





Abstract
During minus-strand DNA synthesis, RNase H degrades viral RNA sequences, generating potential plus-strand DNA primers. However, selection of the 3′ polypurine tract (PPT) as the exclusive primer is required for formation of viral DNA with the correct 5′-end and for subsequent integration. Here we show a new function for the nucleic acid chaperone activity of HIV-1 nucleocapsid protein (NC) in reverse transcription: blocking mispriming by non-PPT RNAs. Three representative 20-nt RNAs from the PPT region were tested for primer extension. Each primer had activity in the absence of NC, but less than the PPT. NC reduced priming by these RNAs to essentially base-line level, whereas PPT priming was unaffected. RNase H cleavage and zinc coordination by NC were required for maximal inhibition of mispriming. Biophysical properties, including thermal stability, helical structure and reverse transcriptase (RT) binding affinity, showed significant differences between PPT and non-PPT duplexes and the trends were generally correlated with the biochemical data. Binding studies in reactions with both NC and RT ruled out a competition binding model to explain NC's observed effects on mispriming efficiency. Taken together, these results demonstrate that NC chaperone activity has a major role in ensuring the fidelity of plus-strand priming.
INTRODUCTION
Reverse transcription consists of a complex series of reactions that result in synthesis of a linear double-stranded DNA copy of the single-stranded viral RNA genome. This process is catalyzed by the virus-encoded enzyme, reverse transcriptase (RT), which exhibits RNA- and DNA-dependent DNA polymerase activities (1–3) as well as RNase H activity, i.e. the ability to degrade the RNA strand of an RNA–DNA hybrid (4).
In one of its key roles, RNase H generates and later removes the polypurine tract (PPT) plus-strand DNA primer, a short, purine-rich sequence present in the viral RNA genome immediately upstream of U3 (3′ PPT). In the case of HIV-1, there is a second copy of the PPT (central PPT) in the integrase coding region (5) that will not be discussed here. Cleavage at the PPT-U3 junction must be precise to permit formation of a viral DNA that has the correct 5′ long terminal repeat (LTR) end and is competent for integration. Moreover, to fulfill this requirement, the PPT must be the exclusive primer for initiation of plus-strand DNA synthesis (6–8).
From available evidence we know that the exclusive use of the PPT is achieved, at least in part, because the PPT duplex, in contrast to all other RNA–DNA hybrids, is resistant to RNase H degradation during reverse transcription (6–8). Several factors are responsible for this unusual property. For example, the PPT has a unique sequence, including six Gs at its 3′-end (Figure 1) that are required for proper RNase H cleavage at the PPT-U3 junction and extension by HIV-1 RT (9) [for further details on mutational analysis of retroviral PPT sequences, see refs. (7,8) and references therein].

Schematic diagram of the RNA primers used in this study. The gray rectangle represents nt 8994–9138 from the 3′-end of the HIV-1 NL4-3 RNA genome (numbering according to GenBank accession number: AF324493) (70). The RNA primers (each 20 nt) are shown beneath the gray rectangle and the tick marks are placed according to the position of the first base in the primer sequence in the viral RNA genome. Note that in the case of the PPT, we used a primer containing the PPT plus the five downstream bases, so that all primers would be the same size. The five additional bases are removed by RNase H to generate the actual PPT primer (9). Symbols: 589R, stippled; 194R (PPT+5), open; 587R, solid; and 591R, hatched. The table below the diagram indicates the nt positions and the sequence (5′ to 3′ direction) of each primer.
In addition to sequence, there are also structural considerations. Based on an X-ray crystal structure, an HIV-1 PPT duplex bound to RT was reported to have significant structural anomalies (10). In addition, structural distortion at the PPT-U3 junction was seen in the absence of RT (11,12). More recently, high-resolution NMR analysis showed that the PPT duplex alone consists entirely of intact, standard Watson–Crick base pairs (bp) (13). Although weakened base pairing could be detected at particular bases (13), it appears that major perturbations in PPT structure are associated with RT binding (10,13,14).
To catalyze cleavage at the junction with U3 and subsequently extend the primer, RT must assume two orientations: one that favors cleavage (see below) and the other, in which the polymerase active site is positioned at the 3′-hydroxyl group of the PPT, as would normally occur with a DNA primer, but not with a non-PPT RNA (15,16). A recent report indicates that a single RT molecule can flip between RNase H cleavage- and DNA polymerase-competent orientations (16).
Interestingly, in the course of reverse transcription, there is a step that could interfere with selection of the PPT as the sole plus-strand primer: during minus-strand DNA synthesis, genomic RNA template sequences that become annealed to the nascent DNA are removed by RNase H cleavage to enable minus-strand transfer to occur and also to allow minus-strand DNA to function as the template for subsequent plus-strand DNA synthesis (6,17). Some of the RNA fragments are large enough to remain in a hybrid structure and could potentially prime DNA synthesis (7). Normally, non-PPT hybrids are further degraded by RNase H-catalyzed 5′-end-directed cleavages. In this case, the polymerase active site is positioned near the 5′-end of the RNA, whereas the RNase H domain is positioned near the 3′-end (15,16,18–24). This orientation of RT is incompatible with DNA synthesis. Thus, the RNA fragments are poorly extended, if at all (9,16,23,25–27).
Nevertheless, there are a few in vivo studies that describe what we refer to as ‘mispriming’, i.e. priming by an RNA other than the PPT, albeit at low levels (28,29). Interestingly, when the 3′ PPT was changed to a completely random sequence, HIV replication occurred, but to only a small extent, e.g. on Day 3, the mutant virus was 1.5 orders of magnitude less infectious than wild-type (WT). By Day 6, however, the mutated PPT sequence had reverted to the WT sequence and the mutant was able to replicate like WT HIV-1 (29). It has also been reported that HIV-1 plus-strand DNA synthesis is discontinuous, presumably reflecting the use of multiple upstream initiation sites (30). In this case as well, the 3′ PPT must be used to initiate plus-strand synthesis so that viral DNA has the correct end for integration. Taken together, these findings emphasize the importance of maintaining the 3′ PPT for successful virus replication.
Based on events occurring during plus-strand DNA transfer, we hypothesized that in addition to RNase H degradation, HIV-1 might also use a complementary mechanism for inhibiting mispriming reactions. Thus, in earlier work, we demonstrated that both secondary RNase H cleavage as well as the destabilizing activity of the HIV-1 nucleocapsid protein (NC) are required for maximal removal of the tRNA3Lys primer still hybridized to (+) SSDNA [for more details, see refs. (31–33)]. We therefore considered the possibility that HIV-1 NC and RNase H activity may both play roles in blocking mispriming and we set out to test this prediction.
HIV-1 NC is a small, highly basic, nucleic acid binding protein with two zinc finger domains that are connected by a short, basic amino acid linker (34–36). Each zinc finger contains the invariant CCHC metal-ion-binding motif (37). The NC protein is a nucleic acid chaperone, which means that it can catalyze nucleic acid conformational rearrangements that lead to the most thermodynamically stable structures (34–36,38–40). Chaperone function consists of two independent activities, which are both essential for NC-dependent reactions in vitro (36) and virus replication in cells (40): (i) aggregation of nucleic acids, which is important for annealing and is localized primarily to the N-terminal basic domain (41–44) and (ii) moderate destabilization of nucleic acid duplexes, an activity that is associated with the zinc fingers (32,33,45–64).
In the present study, we have investigated the influence of HIV-1 NC on the primer extension activities of the PPT and three representative 20-nt purine-rich non-PPT primers having sequences derived from the upstream or downstream regions near the 3′ PPT (Figure 1), in conjunction with authentic HIV-1 minus-strand DNA templates. With WT RT, the non-PPT primers exhibit a range of priming activities in the absence of NC, but addition of NC reduces priming in each case to almost base-line level. RNase H cleavage and zinc coordination by NC are required for maximal inhibition of mispriming, but a modest effect of NC in the absence of RNase H is also observed. In contrast, the PPT duplex is unusually stable and therefore resistant to NC destabilization. The results of the primer extension assays could be correlated with the biophysical properties of the PPT and non-PPT hybrids and all of the assays showed that the PPT duplex is distinct from the non-PPT complexes. Collectively, our findings demonstrate a novel role for NC nucleic acid chaperone activity in reverse transcription, which together with RNase H cleavage dramatically increase the fidelity of plus-strand initiation.
MATERIALS AND METHODS
Materials
RNA oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA) or Dharmacon (Lafayette, CO). DNA oligonucleotides were purchased from Lofstrand (Gaithersburg, MD) or Integrated DNA Technologies. DNA oligonucleotides labeled at the 5′-end with fluorescein and purified by HPLC were obtained from TriLink BioTechnologies (San Diego, CA) (65). [γ-32P]ATP (3000 Ci/mmol) was purchased from GE Healthcare (Piscataway, NJ) and PerkinElmer (Shelton, CT). T4 polynucleotide kinase, SUPERaseIn RNase inhibitor, and Gel Loading Buffer II, were purchased from Applied Biosystems (Foster City, CA). HIV-1 RT was obtained from Worthington Biochemical Corp. (Lakewood, NJ). An RNase H-minus HIV-1 RT, E478Q (66), was a generous gift from Dr Stuart Le Grice (HIV Drug Resistance Program, NCI-Frederick, Frederick, MD).
Methods
Preparation of HIV-1 NC proteins
Recombinant WT NC was prepared as described previously (67,68). The SSHS mutant NC protein, in which all six Cys residues are changed to Ser, was expressed and purified as described in reference (32). Zinc-less WT NC was prepared by solid-phase peptide synthesis (69) and was never exposed to zinc. NC (11–55), which is missing residues 1–10 (36) and was reconstituted with Zn2+, was also prepared by solid-phase chemical synthesis (69).
RNA and DNA oligonucleotides
The HyTher program (http://ozone3.chem.wayne.edu/) was used to predict which 20-nt RNA oligonucleotides in the vicinity of the 3′ PPT would form stable duplexes with Tm values similar to that of the PPT. Experimentally determined Tm values of the duplexes used in this study are given in Table 1. The RNA primers were 194R (the 15-nt PPT with the addition of five bases downstream of the 3′ PPT, i.e. PPT+5), 587R, 589R and 591R. The sequences and positions of the primers on the viral RNA genome are illustrated in Figure 1. We used the 20-nt version of the PPT so that all of the primers would be the same size and also because this oligonucleotide, unlike the 15-nt PPT, displayed only one gel band in the absence of RT (Figure S1). In the case of the 20-nt PPT, the additional 5 nt are removed by RNase H so that the extension products of a 15-nt or 20-nt PPT are identical (9). For biophysical experiments (see below), we used a 15-nt PPT duplex, since the additional 5 nt would not be cleaved under the conditions used.
The minus-strand DNA template (100 nt) for primers 194R, 587R and 591R was 581D (5′-GTG TGT GGT AGA TCC ACA GAT CAA GGA TAT CTT GTC TTC TTT GGG AGT GAA TTA GCC CTT CCA GTC CCC CCT TTT CTT TTA AAA AGT GGC TAA GAT CTA C (nt 9039–9138) and for primer 589R, the 100-nt template was 582D (5′-AGT GAA TTA GCC CTT CCA GTC CCC CCT TTT CTT TTA AAA AGT GGC TAA GAT CTA CAG CTG CCT TGT AAG TCA TTG GTC TTA AAG GTA CCT GAG GTG TGA C (nt 8994–9093). All sequences were derived from the HIV-1 pNL4-3 clone (GenBank accession no: AF324493) (70).
The RNA and DNA oligonucleotides were gel-purified by polyacrylamide gel electrophoresis (PAGE) in 15% or 12% denaturing gels, respectively, followed by excision of the desired band from the gel and further purification with Microcon YM-3 or YM-10 centrifugal filter units (Millipore). RNA primers were 5′-end labeled using T4 polynucleotide kinase and [γ-32P]ATP, as described previously (71), except that labeled primer was separated from unincorporated ATP using a Princeton Separations spin column (Princeton Separations, Adelphia, NJ), following the instructions provided by the manufacturer.
Primer extension assay
RNA-primed plus-strand DNA synthesis was measured in the absence or presence of HIV-1 NC, as specified. Primer extension was assayed with 5′-32P-labeled primer RNAs. Thus, in each case, only extension from the intact primer was detected. Each 5′-32P-labeled primer RNA (0.4 pmol) was heat annealed to its complementary minus-strand DNA template (0.4 pmol); gel-shift assay verified that annealing was complete (data not shown). Reaction mixtures contained the annealed duplex, reaction buffer (50 mM Tris–HCl (pH 8.0), 75 mM KCl, 7 mM MgCl2, 1 mM DTT), SUPERaseIn (final concentration, 0.5 U/µl), HIV-1 RT (1 pmol) and the four dNTPs (100 µM each) in a final volume of 20 µl. Reactions were initiated by addition of MgCl2 and the four dNTPs. After incubation at 37°C for the indicated times, reactions were terminated by addition of 8 µl of Gel Loading Buffer II followed by heating at 95°C for 4 min. Three-microliter samples were subjected to denaturing PAGE in an 8% gel. Radioactivity was quantified by using a Typhoon PhosphorImager (GE Healthcare) and ImageQuant software. For time course experiments, reactions were scaled up and contained 1.7 µM NC (1.45 nt/NC), where specified. Five-microliter aliquots were withdrawn at the indicated times and were processed as described above, except that only 2 µl of loading buffer was added. The amount of full-length (FL) 32P-labeled DNA synthesized in the reaction was expressed as the percentage of total radioactivity in the lane (% FL DNA).
Biophysical assays
The assays, described below, were performed in the absence of Mg2+ (see Supplementary Data, Materials and Methods section).
CD spectroscopy
CD spectra were obtained with a JASCO J710 spectropolarimeter equipped with a water-jacketed cell holder using 1 mM path-length cells. The RNA–DNA hybrid (25 μM) was annealed in buffer containing 50 mM Tris–HCl (pH 8.0) and 75 mM KCl by heating to 80°C for ∼20 min and cooling slowly to room temperature.
UV melting experiments
UV absorption readings at 260 nm were taken as a function of temperature, using a GBC 918 spectrophotometer equipped with thermoelectrically controlled cell holders. Melting studies were performed in the absence or presence of NC as described (60). The concentration of the duplexes was 2 µM and NC was 4 µM (1 NC/strand). The observed melting curves allowed an estimation of melting temperature, Tm, the midpoint temperature of the unfolding process. Under our experimental conditions, no noticeable precipitation (i.e. no light scattering) was observed, as verified by monitoring the UV absorption at 350 nm. Note that for these experiments NC (11–55), which is missing the N-terminal basic residues of NC, was used to minimize aggregation.
Fluorescence anisotropy (FA) measurements
Equilibrium binding constants of RT to RNA–DNA hybrids were determined by measuring the FA of 20 nM duplexes (DNA strands were labeled at their 5′-ends with fluorescein) as a function of increasing concentration of RT or NC. Hybrid duplexes were annealed as described above in buffer containing 50 mM Tris–HCl (pH 8.0), 75 mM KCl and 1 mM DTT. The protein–DNA mixtures were incubated at room temperature for 1 h. Anisotropy measurements were made on a Photon Technology International spectrofluorimeter (Model QM-2000). The excitation and emission wavelengths were 485 and 535 nm, respectively. Data analysis was performed by fitting binding data to a 1:1 binding model as described previously (72). Kd values were determined from two or three independent experiments.
For competition studies, the PPT or 591 duplexes (20 nM) were prebound to either 500 nM RT or 1000 nM NC in 75 mM KCl, 1 mM DTT, 50 mM Tris–HCl (pH 8.0) for 1 h at room temperature. FA was measured as a function of increasing concentration of competing protein (either RT or NC).
RESULTS
HIV-1 NC is required for blockage of mispriming by non-PPT RNA primers
The goal of this work was to determine whether the nucleic acid chaperone activity of NC contributes to selection of the PPT as the exclusive 3′ primer for plus-strand initiation by helping to block mispriming by non-PPT RNAs. Our approach was to test RNA sequences in the vicinity of the 3′ PPT, since suppression of mispriming in this region and in particular, downstream of the PPT, is crucial for producing viral DNA that is competent for integration (6,7). The diagram in Figure 1 shows a schematic representation of the viral RNA genome from nt 8994 to nt 9138 and the positions of the first nt of (i) three representative non-PPT RNA fragments (589R, upstream of the PPT; 587R and 591R, both downstream of the PPT); and (ii) the PPT plus five downstream bases (194R). The nt boundaries and sequence for each primer are given in the table. Note that all of the primers are purine-rich and that 194R has only two pyrimidine bases, which are part of the 5-nt sequence downstream of the PPT.
Each RNA was labeled at its 5′-end with 32P and was then hybridized to a complementary 100-nt DNA template (581D for 194R, 587R and 591R; 582D for 589R). Primer extension was assayed as described in Materials and Methods section and the results are shown in Figure 2. The only gel products detected are extended DNAs that retain the 5′ 32P label, i.e. DNAs still attached to the intact primer, and small labeled RNAs produced by RNase H cleavage of the labeled RNA primer in the RNA–DNA hybrid (bands located beneath the primer band [P]) (Figure 2A). As predicted from the sequence of the templates, the sizes of the FL DNA extension products (including the RNA sequence) for each non-PPT primer were: 587R, 55 nt; 591R, 40 nt; and 589R, 85 nt. For 194R, the FL product was 80 nt. Unannealed primers in reactions without RT and NC migrated as essentially a single band (Figure S1).
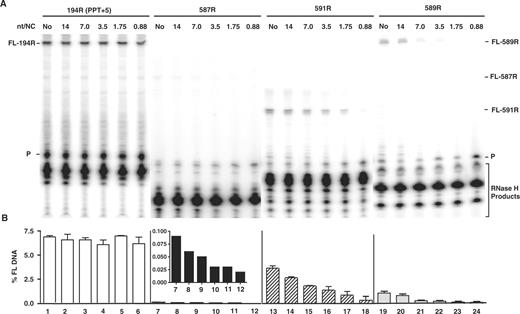
Effect of HIV-1 NC on plus-strand initiation with four RNA primers. The 194R (PPT+5), 587R, 591R and 589R primers were extended by HIV-1 RT in the absence or presence of HIV-1 NC. (A) Gel analysis. FL DNA products synthesized during primer extension after incubation at 37°C for 30 min in the absence (No) (lanes 1, 7, 13, 19) or presence of increasing concentrations of HIV-1 NC as follows: 14 nt/NC (0.17 µM), lanes 2, 8, 14, 20; 7 nt/NC (0.34 µM), lanes 3, 9, 15, 21; 3.5 nt/NC (0.7 µM), lanes 4, 10, 16, 22; 1.75 nt/NC (1.4 µM), lanes 5, 11, 17, 23; 0.88 nt/NC (2.7 µM), lanes 6, 12, 18, 24. The positions of the primer (P) and the FL DNA products formed by 587R (55 nt), 591R (40 nt) and 589R (85 nt) are shown on the right and for 194R (80 nt), on the left. The bracketed bands are RNase H cleavage products. The sizes of the DNA products were verified with appropriate markers. (B) Bar graphs showing the percentage of total radioactivity in a given lane present as the FL 32P-labeled DNA (% FL DNA) as a function of NC concentration. The numbers below each bar in the bar graph also correspond to the lane numbers of the gel. Note that the inset in the bar graph for 587R shows the values for % FL DNA on an expanded scale. Symbols: 194R (PPT+5), open bars; 587R, filled bars; 591R, hatched bars; and 589R, stippled bars.
Gel analysis of primer extension reactions is illustrated in Figure 2A. As expected, 194R was the most efficient primer. The five downstream bases are removed by the RNase H activity of RT (9) leaving a 15-nt RNA; other less prominent RNA bands could represent a small amount of imprecisely cleaved RNA in vitro. In addition, the data showed that RNase H degradation of the non-PPT primers resulted in multiple RNA cleavage products. The most prominent DNA in each case was the FL DNA extension product, although low levels of smaller DNA products were also detected.
What is most striking about the data in Figure 2A is that addition of increasing concentrations of NC resulted in a dramatic reduction of mispriming by the non-PPT primers, but had absolutely no effect on priming by 194R. This is shown most clearly when the data were plotted as bar graphs [percentage FL DNA versus NC concentration (nt/NC)] (Figure 2B). To visualize the effect of NC on priming by 587R, the data in Figure 2B are shown with an expanded scale for the y-axis (see inset of Figure 2).
In the absence of NC, the order of priming efficiency of the RNA primers was as follows: 194R > 591R > 589R > 587R. For example, 591R and 589R had 50% and ∼10–15% of 194R priming activity, respectively, whereas 587R had almost no activity (∼80-fold less activity than 194R). Under these conditions, only RNase H activity can inhibit mispriming of the non-PPT primers. However, when NC was added, the differences between the activities of the non-PPT primers were less dramatic, since in all cases NC further reduced priming activity to essentially base-line levels, particularly at high NC concentrations. The magnitude of NC's inhibitory effect was dependent on NC concentration. The relatively high amount of NC needed for maximal activity in this system, compared to the 7 nt/NC ratio in vivo, reflects the fact that high concentrations of Mg2+ ions are required for in vitro reactions in which both RT polymerase and RNase H activities are required (73). In this case, Mg2+ ions displace some of the NC bound to the nucleic acid (64,74,75). Note too that results obtained from internal labeling experiments (9,76–78), using unlabeled primer, [α-32P]dATP and three unlabeled dNTPs, were consistent with the findings illustrated in Figure 2 (data not shown).
Taken together, the results of Figure 2 show that both RNase H cleavage and NC contribute to blocking mispriming by non-PPT primers during HIV-1 reverse transcription.
Biophysical properties of PPT and non-PPT RNA–DNA hybrids
To gain further insight into the observed differences in priming activity and the effect of NC, it seemed reasonable to assume that differences in the structures of non-PPT primers and the PPT would be of great importance (8) (also, see below). Here, we used several different optical techniques to probe the structure, stability and RT binding affinity of the PPT and non-PPT hybrids: CD spectroscopy, which measures helical structure; UV melting studies (temperature-dependent UV absorption) in the presence and absence of NC; and FA to determine the affinity of RT and NC for the RNA–DNA hybrids. In all of these experiments, the non-PPT hybrids were 20 bp, whereas the PPT hybrid was 15 bp, as described in Materials and Methods section.
CD spectroscopy
In an earlier study of the requirements for PPT priming, we found that helical structure is important for the efficiency of priming activity (9). To determine whether CD spectroscopy might provide a correlation with the observed priming activity of the PPT and non-PPT primers used in this work (Figure 2), we analyzed the spectra of the corresponding RNA–DNA hybrids (Figure 3).
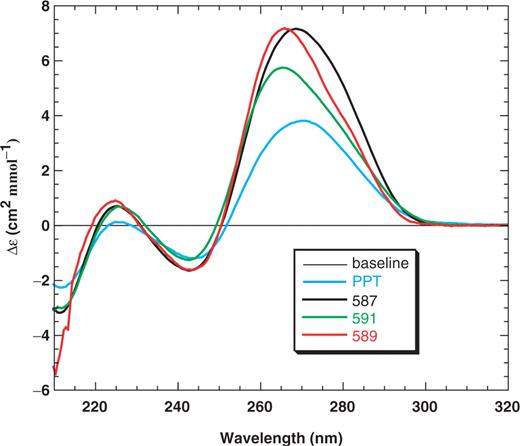
CD spectra of primer RNA–DNA hybrid duplexes. The hybrids were 20 bp in length except for the PPT hybrid, which was 15 bp. The spectra of the PPT (blue), 587 (black), 591 (green) and 589 (red) duplexes were determined as described in Materials and Methods section.
The CD spectrum of a B-form DNA duplex is generally characterized by a positive peak at ∼270 nm and a negative peak of similar size at ∼250 nm. An A-form RNA duplex displays a stronger positive peak at ∼270 nm than a B-form duplex and a relatively weak negative peak at ∼235 nm. In addition, the spectrum of an A-form duplex has a negative peak at 210 nm, whereas this peak is either missing or present as a small positive peak in the spectrum of a DNA–DNA duplex (79,80). CD (79–82) and NMR (83,84) spectroscopy studies have shown that RNA–DNA hybrids are usually intermediate between A- and B-form conformations, although overall, the duplexes studied here appeared to have conformations with more A-like character (Figure 3).
For this work, the positive bands in the spectra at ∼270 nm are of greatest interest. These bands differed in the magnitude and location of their maxima, which is consistent with the different purine content in the RNA strand. For example, the PPT duplex has an all purine RNA strand with the highest G content (47%), and produced a CD spectrum with the smallest positive band intensity at 271 nm. In contrast, the 589 duplex contains 35% Gs and 45% purines in the RNA strand and the spectrum displayed the strongest positive band at 266 nm. Additionally, reduced peak intensity in the 270 nm region is indicative of greater B-form (DNA-like) character. Inspection of Figure 3 indicated that the magnitude of the maxima in this region from smallest to largest was in this order: PPT<591<589∼587. These data support the conclusion that greater B-form character is correlated with increased priming activity (Figure 2). It should be noted that although 589 has more priming activity than 587 (Figure 2), only subtle differences were detected in the CD spectra (Figure 3). This suggests that factors other than B-form character might contribute to the level of priming activity that is observed.
Thermal stability
Melting temperatures of hybrid duplexes were determined in the presence or absence of NC and were derived from UV thermal unfolding experiments (Table 1; Figure S2). For each of the duplexes, the transition started above 40°C, which indicates that at room temperature (used for CD and FA determinations), duplexes were fully folded. In the absence of NC, the hybrid duplexes were characterized by a Tm of 55°C, with the exception of the PPT duplex, which displayed a significantly higher stability (Tm = 61.5°C) despite the fact that it is 5 bp shorter than the other duplexes. In the presence of NC (1 NC/strand), the PPT sequence was not destabilized, whereas the other sequences were destabilized by 2–3°C (Table 1). These small differences in the ΔTm values of the non-PPT duplexes are not significant. However, what is important is that the data are consistent with the results shown in Figure 2 indicating that NC only affects the priming activity of non-PPT RNAs.
RT and NC binding to RNA–DNA hybrid duplexes
In addition to structural considerations, it is possible that differences in the binding affinities of the PPT and non-PPT hybrid duplexes to RT and NC could contribute to the observed differences in priming activity. To address this issue, we initially used FA to measure the apparent equilibrium dissociation constants (Kd values) for RT binding to the PPT, 587 and 591 duplexes (Table 2; Figure S3A). The Kd value for the PPT was ∼140 nM. In contrast, the Kd for 591 was ∼430 nM (3-fold higher than the PPT value), whereas the Kd for 587 was ∼770 nM (almost 6-fold higher than the PPT value). The binding order of each of the duplexes to RT was: PPT>591>587. This result corresponds to the order of priming efficiency in the primer extension experiments (Figure 2). Thus, the data strongly suggest that the affinity of RT to the primer RNA–DNA template duplex also contributes to maintaining the PPT as the exclusive primer for initiation of plus-strand DNA synthesis. Similar experiments were performed to determine the Kd values for NC binding to the PPT and 591 non-PPT duplexes (Figure S3B). In this case, the values were very similar, i.e. ∼650 nM and ∼600 nM, respectively, and were both higher than the values for the binding of these duplexes to RT (Table 2; compare Figure S3B with S3A).
Apparent dissociation constants for RT and NC binding to RNA–DNA hybrids
| Duplex | Kd (nM) | |
|---|---|---|
| RT | NC | |
| PPT | 140 ± 40 | 650 ± 15 |
| 591 | 430 ± 20 | 600 ± 20 |
| 587 | 770 ± 40 | n.d. |
| Duplex | Kd (nM) | |
|---|---|---|
| RT | NC | |
| PPT | 140 ± 40 | 650 ± 15 |
| 591 | 430 ± 20 | 600 ± 20 |
| 587 | 770 ± 40 | n.d. |
The error determinations represent the SD.
n.d., not determined.
Apparent dissociation constants for RT and NC binding to RNA–DNA hybrids
| Duplex | Kd (nM) | |
|---|---|---|
| RT | NC | |
| PPT | 140 ± 40 | 650 ± 15 |
| 591 | 430 ± 20 | 600 ± 20 |
| 587 | 770 ± 40 | n.d. |
| Duplex | Kd (nM) | |
|---|---|---|
| RT | NC | |
| PPT | 140 ± 40 | 650 ± 15 |
| 591 | 430 ± 20 | 600 ± 20 |
| 587 | 770 ± 40 | n.d. |
The error determinations represent the SD.
n.d., not determined.
The results of these biophysical studies show that the PPT duplex is distinct from the non-PPT duplexes. Thus, although the PPT duplex is 5 bp shorter than the other duplexes studied here, it is characterized by high thermal stability, lack of NC-induced destabilization and high binding affinity to RT. Yet, despite these differences, the PPT and 591 duplexes have equivalent binding affinity to NC. This result suggests that the stability of the PPT duplex in the presence of NC is not due to an NC preference for binding to the non-PPT duplexes.
To investigate the possibility that NC might be inhibiting mispriming by blocking RT from binding to the 3′-terminus of the primer (78), we performed competition experiments (Figure 4). Either RT or NC was prebound to the PPT or 591 duplexes and then increasing concentrations of the competing protein were added. Binding was evaluated by measuring FA. When NC was the competing protein, FA of both the PPT and 591 duplexes was unchanged over a broad range of NC concentration. This indicates that NC was unable to displace RT that was bound to the nucleic acid hybrids (Figure 4A). In contrast, when RT was the competing protein, FA of both duplexes was increased with increasing concentrations of RT (Figure 4B). These data show that RT competed effectively for binding to RNA–DNA hybrids prebound to NC in a sequence-independent fashion, i.e. the results were the same with the PPT and non-PPT duplexes. Collectively, these findings rule out a direct binding competition model to account for the ability of NC to specifically inhibit mispriming of non-PPT primers. The results are also in agreement with the measured dissociation constants (Table 2; Figure S3).
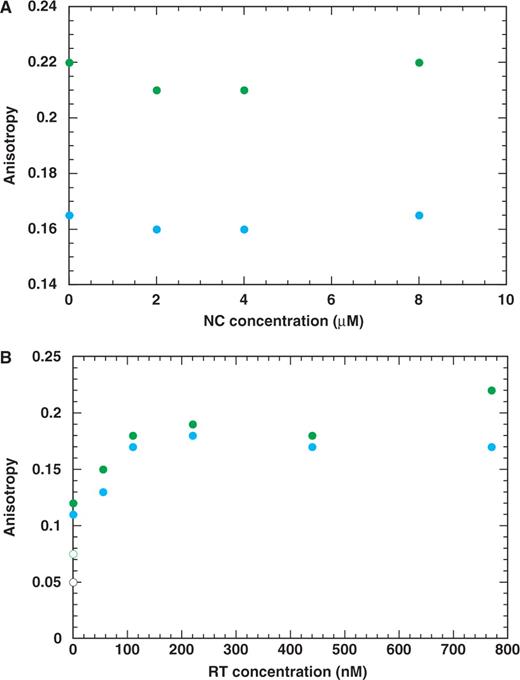
Competition binding experiments with RT and NC to PPT (blue) and 591 (green) duplexes. The duplexes (20 nM) were prebound to 500 nM RT (A) and to 1000 nM NC (B). FA values for the protein-free duplexes were approximately 0.05 and 0.08 for the PPT and 591 duplexes, respectively.
RNase H requirement for blocking mispriming activity
The results presented thus far have demonstrated that the biophysical properties of the duplexes as well as the activities of RNase H and NC are crucial determinants for inhibition of non-PPT primer usage. To obtain a greater understanding of the mechanism underlying this phenomenon and to provide further evidence for the RNase H and NC contributions, we decided on a genetic approach that would take advantage of available RNase H and NC mutants. The relatively high priming activity of 591R (Figure 2) makes the 591 RNA–DNA hybrid an especially good substrate for such experiments.
To probe RNase H function, we compared the time course of 591R priming activity with WT RT or an RNase H-minus RT mutant (E478Q) (66) in the presence and absence of NC (Figure 5). The WT reaction without NC proceeded at a slow rate and significant extension was first detected at 30 min. By 1 h and 3 h, 8% and 15% of the total radioactivity were present as the 32P-labeled DNA, respectively. With addition of NC, extension was very poor at both the early and late time points: at 1 h and 3 h, the extended DNA product was only 1% and 3% of the total radioactivity, respectively. This indicates that the extent of synthesis of the 5′ labeled DNA product was reduced by 5–8-fold in the presence of NC.
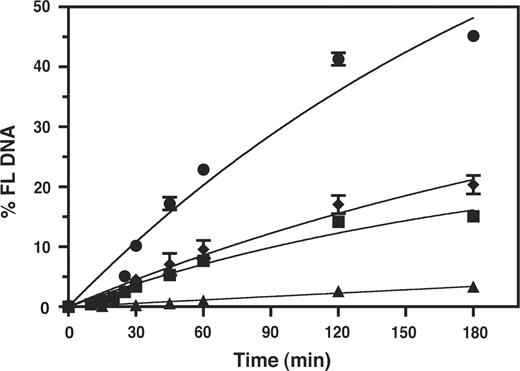
Effect of HIV-1 NC on the kinetics of 591R primer extension catalyzed by WT RT and RNase H-minus RT. The data were plotted as % FL DNA versus Time (min). Symbols. WT RT: minus NC, squares; plus NC, triangles. RNase H-minus RT: minus NC, circles; plus NC, diamonds.
In contrast to these results, when reactions were performed with the RNase H-minus RT in the absence of NC, extension was more efficient than that observed with WT RT. The rate of synthesis was higher and by 3 h, 45% of the total radioactivity was present in the extended DNA. This indicates that RNase H activity is critical for inhibition of mispriming. Interestingly, even in the absence of RNase H activity, addition of NC resulted in a reduction in the rate and extent of the reaction. At 3 h, 20% of the primer was extended, which corresponds to a 2.5-fold reduction in the level of priming activity compared with that seen in reactions without NC.
Taken together these results demonstrate that RNase H plays a major role in blocking mispriming by a non-PPT primer. In RNase H-minus RT reactions, NC alone also had an inhibitory effect on the rate and extent of mispriming. However, the greatest effect was observed when RNase H and NC activities were both present, consistent with the results of Figure 2 and previous studies of plus-strand DNA transfer (31–33).
NC coordination of zinc is required to block mispriming
It was also of interest to determine whether NC's ability to coordinate zinc is required for inhibition of mispriming. Since NC's destabilization activity is associated with the zinc finger domains [for references before 2005, see ref. (36); (59,62)], this question is of great importance for elucidating the mechanism of NC activity in our system. To address this issue, we tested two NC proteins that do not have zinc finger structures: (i) the SSHS mutant, which has all six Cys residues changed to Ser (32) and (ii) chemically synthesized WT NC that was never exposed to zinc (zinc-less NC) (69) (Figure 6). The time course of 591R priming activity was assessed with the two NC variants and was compared with activity in the absence and presence of WT NC. Only WT RT was used in these experiments. The data for reactions with and without WT NC are taken from Figure 5, except that in this case, the values for percentage FL DNA are displayed on an expanded y-axis.
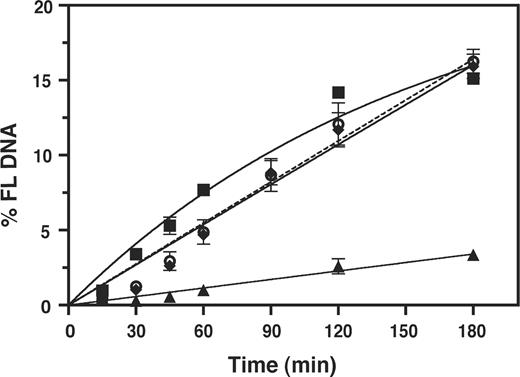
Effect of zinc coordinating activity of NC on the kinetics of 591R primer extension. Reactions were performed with RT in the presence or absence of WT NC, SSHS NC (32), or zinc-less NC (69). The data were plotted as % FL DNA versus Time (min). Symbols: minus NC, squares; WT NC, triangles; SSHS NC, open circles, dashed line; zinc-less NC, diamonds. Note that the minus NC and WT NC curves are the same as those shown in Figure 5 for reactions with WT RT, but the scale on the y-axis is expanded here.
Examination of the data presented in Figure 6 shows that both SSHS NC and zinc-less NC behaved in an identical manner. Throughout the 3 h incubation period, primer extension was ∼1.7–2-fold lower than extension in the absence of NC, but by 3 h, there was no difference between the three curves. This result indicates that SSHS NC and zinc-less NC inhibited mispriming to only a very small extent. In support of this conclusion, we also observed that extent of mispriming was reduced ∼5-fold more efficiently with WT NC than with the NCs that do not coordinate zinc. Thus, the data of Figure 6 demonstrate that zinc coordination is essential for maximal NC function in our system. The results also imply that NC nucleic acid chaperone activity is responsible for destabilization of the non-PPT duplexes.
DISCUSSION
In the present study, we have demonstrated a new role for the nucleic acid chaperone activity of HIV-1 NC in reverse transcription, i.e. the ability of NC to block mispriming by non-PPT primers and ensure selection of the PPT as the sole primer for initiation of plus-strand DNA synthesis at the 3′-end of the genome. This activity, which functions together with RNase H, is critical for synthesis of integration-competent HIV-1 DNA and ultimately for successful replication of the virus.
By using a genetic approach, we could formally establish that RNase H and NC are essential for reduction of mispriming in our reconstituted primer extension system. Indeed, with an RNase H-minus RT, mispriming by 591R RNA was increased by 3-fold in the absence of NC (Figure 5). However, the fact that NC could inhibit primer extension even when RNase H was absent (Figure 5) demonstrates that the inhibitory effects of NC and RNase H can be uncoupled.
A major issue that we address concerns how differences in the biochemical and biophysical properties of the PPT and non-PPT primers affect priming activity. It has long been recognized that the PPT duplex has unique sequence and structural features that distinguish it from other RNA–DNA hybrids (7,8,10,12,13,85) and contribute to its being RNase H-resistant in biologically relevant reactions (6,7,15,16) (also, see above).
Here, we find differences between the CD spectra of PPT and non-PPT duplexes, which reflect differences in helical structure. In particular, the observed differences in the magnitude of the ∼270 nm peak (Figure 3), are generally correlated with the order of priming efficiencies (Figure 2) and B-form (i.e. DNA-like) character. These results are in accord with the known preference of RT for extending DNA primers (16,26,86–88). The data are also consistent with NMR analysis indicating that the major groove of a PPT-related duplex is different from that of other RNA–DNA hybrids and has the shape and relative width more characteristic of a B-form DNA duplex (11).
We also determined Kd values for binding of RT to the PPT and non-PPT hybrids (Table 2; Figure S3A) and found that the data could be correlated with the order of priming activity (Figure 2). For example, the high binding affinity of the PPT duplex to RT is consistent with the fact that of all the primers examined here, the PPT exhibits the most efficient plus-strand priming activity. These data are also in agreement with a study demonstrating that similar to the PPT, primers selected for high binding affinity to RT have at least 6–8 Gs at their 3′-end (89). Interestingly, although PPT priming is not affected by NC (Figure 2), FA measurements demonstrate that the PPT and 591 non-PPT duplexes bind to NC with the same affinity (Table 2; Figure S3B). Thus, binding affinity cannot explain the ability of NC to inhibit extension exclusively from non-PPT primers.
While this manuscript was in preparation, a paper by Jacob and Destefano appeared demonstrating that NC inhibits priming by non-PPT RNAs (two random primers and a primer from the gag-pol region of HIV-1), while having no effect on PPT priming (78), in agreement with our results. The authors postulate that the NC effect on non-PPT priming reflects a competition between RT and NC for binding to the 3′ primer terminus. However, as discussed above, the competition experiments shown in Figure 4 do not support this hypothesis. Additionally, FA measurements show that Kd values for RT binding to the PPT and 591 hybrids are lower than the corresponding values for NC binding (Table 2; Figure S3). This result reflects RT's preference for binding ds nucleic acids (65,90) and NC's preference for binding ss nucleic acids (36).
It is well known that NC's nucleic acid chaperone activity is required for almost every reaction that occurs during reverse transcription (36,40). Since differences in binding affinity and a direct binding competition model do not appear to be responsible for our results, NC's ability to destabilize nucleic acid secondary structures is likely to be involved in blocking mispriming. Several lines of evidence support this idea.
First, we find that NC lowers the Tm of non-PPT duplexes despite these values being relatively high, whereas it has no effect on the Tm of the PPT duplex (Table 1, Figure S2). The resistance of the PPT duplex to NC reflects its strong secondary structure (e.g. see Tm value in Table 1), which is not susceptible to the moderate destabilizing activity of NC (36,57,58,63,64). Note that control experiments demonstrated that PPT priming was also unaffected by addition of either SSHS NC (32), zinc-less NC (69) or RNase H-minus RT (66) (data not shown). Although the ΔTm values for UV melting of the non-PPT hybrids are small (2–3°C range) (Table 1), these results are nevertheless suggestive of a role for NC's nucleic acid chaperone activity in preventing mispriming by non-PPT primers.
Using an RNase H-minus RT (66), we have shown that in the absence of RNase H, NC still has an effect on mispriming (Figure 5). Since competition between RT and NC for binding to the primer does not appear to be a factor (Figure 4), it is more likely that NC destabilizes the non-PPT hybrids. Most importantly, we have found that SSHS NC and zinc-less WT NC, which have been shown to be poor nucleic acid chaperones (36,59) (M. Mitra, D. Mullen, G. Barany, I. Rouzina and K. Musier-Forsyth, manuscript in preparation), are unable to complement the inhibitory effect of RNase H on mispriming (Figure 6). This result does not reflect inefficient nucleic acid binding by these two NCs. In fact, SSHS NC (59) and zinc-less NC (M. Mitra, D. Mullen, G. Barany, I. Rouzina and K. Musier-Forsyth, manuscript in preparation) bind to nucleic acids with even higher affinity than WT NC. The lack of activity in the mispriming assays is therefore most likely due to the severely reduced duplex destabilizing activity of these NC proteins. This conclusion is consistent with many other studies showing that NC-dependent destabilization of nucleic acid duplexes is associated with zinc finger function (32,33,45–47,49,52,53,59–62).
Thus, taken together, our results strongly support a mechanism by which NC's nucleic acid chaperone activity destabilizes the moderately stable non-PPT hybrids, which could lead to dissociation of the primer RNA. It should be noted that when RNase H and NC are both present, the helix destabilizing activity of NC most likely results in displacement of RNase H cleavage products that are still annealed to template DNA.
In summary, we have uncovered a new role for NC's nucleic acid chaperone activity that together with RNase H assures the specific selection of the PPT as the primer for plus-strand DNA synthesis. We show that significant differences in the biophysical properties of PPT and non-PPT duplexes correlate with differences in priming activity. Moreover, the requirement for the coordination of zinc by the CCHC residues indicates that the mechanism for NC's role in blocking mispriming is its ability to destabilize nucleic acid duplexes that lack a highly stable secondary structure. In turn, this also explains why the unique structure of the PPT duplex renders it resistant to NC. Finally, these findings reveal a novel target for development of new strategies for anti-HIV therapy.
FUNDING
This research was supported in part by the Intramural Research Program of the NIH, Eunice Kennedy Shriver National Institute of Child Health and Human Development (to J.G.L.) and NIH grant GM065056 (to K.M.F.) and was also funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400 (to R.J.G.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. Funding for open access charge: Intramural Research Program of the NIH, Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Drs Daniel G. Mullen, Brandie J. Kovaleski and George Barany for synthesis of NC (11–55) and zinc-less NC, Dr Abbey Rosen for performing the initial CD experiments, Amber Hertz for outstanding assistance with some of the biochemical experiments, and Dr Stuart LeGrice for his generous gift of RNase H-minus RT. We are also grateful to Drs Mithun Mitra, James A. Thomas and Tiyun Wu for valuable discussion and critical reading of the manuscript.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments