.
deepBase provides a variety of search functions, including keyword function for searching small RNA, ncRNA and RNA cluster information, and a BLAST (41) function for performing searches against sets of small RNA sequences. The search results are linked to the full database records.
DISCUSSION AND CONCLUSIONS
By mapping and annotating ∼66 million unique sequences derived from 185 small RNA libraries of diverse tissues and cell lines from seven organisms (Supplementary Data), we have provided a comprehensive integrated map of the diverse small RNAs, including miRNAs, piRNAs, endo-siRNAs, nasRNAs, pasRNAs, easRNAs and rasRNAs, in these genomes. In addition to recapitulating known small RNAs, we provide enhanced resolution and novel findings owing to the integration of the large number of small RNA libraries of diverse tissues and cell lines. Moreover, the ∼1.2 million RNA clusters identified in this study have shown an extensive and complex transcriptional map in the seven genomes.
Our initial analysis of these RNA clusters reveals that (i) these clusters cover thousands of known ncRNAs and protein exons (Supplementary Data) and (ii) additional members of known ncRNA (miRNA and snoRNA) families were identified from deep sequencing data using miRDeep (5) and snoSeeker (30). However, the most intriguing result of our study is the numerous predicted RNA clusters that could not be assigned to known annotated RNAs. Some of these overlapped with the evolutionarily conserved phastCons elements (62), indicating their important functions. By contrast, many of these RNA clusters might not be functional, but rather ‘junk’ RNA generated as a by-product of cellular activities. To determine whether these RNA clusters are evidence of important new biochemical pathways, it will ultimately be necessary to test their function by new experimental or computational methods. Nevertheless, our findings indicate that future investigation of the RNA clusters is worthwhile for discovering novel ncRNAs and even novel ncRNA classes.
In comparison to the other databases related to deep sequencing small RNA data sets including FANTOM4 (29,63) and Gene Expression Omnibus (GEO) Short Read Archive (SRA) (31), deepBase aims on the mapping, annotation, mining and visualization of deep sequencing data from multiple technological platforms, tissues and cell lines of different organisms, and customizing the analysis so that a variety of biological questions can be addressed. The GEO SRA mainly offers the submission, storage and retrieval of deep sequencing data (31), whereas the FANTOM4 currently provides a genome browser for displaying all their own data and only contains the deep sequencing data from a human monocytic cell line THP-1 (29,63). Finally, the data and the integrative, interactive and versatile display provided by the deepBase database will aid future experimental and computational studies in the discovery of novel ncRNAs and transcriptomes.
FUTURE DIRECTIONS
Next-generation sequencing technologies have played a vital role in improving our understanding of functional genomics. As new genome builds and genome-wide high-throughput deep sequencing data from different species, cell lines, tissues and conditions become available, we will continuously maintain and update the database. The Automatic Mapping, Annotating and Mining Tools (AutoMAMT) in deepBase are run in our high-performance computer servers. Indeed, we have updated the deepBase for human genome (hg19 version) using AutoMAMT. At present, deepBase has integrated additional 52 small RNA libraries which are annotated and mapped to the latest human assemble version (hg19). We will continue to extend the volume on the current disk and improve the performance of our computer servers for storing the new sequencing data. The stand-alone graphical user interface (GUI) softwares (http://deepbase.sysu.edu.cn/deepTools.php) will be continuously released in deepBase. Bench biologists can use these stand-alone GUI softwares to manipulate and analyze their own data or data downloaded from deepBase locally on personal computers. The integration of transcriptome datasets from the deepBase database with other deep sequencing research (1–3), such as genomic mRNA-Seq, methylC-Seq and ChIP-Seq, will contribute to functional annotation of the genome and to a deeper understanding of genomic and cellular dynamics and features.
AVAILABILITY
deepBase is freely available at http://deepbase.sysu.edu.cn/. The deepBase data files can be freely downloaded and used according to the GNU Public License.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors are grateful to Dr Daniel Gautheret for his useful communications
FUNDING
National Natural Science Foundation of China (No. 30830066, 30771151, 30900820); National Basic Research Program (No. 2005CB724600) from the Ministry of Science and Technology of China, the funds from the Ministry of Education of China and Guangdong Province (No. IRT0447, NSF05200303, 9451027501002591); China Postdoctoral Science Foundation (No. 4109898); Young Teacher Fund of Sun Yat-sen University (No. 3171917). Funding for open access charge: National Basic Research Program (No. 2005CB724600) from the Ministry of Science and Technology of China.
Conflict of interest statement. None declared.
REFERENCES

{kind=link}
{kind=link}
Comments