





Abstract
U1 snRNP plays a crucial role in the 5′ splice site recognition during splicing. Here we report the first example of naturally occurring U1-independent U2-type splicing in humans. The U1 components were not included in the pre-spliceosomal E complex formed on the human F1γ (hF1γ) intron 9 in vitro. Moreover, hF1γ intron 9 was efficiently spliced even in U1-disrupted Xenopus oocytes as well as in U1-inactivated HeLa nuclear extracts. Finally, hF1γ exon 9 skipping induced by an alternative splicing regulator Fox-1 was impaired when intron 9 was changed to the U1-dependent one. Our results suggest that U1-independent splicing contributes to the regulation of alternative splicing of a class of pre-mRNAs.
INTRODUCTION
Alternative pre-mRNA splicing is one of the central mechanisms for the regulation of gene expression in eukaryotic cells. It allows the generation of multiple proteins from a single gene. It has been estimated that more than 70% of human gene are alternatively spliced. Moreover, alternative splicing is often regulated in a spatio-temporal manner (1–4).
Pre-mRNA splicing is catalyzed by a large ribonucleoprotein complex called the spliceosome, which contains five small nuclear ribonucleoproteins (snRNPs), U1, U2, U4, U5 and U6, as well as many protein-splicing factors. Spliceosome assembly occurs in an ordered manner within each intron. The initial step for spliceosome formation is assembly of the E complex: U1 snRNP binds to the 5′ splice site, SF1 (BBP) binds to the branchpoint, U2AF binds to the 3′ splice site and U2 snRNP loosely associates (5). As the E complex assembly is a key step for exon definition, it is regulated by several nonspliceosomal RNA-binding proteins, including SR proteins, hnRNP proteins and tissue-specific splicing regulators. These RNA-binding proteins bind to various types of exonic and intronic elements and modulate the use of nearby splice sites. SR proteins bind to purine-rich exonic splicing enhancers (ESEs) to activate the splicing reaction. They stimulate recognition of the 5′ splice site by recruitment of U1 snRNP and also increase the efficiency of recruiting U2AF to the 3′ splice site, leading to U2 snRNP association (6). In contrast, hnRNP proteins bind to exonic splicing silencers (ESSs) and intronic splicing silencers (ISSs), and repress the splicing reaction by interfering with the binding of SR proteins, U1 snRNP or U2AF (7). In addition, several tissue-specific splicing regulators affect the E complex formation (8,9).
We have focused on the mechanism of tissue-specific splicing that is controlled by Fox-1. Fox-1 is expressed in the brain, heart and skeletal muscle in mammals and regulates tissue-specific splicing of several genes including human mitochondrial ATP synthase γ subunit gene (hF1γ), nonmuscle myosin heavy chain (NMHC-B) and c-src via binding to the (U)GCAUG element (10–12). In a previous study using in vivo and in vitro splicing assays, we showed that Fox-1 induces hF1γ exon 9 skipping by interfering with the formation of the E complex on intron 9 through binding to the GCAUG element in intron 8 (13).
In this study, we performed affinity purification of the E complex on hF1γ intron 9 (hF1γ exon 9–10) to clarify the mechanism of how the E complex formation is inhibited by Fox-1. Unexpectedly, we found that the E complex formed on the hF1γ exon 9–10 pre-mRNA did not contain U1 snRNP even in the absence of Fox-1, although the pre-mRNA was spliced with high efficiency. Moreover, hF1γ intron 9 was successfully spliced in U1-disrupted Xenopus oocyte as well as in U1-inactivated HeLa cell nuclear extracts, indicating that hF1γ exon 9–10 is a natural U1-independent splicing substrate. It has been reported that a class of pre-mRNAs can be spliced in U1 snRNP-depleted HeLa nuclear extracts with enriched SR protein (14,15). In addition, Drosophila fushi-tarazu (ftz) pre-mRNA can be spliced in the U1-depleted HeLa nuclear extract even without addition of SR protein (16). However, the mechanism and biological significance of naturally occurring U1-independent splicing is unclear. As compared with the consensus of the canonical 5′ splice site, the positions –3 and +5 of the 5′ splice site of hF1γ intron 9 are different. If these different nucleotides were substituted to match the consensus sequence, the splicing of hF1γ intron 9 was completely shifted to the U1-dependent splicing. Finally, in vivo splicing assay revealed that Fox-1 failed to induce hF1γ exon 9 skipping if the 5′ splice site of intron 9 was mutated to the U1-dependent type, or if a suppressor U1 snRNA that is expected to base-pair with the 5′ splice site was expressed. Our results suggest that U1-independent splicing contributes to the regulation of alternative splicing of a class of pre-mRNAs.
MATERIALS AND METHODS
In vitro splicing and spliceosome purification
32P-labeled pre-mRNAs (2.5 × 104 c.p.m.) were incubated in 10 µl of reaction mixture containing 1.6 mM MgCl2, 0.5 mM ATP, 20 mM creatine phosphate and 30% HeLa nuclear extracts and then terminated with proteinase K. The splicing products were extracted and separated by electrophoresis on 6% denaturing PAGE. For the purification of the E complex, 32P-labeled pre-mRNAs (12.5 × 104 c.p.m.) were incubated in 30 µl mixture containing 30% ATP-depleted nuclear extracts with 1 µg of GST-MS2 protein. After 15 min incubation at 30°C, 3 µl of the reaction mixture was set aside as the input control; 20 µl of glutathione beads was added to the remaining mixture, and the final volume was brought to 200 µl by the addition of NET2 buffer [50 mM Tris (pH 7.5), 150 mM NaCl and 0.05% NP-40]. After a 2-h incubation at 4°C, the beads were washed six times with cold NET2 buffer and treated with proteinase K for RNA analysis. RNA was recovered by phenol extraction and ethanol precipitation, fractionated by 6% denaturing PAGE and transferred to Biodyne Plus membrane (Pall). Hybridization with 32P-labeled RNA probes for U1 and U2 snRNAs were transcribed in vitro. For protein analysis, the beads were boiled in SDS–PAGE sample buffer and used for western blotting with anti-U1-70K anti-body (17) or anti-U2AF65 anti-body (Sigma).
RNA microinjection into Xenopus oocytes
RNA microinjection experiments into Xenopus oocytes were performed as described previously (18,19). Total RNAs were recovered from the injected oocytes. Splicing products were analyzed on the denatured gel and detected by autoradiography. Northern blotting was also performed to detect endogenous U1 and U2 snRNAs.
Transfection experiments
HeLa cells were grown in DMEM containing 10% FBS. Transfection was performed by the Polyfect reagent (QIAGEN). Transfected cells were usually incubated for 18 h before RNA extraction, except U1 suppressor experiments in which the transfected cells were incubated for 48 h. The myc fusion proteins expressed in transfected cells were examined by western blotting using anti-myc antibody (cMyc 9E10; Santa Cruz Biotechnology). As a loading control of western blotting, U2AF65 was detected by anti-U2AF65 antibody (Sigma). To analyze splicing products from hF1γ mini-gene by RT-PCR, F1-2903 and T7 primers were used as described previously (13). Splicing efficiencies of exon 8–9 and exon 9–10 were analyzed by PCR using the same cDNA with SP6 and exon 8–9-AS primers and exon 9–10-S and T7 primers, respectively. PCR products were electrophoresed in 5% native PAGE, and visualized by a phosphorimager. Splicing products were quantified using NIH Image J software.
RESULTS AND DISCUSSION
U1 snRNP-independent E complex is formed on the hF1γ exon 9–10
In our previous study, we showed that Fox-1 induces the hF1γ exon 9 skipping by interfering with the pre-spliceosomal E complex formation on intron 9 (exon 9–10) (13). To further clarify the mechanism, we performed affinity purification of the E complex formed on hF1γ exon 9–10 pre-mRNA fused with two MS2 binding sites at the terminus of 3′ exon (Figure 1A). The hF1γ3GCAUG pre-mRNA contains a portion of the preceding intron 8 with three copies of the GCAUG element (Fox-1 binding element). Note that the branch point in intron 8 was disrupted by base-substitution mutations. The GCAUG elements were deleted in hF1γΔGCAUG pre-mRNA (Figure 1A). We also used CDC14-15 (chicken δ crystallin exon 14–15) pre-mRNA with MS2-binding sites as a control substrate, which is often used for in vitro splicing analyses (20,21). Under our in vitro splicing conditions, hF1γ pre-mRNAs were spliced as efficiently as CDC14-15 (see Supplementary Figure S1).
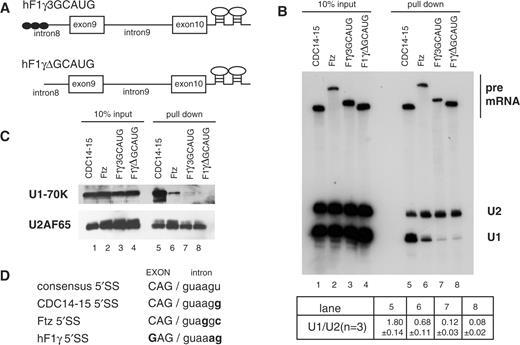
Purification of the pre-spliceosomal E complex formed on hF1γ exon 9–10 pre-mRNA in vitro. (A) The schematic representation of pre-mRNAs, CDC14-15, ftz, hF1γ3GCAUG and hF1γΔGCAUG, fused with two MS2-binding sites at the 3′ exons. Boxes and lines represent exons and introns, respectively. The Fox-1 binding element GCAUG is shown as a closed circle. (B) Northern blotting of the purified E complexes with U1 and U2 snRNA probes (lanes 5–8) and aliquots of the reaction mixtures (lanes 1–4). Average and standard deviation (SD) of the ratio of U1 snRNA to U2 snRNA from three independent experiments are shown at the bottom. (C) Western blotting of the purified E complexes on the pre-mRNAs using U1-70K and U2AF65 antibodies (lanes 5–8) and aliquots of the reaction mixtures (lanes 1–4). (D) The sequences of 5′ splice sites of four kinds of pre-mRNAs as shown in (A). Capital and lowercase letters correspond to exonic and intronic residues, respectively. The nucleotide different from the consensus site is shown in bold.
For the pre-spliceosomal E complex purification, the pre-mRNAs were incubated in HeLa cell nuclear extracts in the absence of ATP since the E complex formation does not require ATP. The E complex formed on the substrate RNA was then pulled down with GST-MS2 fusion protein. We analyzed the purified complexes by northern blotting for detection of U1 and U2 snRNAs and also by western blotting with U1-70K and U2AF65 antibodies. As expected, a typical E complex, containing U1 snRNP, U2AF and U2 snRNP (5), was formed on the CDC14-15 pre-mRNA (Figure 1B and C, lane 5). To our surprise, however, U1 snRNP components were barely detectable in the E complex formed on the hF1γ pre-mRNAs, although the complex contained U2AF and U2 snRNP (Figure 1B and C, lanes 7 and 8). We confirmed that U4 and U6 were not included in the E complex of hF1γ, whereas these snRNAs as well as U2 were present in the late splicing complexes that formed in the presence of ATP (Supplementary Figure S2). It has been reported that Drosophila ftz pre-mRNA is spliced efficiently in the absence of U1 snRNP in vitro (16). Consistently, we found that U1 snRNP was much less detected in the E complex formed on the ftz pre-mRNA (Figure 1B and C, lane 6), although the pre-mRNA was spliced efficiently (Supplementary Figure S1). These results indicated that U1-independent E complex (hereinafter ΔU1-E complex) is formed on the hF1γ exon 9–10 irrespective of the presence of Fox-1-binding elements.
We compared the nucleotide sequences from –3 to +6 relative to the 5′ splice site of these pre-mRNAs, which is thought to affect binding of U1 snRNA (Figure 1D). The CDC14-15 has the same sequence with the consensus of the canonical 5′ splice site except the nucleotide at position +6. As compared with CDC14-15, position +4 of the ftz 5′ splice site is substituted by guanosine, and the positions –3 and +5 of the hF1γ 5′ splice site are substituted by guanosine and adenosine, respectively (Figure 1D).
Splicing of the hF1γ exon 9–10 pre-mRNA is naturally U1-independent
The hF1γ exon 9–10 pre-mRNA formed ΔU1-E complex in vitro. Therefore, we assumed that it is spliced even in the absence of U1 snRNP. At first, we employed an in vitro splicing system using U1 snRNP-inactivated HeLa cell nuclear extracts. To inactivate U1 snRNP, HeLa nuclear extracts were pretreated with antisense 2′-O-methyl oligonucleotide that was complementary to the 5′ end of U1 snRNA (22). We found that the AdML pre-mRNA was not spliced in the HeLa nuclear extracts lacking U1 snRNP as described previously (22–24) (Figure 2, lanes 1–4). Splicing of CDC14-15 pre-mRNA was also reduced, although the pre-mRNA was less sensitive to U1-inactivation (Figure 2, lanes 5–8). We confirmed that addition of antisense oligonucleotide against U1 snRNA strongly inhibited the binding of U1 with the pre-mRNAs (Supplementary Figure S3). Under the same condition, however, ftz and hF1γ exon 9–10 pre-mRNAs were spliced efficiently (Figure 2, lanes 9–16).
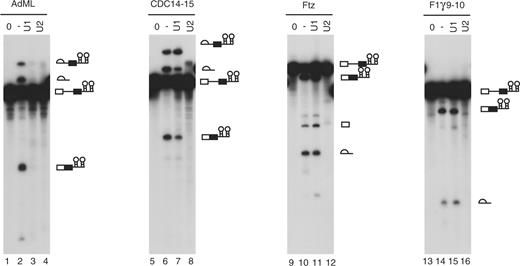
hF1γ exon 9–10 pre-mRNA is spliced efficiently in U1-inactivated HeLa nuclear extracts in vitro. Splicing reaction of AdML (lanes 1–4), CDC14-15 (lanes 5–8), ftz (lanes 9–12) and hF1γ 9–10 (lanes 13–16) with the normal HeLa cell nuclear extracts (lanes 2, 6, 10 and 14), the U1- (lanes 3, 7, 11 and 15) or U2-inactivated (lanes 4, 8, 12 and 16) extracts, as well as transcripts without incubation (lanes 1, 5, 9 and 13). Pre-mRNA, intermediates and splicing products are indicated schematically on the right.
To further examine if hF1γ intron 9 is spliced in a U1-independent manner, we performed microinjection experiments of the RNA substrates into Xenopus oocytes, in which U1 or U2 snRNP had been disrupted by the pre-injection of an antisense oligonucleotide against U1 or U2 snRNA (18,19). Destruction of U1 and U2 by endogenous RNase H activity was confirmed by northern blotting. U1 snRNA was truncated, whereas U2 was completely destroyed (Figure 3A). Under these conditions, three kinds of in vitro transcribed 32P-labeled pre-mRNAs, CDC14-15, ftz and hF1γ 9–10, were individually injected into the nucleus of Xenopus oocytes. When U2 was disrupted, splicing of these three pre-mRNAs was drastically inhibited (Figure 3B, lanes 4, 8 and 12). In the case of U1 disruption, splicing of CDC14-15 pre-mRNA was strongly inhibited (Figure 3B, lane 3). Nevertheless, ftz and hF1γ pre-mRNAs were efficiently spliced (Figure 3B, lanes 7 and 11). These results led us to conclude that hF1γ exon 9–10 is a natural substrate of U1-independnt splicing and that the U1-independece in the splicing reaction can be correctly judged by the formation of ΔU1-E complex.

Splicing of hF1γ exon 9–10 pre-mRNA in U1- or U2-disrupted Xenopus oocytes. (A) Northern blotting of endogenous U1 and U2 snRNAs in Xenopus oocytes. Antisense oligonucleotide against U1 or U2 snRNA was injected into the cytoplasm of Xenopus oocytes to disrupt endogenous U1 or U2. The same volume of water was injected as a control (–). Note that U1 snRNA was truncated by the injection of the U1 antisense oligo (the fast migrating band indicated as U1*), since the oligo targets the 5′ end of U1 snRNA. (B) Splicing of the 32P-labeled CDC14-15, ftz and hF1γ 9–10 pre-mRNAs in the nuclei of control (lanes 2, 6 and 10), U1-disrupted (lanes 3, 7 and 11), or U2-disrupted oocytes (lanes 4, 8 and 12). The hF1γ 9–10 transcript lacks the intron 8 sequence. RNA was analyzed immediately (lanes 1, 5 and 9) or at 70 min (lanes 2–4, 6–8 and 10–12) after injection.
The suboptimal 5′ splice site plays a key role in U1-independent splicing
To identify a cis-element that is required for U1-independnet splicing, we constructed two chimeric constructs of hF1γ and CDC14-15, designated as 9–15 and 14–10, in which the 5′ and 3′ halves of hF1γ exon 9–10 were swapped with those of CDC14-15 in the center of their introns (Figure S4A). The two MS2-binding sites were also fused to the 3′ termini of transcripts. In vitro splicing experiments showed that the chimeric pre-mRNAs as well as CDC14-15, hF1γΔGCAUG, and hF1γ exon 9–10 were spliced with a comparable efficiency (Supplementary Figure S4C). Subsequently, the E complex formed on these pre-mRNAs were purified and subjected to northern blotting with probes for U1 and U2 snRNAs. We found that the E complexes of CDC14-15 and 14-10 contained U1 snRNA, whereas no U1 association was observed with hF1γΔGCAUG, hF1γ exon 9–10 and 9–15 pre-mRNAs, suggesting that the cis-element(s) for ΔU1-E complex formation lies in the 5′ half region of hF1γ exon 9–10 (Supplementary Figure S4B). By introduction of base-substitution mutations into the region except the 5′ splice site, however, we could not find out a specific cis-element that is indispensable for U1-independent splicing (data not shown). Multiple elements may be involved.
The hF1γ intron 9 possesses the typical GT-AG boundaries, although the 5′ splice site has a suboptimal sequence as compared with the consensus sequence, CAG/guaagu (Figure 1D). The 5′ splice site sequence of intron 9 has a conserved sequence at least in humans, mice and bovine (25). To examine if the 5′ splice site sequence is crucial for U1-independent splicing, two base-substitutions were introduced (–3G>C and +5a>g) to change the hF1γ 5′ splice site sequence to that of CDC14-15 (Figure 4A). The mutant was spliced efficiently as the parent substrate in vitro (Figure 4C). Northern blotting showed that the base-substitutions into the 5′ splice site drastically led to the normal E complex formation containing U1 snRNP (Figure 4B). These results strongly suggested that the positions at –3C and +5G of 5′ splice site are important for U1 snRNP binding.
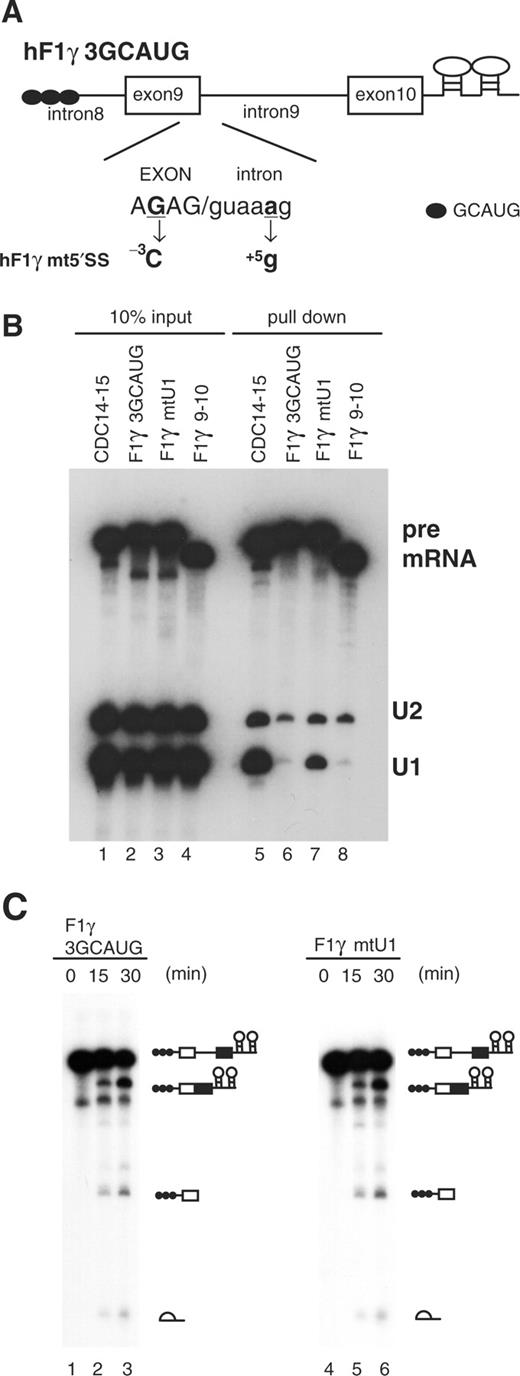
Role of the 5′ splice site sequence in U1-independent splicing. (A) Base-substitutions introduced into the 5′ splice site of hF1γ3GCAUG pre-mRNA are shown (–3G > C and +5a > g). Capital and lowercase letters correspond to exonic and intronic nucleotides, respectively. Boxes and lines represent exons and introns, respectively. The Fox-1 binding element GCAUG is shown as a closed circle. (B) Northern blotting of the purified E complexes with U1 and U2 snRNA probes (lanes 5–8) and aliquots of the reaction mixtures (lanes 1–4). (C) Splicing reaction of hF1γ3GCAUG (lanes 1–3) and hF1γmt5′SS (lanes 4–6). These pre-mRNAs were incubated under the standard in vitro splicing condition for indicated time above each lane. Pre-mRNA, intermediates and splicing products are indicated schematically on the right.
In the 5′ splice site sequence, the nucleotide at position +5 is suggested to be important for base-pairing with U1 snRNA as well as U6 snRNA (26–30). In the case of Drosophila ftz pre-mRNA, which seems to be spliced in a manner partly independent of U1 snRNP, position +4, instead of +5, is substituted from the consensus sequence (Figure 1D). Thus, it is unlikely that the primary sequence of the 5′ splice site is the only determinant for U1-independent splicing. Crispino et al. (16) showed that the sequences flanking the 5′ splice site of the ftz pre-mRNA are necessary but not sufficient for U1-independent splicing. Taken together, our current model for U1-independent splicing is as follows. First, the suboptimal 5′ splice site is necessary for avoiding tight base-pairing with U1 snRNA. Second, some trans-acting factor may associate with the nearby cis-element for definition of the 5′ splice site, and then inducing ΔU1-E complex formation. However, we cannot exclude the possibility that the flanking sequence functions to prevent U1 binding, for example, through a secondary structure (31).
Possible candidates for trans-acting factors in U1-independent splicing include U6 snRNA and SR proteins. It has been shown that several pre-mRNAs can be spliced with high efficiency in U1-depleted nuclear extracts enriched in SR proteins. Excess SR proteins form a complex with the pre-mRNA and then promote the association of U2 snRNP with the branch site in the absence of U1 snRNP (14,15). However, our results show no enhanced association of SR proteins with the hF1γ and ftz pre-mRNAs (data not shown). Moreover, U6 snRNP was not detected in the E complex formed on the hF1γ, although it was included in the late splicing complexes in vitro. Crispino and Sharp (32) reported that the base pairing of the pre-mRNA with U6 snRNA, probably during the binding of the U4/U5/U6 tri-snRNP, is a rate-limiting step in U1-independent splicing. Thus, it is unlikely that U6 functions as a cue for ΔU1-E complex formation, the initial step of U1-independent splicing. Further investigation to identify the trans-acting factors involved in U1-independent splicing will be necessary.
U1-independent splicing is indispensable for hF1γ exon 9 skipping by Fox-1
The biological role of U1-independent splicing remains totally unknown. Previously, we showed that Fox-1 induced hF1γ exon 9 skipping by repressing the splicing of intron 9. Therefore we assumed that U1-independent splicing plays a role in exon 9 skipping induced by Fox-1. We verified this hypothesis by the following two kinds of experiment.
First, we performed transfection experiments using a mutant hF1γ mini-gene, hF1γS3GCAUG (5′SSmt), in which the 5′ splice site of intron 9 had been substituted to that of CDC14-15, namely the U1-dependent splice site (Figure 5A). Transfection assay of wild-type hF1γ mini-gene into HeLa cells showed that Fox-1 proteins promoted exon 9 exclusion as described in our previous report (13) (Figure 5B, lanes 1 and 2). It has been shown that endogenous Fox-1 is undetectable in HeLa cells (11). In contrast, when the mutant mini-gene hF1γS3GCAUG (5′SSmt) containing the U1-dependent splice site was transfected, exon 9 exclusion was barely observed even in the presence of Fox-1 (Figure 5B, lanes 3 and 4). Note that the splicing efficiencies of introns 8 and 9 were not largely affected by the mutation into the splice site in the absence of Fox-1 (lanes 5, 7, 9 and 11). Importantly, when the Fox-1 protein was co-expressed (Figure 5D), splicing of the downstream intron 9 (E9–E10) of wild-type hF1γ mini-gene was inhibited by Fox-1 as expected (13), whereas that of the mutant mini-gene hF1γS3GCAUG (5′SSmt) was not (Figure 5B, lanes 9–12). Irrespective of the mutation into the 5′ splice site, splicing of intron 8 (E8–E9) was not affected by the Fox-1 expression (lanes 5–8). In addition, we confirmed that the expressed Fox-1 protein was properly localized to the nucleus (data not shown).
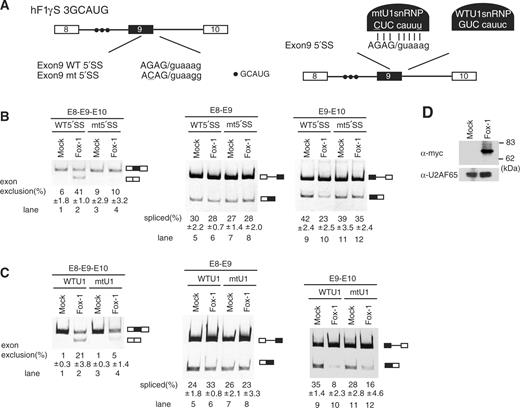
U1-independent splicing is indispensable for the regulation of alternative splicing of hF1γ by Fox-1. (A) The schematic representation of wild-type (WT) and mutant (mtU1) hF1γ mini-genes (left) and the suppressor U1 snRNP (right). The mutations introduced into the 5′ splice site of exon 9 are underlined. Closed circles indicate the Fox-1 binding element. (B) Transfection assay of hF1γ WT and 5′SSmt mini-genes co-expressed with pCS2+MT vector or pCS+MT-Fox-1 in HeLa cells. The left panel shows spliced products amplified by the oligonucleotides annealed with exons 8 and 10. The middle and right panels show the splicing reaction between exons 8 and 9 and between exons 9 and 10. Positions of splicing products and unspliced transcripts are schematically shown on the right. All experiments were independently performed four times. Average and standard deviation of exon 9 exclusion and splicing efficiency are shown at the bottom of each lane. (C) Suppressor U1 snRNA experiments in HeLa cells. The wild-type or mutant U1 snRNA was co-expressed with wild-type hF1γ mini-gene and pCS2+MT vector or pCS+MT-Fox-1. The spliced products were analyzed by RT-PCR using the oligonucleotides annealed with exons 8 and 10 (left panel), exons 8 and 9 (middle panel) and exons 9 and 10 (right panel). Positions of splicing products and unspliced transcripts are schematically shown on the right. All experiments were independently performed three times. Average and standard deviation of exon 9 exclusion and splicing efficiency are shown at the bottom of each lane. (D) Western blotting of cell extracts using the anti-Myc antibody (upper panel) and anti-U2AF65 antibody as a loading control (lower panel). Molecular size markers are shown on the right.
Second, we examined the effect of a suppressor U1 snRNA (30,33), which is expected to stably base pair with the intact 5′ splice site in intron 9 (Figure 5A). In the absence of Fox-1, the suppressor U1 snRNA did not largely affect the spicing efficiencies of introns 8 and 9 as compared with the wild-type U1 snRNA expression (Figure 5C, lanes 1, 3, 5, 7, 9 and 11). However, the exon 9 skipping induced by Fox-1 was disrupted to some extent by the suppressor U1 snRNA expression (Figure 5C, lanes 4, 8 and 12).
These results led us to conclude that U1-independent splicing of hF1γ intron 9 is indispensable for induction of exon 9 skipping by Fox-1. It is possible that Fox-1 inhibits the association of some trans-acting factor for ΔU1-E complex formation with the hF1γ pre-mRNA.
Role of U1-independent splicing
Our study implies that there exist a number of U1-independent introns in mammalian genomes. Recently, Raponi and Baralle (34) proposed a possibility that U1-independent splicing could be disrupted by some disease-causing mutations in the 5′ splice site.
Our study showed that U1-independent splicing of hFγ intron 9 requires U2 snRNP. A class of minor introns, AT-AC introns, having noncanonical splice sites are spliced by U12-type spliceosome (35). The 5′ splice site of AT-AC intron was recognized by base pairing with U11 snRNP that is different from U1 snRNP but functionally analogous to the U1 snRNP. Thus, it is possible that U11-independent splicing may take place in a subset of AT-AC introns.
U1-independent splicing may be necessary for the splicing regulation of other genes by Fox-1 and also by other splicing regulators. Fox-1 and Fox-2 function as both negative and positive splicing regulators (10–12,36). Our study suggests that U1-independent splicing may be involved at least in the negative regulation of alternative splicing by Fox proteins.
It has been shown that U1 snRNP often plays key roles in the splicing regulation. The association of U1 snRNP to the alternative exons is promoted by SR proteins and several regulators such as TIA-1 and HMGA-1 (6,8,37). Shin et al. (38,39) reported that mRNA splicing is downregulated through the inhibition of U1 snRNP function by SRp38 dephoshorylated during mitosis and also in response to heat shock (37,38). It should be clarified if U1-independent splicing is also inhibited under such conditions.
FUNDING
Grants-in-Aid for Scientific Research from JSPS and MEXT; and by the Mitsubishi Foundation, partial. Funding for open access charge: Mitsubishi Foundation.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Samuel I. Gunderson for U1-70K anti-bodies, Alan M. Weiner and Kinji Ohno for U1 snRNA expression plasmid, Naoyuki Kataoka, Ray Yoshimoto and Toshinobu Fujiwara for their valuable advice on the splicing complex purification using the GST-MS2 fusion protein and our laboratory members, especially Tomoko Yamada and Izumi Koga, for experimental assistance.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments