





Abstract
Mammalian tissue- and/or time-specific transcription is primarily regulated in a combinatorial fashion through interactions between a specific set of transcriptional regulatory factors (TRFs) and their cognate cis-regulatory elements located in the regulatory regions. In exploring the DNA regions and TRFs involved in combinatorial transcriptional regulation, we noted that individual knockdown of a set of human liver-enriched TRFs such as HNF1A, HNF3A, HNF3B, HNF3G and HNF4A resulted in perturbation of the expression of several single TRF genes, such as HNF1A, HNF3G and CEBPA genes. We thus searched the potential binding sites for these five TRFs in the highly conserved genomic regions around these three TRF genes and found several putative combinatorial regulatory regions. Chromatin immunoprecipitation analysis revealed that almost all of the putative regulatory DNA regions were bound by the TRFs as well as two coactivators (CBP and p300). The strong transcription-enhancing activity of the putative combinatorial regulatory region located downstream of the CEBPA gene was confirmed. EMSA demonstrated specific bindings of these HNFs to the target DNA region. Finally, co-transfection reporter assays with various combinations of expression vectors for these HNF genes demonstrated the transcriptional activation of the CEBPA gene in a combinatorial manner by these TRFs.
INTRODUCTION
In mammals, tens of thousands of protein-coding and nonprotein-coding genes are differentially expressed in specific cell types, during development, in the process of diseases and in response to various extracellular signals (1). Accumulating evidence indicates that the complex transcriptional regulatory patterns of mammalian genes are mediated via interactions of a set of transcriptional regulatory factors (TRFs) with their cognate cis-regulatory elements (2,3) (combinatorial transcriptional regulation). Combinatorial arrangement of multiple TRFs allows cells to finely control gene transcription and integrate multiple signal transduction pathways (4,5). Therefore, identification of the set of TRFs and their binding sites is essential for understanding the mechanism of combinatorial transcriptional regulation relevant to the biological event in question.
Current approaches to predicting and/or identifying combinatorial transcriptional regulatory sites have stemmed from coexpression analysis of TRFs and their target genes in a given tissue or cell, TRF-chromatin binding analysis and a computational search for possible TRF binding sites (TFBSs). Although several combinations of these methods and criteria can be applied to probe regulatory sites, highly reliable methods are needed to identify TRFs as well as their binding sites that are involved in the combinatorial regulation of a given gene.
In contrast to the proximal regulatory region located near the transcription start site (TSS) of each gene to be regulated, the distal regulatory regions such as enhancers and locus control regions (LCRs) are located at 5′- or 3′-flanking regions, intronic or even intergenic regions, which have often a few hundred kilo base pairs distant from the core promoter of the regulated gene (6,7), requires sophisticated strategies for their probing. Recent work has confirmed that, in general, individual TFBSs are conserved more than their surrounding DNA (8) and thus conserved noncoding sequences are good candidates for transcriptional regulatory elements (9). Hence, evolutionary sequence conservation has been recognized as an informative indicator of biological significance (10).
Another critical issue is to identify the TRFs engaged in combinatorial regulation. Knockdown of a specific TRF gene leads to perturbation of expression of its downstream genes and hence is a powerful search method (11–13). We have recently performed RNAi experiments in which TRF genes in a group are individually knocked down by specific respective siRNAs and the changes in their expression levels are quantified (submitted for publication). In these experiments, several TRF genes were simultaneously down- or upregulated accompanied by knockdown of each of a set of several TRFs. This finding suggested that it might be possible to predict the TRFs involved in combinatorial regulation of a given gene by examining its coincidental perturbation upon individual RNAi knockdown of a set of TRF genes.
Here, we present a successful application of this approach in combination with conventional approaches such as bioinformatic and TRF location analysis to characterize the combinatorial regulation of human TRF gene transcription. This approach led to detection of both known and/or novel combinatorial regulatory sites, which were targeted by several liver-enriched TRFs [HNF1A, HNF3A (FOXA1, HGNC symbol), HNF3B (FOXA2), HNF3G (FOXA3) and HNF4A] in the three liver-enriched TRF genes, HNF1A, CEBPA and HNF3G. The transcription-enhancing activity of and the specific TRF bindings to the deduced combinatorial regulatory region that was located downstream of the CEBPA gene were demonstrated by luciferase reporter assays, EMSA and co-transfection reporter assays.
MATERIALS AND METHODS
Cells and medium
HepG2 cells were obtained from the Japanese Collection of Research Bioresources (Osaka, Japan) and cultured in Minimum Essential Medium Eagle (Sigma, St Louis, MO) with 1 mM sodium pyruvate (Sigma) supplemented with 10% fetal bovine serum at 37°C in a 5% CO2 and 95% atmosphere(13).
Prediction of combinatorial transcriptional regulatory sites
To search for the potential TFBSs, we used PReMod(genomequebec.mcgill.ca/PReMod), a cis-regulatory module (CRM) prediction database (14,15), and rVISTA 2.0(/rVISTA.dcode.org/), a tool for analyzing the regulatory potential of noncoding sequences (16,17). We restricted our search for CRMs to regions that did not overlap with other target genes. HNF1A and 1B share the same recognition sequences. Similarly, HNF3 family members (3A, 3B and 3G) and HNF4 family members (4A and 4G) recognize their respective cognate sequences. In PReMod analysis, we searched for CRMs containing the HNF1, HNF3, HNF4 matrices in the region that PReMod database predicted on each of the target gene loci (HNF1A, NM_000545; HNF3G, NM_004497; CEBPA, NM_004364). In the PReMod analysis, putative human TFBSs are scored based on orthology with mouse and rat genomic sequences, which is assessed by the Multiz program (18).
We used precomputed pairwise alignments between human (hg17) and mouse (mm7) genomes available from the ECR Browser and GALA database (19–21) to extract the evolutionary conserved regions. In the conserved regions extracted, TFBSs that showed more than 90% sequence similarity were selected by rVISTA. To probe the TRF binding modules with rVISTA, we used a matrix similarity threshold of 0.85 for all of the HNF1, HNF3 and HNF4 matrices in the TRANSFAC v10.4 MATCH program (22,23). The used matrices were as follows: V$HNF1_01, V$HNF1_C, V$HNF1_Q6, V$HNF1_Q6_01, V$HNF1_C, V$HNF3ALPHA_Q6, VHNF3B_01, V$HNF3_Q6, V$HNF3_Q6_01, V$HNF4_01, V$HNF4_01_B, V$HNF4ALPHA_Q6, V$HNF4_DR1_Q3 for HNF1, HNF3, and HNF4, respectively. We selected regions including more than two kinds of TRF matrix in the target ones predicted by either of the two tools as potential combinatorial regulatory sites.
X-ChIP/qPCR
The HepG2 cell line was cultured to 70–80% confluency in MEM supplemented with 10% FBS and 1 × 108 cells were fixed with 1% formaldehyde at room temperature for 10 min. The reaction was stopped by incubation with 125 mM glycine for 5 min. The fixed cells were washed twice with PBS and resuspended in LB1 buffer as previously reported (24,25). After treatment with LB2 buffer, the cell pellets were resuspended in 5 ml of LB3 buffer. The resultant lysate was placed on ice and sonicated for 10 min with BranSon 450 Sonifier equipped with a cup horn filled with cold water (4°C) at 100% duty cycle. This yielded DNA fragments of 500 bp average length. The sonicated sample was diluted 2-fold with LB3 buffer and incubated at 4°C overnight with 5 μg of each of the TRF-specific antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, USA): HNF1A (sc-6547), HNF3A (sc-22841), HNF3B (sc-9187), HNF3G (sc-5361), HNF4A (sc-6556) and CBP (sc-369) and p300 (sc-584). All of these antibodies have been reported to be effective for X-ChIP experiments (24,26–29). Fifty microliters of protein-G magnetic beads (Dynal AS, Oslo, Norway) were added to the chromatin-antibody mixture and incubated for 2 h. The magnetic beads were washed once with low salt wash buffer, once with high salt wash buffer, once with LiCl wash buffer and twice with TE buffer as previously reported (30). The chromatin–antibody complexes were eluted twice with 1% SDS and 100 mM NaHCO3 at room temperature by 20-min vortexing and then mixed with 20 μl of 5 M NaCl. Reversal of the crosslinking and treatments with RNase and then proteinase K were carried out as previously described (11,30). The DNA samples were purified twice with phenol–chloroform–isoamylalcohol and chloroform–isoamylalcohol, and then the purified DNA was precipitated by isopropanol. The purified DNA was dissolved in 150 μl of distilled water for the qPCR reaction.
PCR primer sequences are described in Supplementary Table 1. All the primers were designed by Primer 3 (31). The DNA segments (2 μl) were quantified by qPCR in a 10 -μl reaction mixture containing SYBR green perfect real-time PCR mix (Takara, Japan) in an ABI 7500 fast system (ABI). PCR reaction was carried out on an ABI PRISM 7500 Fast real-time PCR System (Applied Biosystems) by denaturation at 95°C for 10 s, followed by running at 95°C for 3 s and 62.5°C for 20 s for 40 cycles. As controls, we amplified 1 ng of input DNA and an exon of the human GAPDH gene was amplified. The enrichment of the DNA fragments corresponding to the TRF binding regions was evaluated in triplicate by calculating ΔΔCT values as follows. The difference (ΔCTsample) in CT values observed with input DNA (1 ng of chromatin DNA) and with the immunoprecipitates obtained by ChIP in the qPCR by using the target site-specific primers was first calculated. Similarly, the difference in CT values (ΔCTcalibrator) was calculated by subtracting the CT observed with input DNA from CT observed with ChIP sample by using the calibrator (GAPDH)-specific primers. Finally, the difference between ΔCTsample and ΔCTcalibrator (ΔΔCT) was calculated. We carried out three independent experiments for each region with duplicate PCR reactions. The threshold criteria used were ΔΔCT ≥ 1, average ΔΔCT ≥ 2× standard deviation (SD) and P-value <0.05 in Student's t-test.
Comparative PCR
Purified DNA from X-ChIP as in X-ChIP/qPCR methods was used for comparative PCR with specific primers (CEBPA_4) described in Supplementary Table 1 to amplify the fragments containing our putative region. PCR was conducted in a mixture containing 5 μl of purified DNA (totally 150 μl) or input DNA in several dilutions, 0.2 μM of each primer, 1 mM dNTP mixture, 1.5 mM MgCl2 and 1.25 U of Ex Taq DNA polymerase (Takara) in a total volume of 50 μl. The PCR condition was as follows: a denaturation by 95°C for 30 s followed by 95°C for 10 s and 68°C 30 s for 25 to 40 cycles in stepwise to check the optical PCR cycles. Amplified DNA (34 cycles) was electrophoresed in a 3% agarose gel. The precipitates obtained without antibodies (no anti) and genome DNA (input) with several dilutions were also used for PCR amplification as the controls.
ChIP–chip analysis
We used 3 × 106 cells of HepG2 cells for X-ChIP with 10 μg of HNF3A/B antibody (sc-6553). Amplification, hybridization with an Affymetrix tiling array (GeneChip Chromosome 21/22 1.0 Array Set, c-chip), and data extraction protocol were previously described (32). In qPCR experiments, 1 ng of the X-ChIP samples and 1 ng of the input chromatin samples were used for amplification of the target DNA regions with specific primer sets. The enrichment of DNA fragments (ΔCT) was calculated by subtracting the CT observed with the ChIP sample from CT with the input chromatin DNA. A total of 35 HNF3 binding sites with a P-value <10–4 were assigned to the conserved and nonconserved DNA regions on a part of the human chromosomes 22. For the sequence alignment to find conserved regions meeting criteria for the conservation (minimal identity of 70% in minimum length of 100 bp), the ECR browser (19) was used.
Luciferase reporter assay and co-transfection assay
The CEBPA promoter region (–426 to +7 relative to the transcription start site) was amplified by PCR from HepG2 genomic DNA using promoter_XhoI_f (5′-AAACTCGAGGACTCCATGGGGGAGTTAGAG-3′) flanked by an XhoI site and promoter_SacI_r (5′-AAAGAGCTCTAGGGCAGGCAGGAGGAG-3′) flanked by a SacI site as primers. PCR parameters consisted of 5-min incubation at 95°C for denaturation, followed by 30 cycles of 95°C for 30 s and 68°C for 30 s using KOD-Plus- (TOYOBO) with a final concentration of 2% DMSO. PCR products were electrophoresed in agarose gels and purified. After digestion of the PCR product by XhoI and SalI, the fragment was ligated to the luciferase gene in pGL4.12 [luc2CP] Vector (Promega) to construct a ‘promoter vector’. To construct the plasmid for estimating the enhancing activity of the combinatorial regulatory region (CEBPA_4; +26094–+26416, relative to the TSS) deduced, we placed it downstream of the reporter gene that was expressed under The deduced regulatory region was amplified by PCR using infusion_f (5′-AAATCGATAAGGATCCTCCACCTCAGCAGCAAAG-3′) and infusion_r (5′-ATCGGTCGACGGATCGTGGGGAAAGACGATGTCAG-3′) as primers and KOD-Plus-. The thermal profile for PCR was 95°C for 5 min, followed by 30 cycles of 95°C for 30 s and 68°C for 30 s. The PCR product (353 bp) was purified as described above and cloned into the BamHI site of the ‘promoter vector’ using In-Fusion PCR Cloning Kit (ClonTech) so as to be flanked by the end of the luciferase gene to construct a ‘promoter-enhancer vector’. To generate three kinds of mutant ‘promoter-enhancer vectors’ for each of the HNF1, 3, 4 putative binding sites we used GeneTailor™ Site-Directed Mutagenesis System (Invitrogen). All of the primer sequences used for mutagenesis are described in Supplementary Table 2. Every mutant was sequenced for verification.
Luciferase reporter assays were performed in HepG2 cells. Cells (1 × 104) were seeded onto each well of 96-well plates and transfected with 200 ng of the plasmids on the following day using Lipofectamine2000 (Invitrogen). Cells were harvested 48 h after transfection and the luciferase activities were determined by a luminometer using Steady-Glo® Luciferase Assay System (Promega).
Human HNF1A, HNF3B, HNF4A cDNAs were PCR amplified from HepG2 transcripts and cloned into NheI site of pcDNA3.1(+) vector (Invitrogen) using In-Fusion PCR Cloning Kit (ClonTech). Positive clones were sequenced for verification. Co-transfection assays were performed in HeLa cells. Cells (1 × 104) were seeded onto each well of 96-well plates and transfected with 200 ng of the ‘promoter-enhancer vector’ and various combinations of HNF expression vectors for HNF1A, HNF3B, and HNF4A (50 ng) or pcDNA3.1(+) vector without any insert (control) with Lipofectamine2000 (Invitrogen) on the following day. Cells were harvested 48 h after transfection and the luciferase activities were determined as described above.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared from HepG2 cells by using the NE-PER nuclear and cytoplasmic extraction reagent according to the manufacturer's protocol (Pierce). The total protein concentration was determined by the Bradford assay (Bio-Rad) with BSA as standard. EMSA was performed with a series of double-stranded (ds-) oligonucleotides containing all (70 bp) or some of the HNF1, HNF3 and HNF4 recognition sequences derived from the deduced CEBPA combinatorial regulatory region (Supplementary Table 2) as probes. The 70-bp probe was PCR-amplified with a specific primer set (Supplementary Table 2) and purified by gel extraction. Mutant oligonucleotides that had the same nucleotide substitutions as those tested in the luciferase assay were used as competitive inhibitors. In addition, the consensus binding sequences for HNF1, HNF3 and HNF4 (33) were prepared to be used as additional specific competitors. To obtain ds-oligonucleotides, two complementary oligonucleotides (2.5 μM each) were annealed by heating at 80°C for 10 min and slowly cooling down to room temperature. Annealed ds-oligonucleotides were 5′-end-labeled at 37°C for 2 h in the reaction mixture (20 μl) containing 1× T4 polynucleotide kinase (PNK) buffer (Takara), 0.25 μM ds-oligonucleotides or 70-bp probe, 0.74 MBq [γ-32P]ATP (111 TBq/mmol), 10 U of T4 PNK. The labeled probes were purified by using Nick columns(GE Healthcare). Approximately 5 μg of the nuclear extracts were incubated for 30 min at room temperature (for 1 h at 4°C for the EMSA with HNF4 probes) in 1× binding buffer containing 20 mM HEPES (pH 7.9), 12% glycerol, 40 mM KCl, 2 mM EDTA, 0.5 mM DTT, 0.5 μg of poly (dI–dC) and the labeled probe (0.05 pmol) in a final volume of 10 μl. In competition or supershift assays, 100-fold molar excess of cold (non-labeled) ds-oligonucleotides or 4 μg of the specific antibody raised against each of the HNFs (described in X-ChIP/qPCR) were added. The incubation mixtures were electrophoresed at 4°C on 7% polyacrylamide Tris–glycine gels, which were precooled at 4°C, at 200 V for 2.5–3 h (at 300 V for 80 min for the EMSA with HNF4 probes). Gels were dried, exposed to BAS2000 filters with BAStation software (Fuji Photo Film, Kanagawa, Japan).
RESULTS
Prediction of multiple TRF binding regions
In our recent study on RNAi knockdown of liver-enriched TRFs, we observed that suppression of liver-enriched TRFs (HNF1A, HNF3A, HNF3B, HNF3G and HNF4A) induced clear downregulation of HNF1A, HNF3G and CEBPA genes in HepG2 cells (submitted). Because such multiple inputting hubs appear to be a good indicator of combinatorial transcriptional regulation, a set of these TRFs and TRF genes were chosen for the subsequent analysis. Various experimental and computational studies have suggested that conserved noncoding sequences are good candidates for transcriptional regulatory elements (17). To test this, we first analyzed the in vivo binding of HNF3A/HNF3B as a model by using a tiling array.
A ChIP–chip experiment with an antibody specific for HNF3A/HNF3B detected 35 peaks (sites) in a 35-Mb region, a portion of the human chromosome 22, with a P-value < 10–4. We then compared the enrichment between DNA fragments in the X-ChIP immunoprecipitates derived from the conserved regions and those from the nonconserved regions with qPCR. Proper primers could not be designed for two out of the 35 sites. Human–mouse genome comparison analysis revealed that 19 and 14 sites were located in a conserved and a nonconserved region, respectively, and mostly in intronic as well as intergenic conserved regions. Figure 1 clearly shows more enrichment of DNA fragments from the conserved regions over those from the nonconserved regions with the P-value of 0.009 for the paired t-test. This finding drove us to develop a system for identifying regulatory sites including combinatorial regulatory ones in evolutionarily conserved noncoding regions in the human genome.
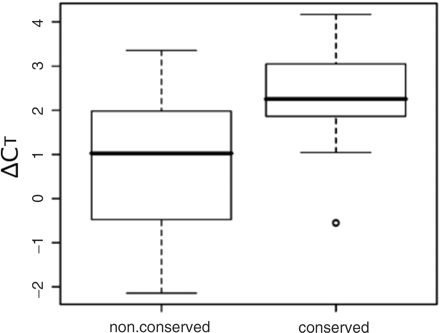
Comparison of enrichment of HNF3A/HNF3B targets between the TFBSs in the conserved and nonconserved regions on a part of the human chromosome 22. ChIP–chip analysis was performed by using an Affymetrix tiling array (GeneChip Chromosome 21/22 1.0 Array Set, c-chip). A total of 33 sites that exhibited a P-value <10–4 were detected and they were divided in two groups according to their localization within conserved (19 sites) and nonconserved (14 sites) regions. The conserved and nonconserved regions between human and mouse genome were defined by the analysis with zPicture program, an option of the ECR browser. The enrichment of each target site was assessed by X-ChIP/qPCR with specific primer sets measuring ΔCT, the difference between the threshold cycles observed with input chromatin DNA (1 ng) and with ChIP sample recovered by using anti-HNF3 antibody recognizing HNF3A and HNF3B. The median, upper and lower quartiles and minimum and maximum values of ΔCT are shown in the boxplot. The outlier value is plotted with a small open circle.
Next, we analyzed the potential HNF binding sites in the DNA regions conserved in and around the human HNF1A, HNF3G and CEBPA genes. Many prediction tools have been developed for searching the TFBSs in combination with genomic conservation analysis. Among the currently available tools, we first comparatively examined two methods, PReMod and rVISTA, to predict the combinatorial regulatory regions because both data sets are not restricted to modules or regions located proximal to genes, but mostly contain distal predicted CRMs or regions. Both PReMod and rVISTA use position weight matrices (PWMs) of TRANSFAC for probing TFBSs. PReMod is a database of genome-wide CRM predictions within the human genome and 481 TRANSFAC version 7.2 PWMs for vertebrate TRFs are used to score putative TFBSs based on how well the human site and its orthologs in mouse and rat match the matrix (14,15). rVISTA combines clustering of the predicted TFBSs and analysis of interspecies sequence conservation to maximize the identification of functional regulatory sites (17). In the present study, we used human- and mouse-genome comparison to analyze conserved DNA regions related to combinatorial transcriptional regulation.
To predict the transcriptional regulatory regions by PReMod, we used data sets of the UCSC hg17 human genome assembly. We conducted locus-wide searches for modules in the DNA regions of 98 kb (chr12:119804871-119903567 in hg17), 130 kb (chr19:38408548-38545500) and 20 kb (chr19:51058207-51078705) that were predicted by PReMod in or around each of HNF1A, HNF3G, and CEBPA genes, respectively. Next, we probed each module which included the consensus matrices of HNF1, HNF3 and HNF4. In the rVISTA analysis, we also checked the HNF1, HNF3 and HNF4 matrices in the conserved regions of the three target genes that were predicted by the ECR Browser alignment analysis (see Materials and methods section).
The results of the analysis of TFBSs in the conserved regions of the HNF1A, HNF3G and CEBPA genes are summarized in Table 1. A typical transcriptional regulatory region, having three potential binding sequences for the HNF1, HNF3 and HNF4, was predicted in the proximal promoter of the HNF1A gene by both PReMod and rVISTA (mod028501 and HNF1A_1). In addition, two possible conserved regulatory regions were also detected in the downstream regions of the HNF1A gene, and these regions predicted by PReMod and rVISTA were consistent with each other in terms of their locations and TFBSs (mod028503 and HNF1A_2; mod028502 and HNF1A_4). On the other hand, either PReMod or rVISTA detected a set of potential regulatory regions around the CEBPA gene but their respective locations were quite different and did not overlap with each other. PReMod detected three potential regulatory sites: one is located in the upstream region and the other two in the downstream region of the human CEBPA gene (mod054831). rVISTA detected two sites containing HNF3 and either HNF4 or HNF1 consensus sequences in the downstream region of the CEBPA gene (CEBPA_1, CEBPA_4).
Conserved DNA regions and HNF binding sites predicted in and around the human HNF1A, HNF3G and CEBPA genes
| Predicted | Validated | Predicted | Validated | Validated | Validated | Predicted | Validated | Validated | Validated | Location | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Site | HNF1 | HNF1A | HNF3 | HNF3A | HNF3B | HNF3G | HNF4 | HNF4A | CBP | P300 | from TSS (bp) |
| HNF1A | HNF1AJ | + | + | + | + | + | + | + | + | + | −441 ∼ +424 | |
| HNF1A4 | + | + | + | + | + | +2176 ∼ +2508 | ||||||
| mod028501 | + | + | + | + | + | + | + | + | + | −459 ∼ −99 | ||
| mod028503 | + | + | + | +1059 ∼ +1161 | ||||||||
| mod028502 | + | + | + | + | + | +2209 ∼ +2350 | ||||||
| HNF3G | HNF3GJ | + | + | + | + | + | + | +9430 ∼ +9808 | ||||
| HNF3G 2 | + | + | + | + | + | + | + | + | + | +17703 ∼ +18018 | ||
| CEBPA | CEBPA_1 | + | + | + | + | + | + | + | + | + | +55013 ∼ +54674 | |
| CEBPA_4 | + | + | + | + | + | + | + | + | + | + | +26338 ∼ +26111 | |
| mod054825 | + | + | + | + | +20066 ∼ +19896 | |||||||
| mod054829 | + | + | + | + | + | +2666 ∼ +1978 | ||||||
| mod054831 | + | + | + | −1230 ∼ −1983 |
| Predicted | Validated | Predicted | Validated | Validated | Validated | Predicted | Validated | Validated | Validated | Location | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Site | HNF1 | HNF1A | HNF3 | HNF3A | HNF3B | HNF3G | HNF4 | HNF4A | CBP | P300 | from TSS (bp) |
| HNF1A | HNF1AJ | + | + | + | + | + | + | + | + | + | −441 ∼ +424 | |
| HNF1A4 | + | + | + | + | + | +2176 ∼ +2508 | ||||||
| mod028501 | + | + | + | + | + | + | + | + | + | −459 ∼ −99 | ||
| mod028503 | + | + | + | +1059 ∼ +1161 | ||||||||
| mod028502 | + | + | + | + | + | +2209 ∼ +2350 | ||||||
| HNF3G | HNF3GJ | + | + | + | + | + | + | +9430 ∼ +9808 | ||||
| HNF3G 2 | + | + | + | + | + | + | + | + | + | +17703 ∼ +18018 | ||
| CEBPA | CEBPA_1 | + | + | + | + | + | + | + | + | + | +55013 ∼ +54674 | |
| CEBPA_4 | + | + | + | + | + | + | + | + | + | + | +26338 ∼ +26111 | |
| mod054825 | + | + | + | + | +20066 ∼ +19896 | |||||||
| mod054829 | + | + | + | + | + | +2666 ∼ +1978 | ||||||
| mod054831 | + | + | + | −1230 ∼ −1983 |
Both of PReMod and rVISTA were used to predict the HNF1A, FOXA and HNF4A binding sites in each of the evolutionary conserved regions. + in the predicted TRF columns indicates that the potential TFBS was detected, – indicates no potential TFBS. + in the validated TRF and cofactor columns indicates that chromatin binding was demonstrated, – indicates no significant binding detected. Location relative to the TSS is indicated in base pair.
Conserved DNA regions and HNF binding sites predicted in and around the human HNF1A, HNF3G and CEBPA genes
| Predicted | Validated | Predicted | Validated | Validated | Validated | Predicted | Validated | Validated | Validated | Location | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Site | HNF1 | HNF1A | HNF3 | HNF3A | HNF3B | HNF3G | HNF4 | HNF4A | CBP | P300 | from TSS (bp) |
| HNF1A | HNF1AJ | + | + | + | + | + | + | + | + | + | −441 ∼ +424 | |
| HNF1A4 | + | + | + | + | + | +2176 ∼ +2508 | ||||||
| mod028501 | + | + | + | + | + | + | + | + | + | −459 ∼ −99 | ||
| mod028503 | + | + | + | +1059 ∼ +1161 | ||||||||
| mod028502 | + | + | + | + | + | +2209 ∼ +2350 | ||||||
| HNF3G | HNF3GJ | + | + | + | + | + | + | +9430 ∼ +9808 | ||||
| HNF3G 2 | + | + | + | + | + | + | + | + | + | +17703 ∼ +18018 | ||
| CEBPA | CEBPA_1 | + | + | + | + | + | + | + | + | + | +55013 ∼ +54674 | |
| CEBPA_4 | + | + | + | + | + | + | + | + | + | + | +26338 ∼ +26111 | |
| mod054825 | + | + | + | + | +20066 ∼ +19896 | |||||||
| mod054829 | + | + | + | + | + | +2666 ∼ +1978 | ||||||
| mod054831 | + | + | + | −1230 ∼ −1983 |
| Predicted | Validated | Predicted | Validated | Validated | Validated | Predicted | Validated | Validated | Validated | Location | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Site | HNF1 | HNF1A | HNF3 | HNF3A | HNF3B | HNF3G | HNF4 | HNF4A | CBP | P300 | from TSS (bp) |
| HNF1A | HNF1AJ | + | + | + | + | + | + | + | + | + | −441 ∼ +424 | |
| HNF1A4 | + | + | + | + | + | +2176 ∼ +2508 | ||||||
| mod028501 | + | + | + | + | + | + | + | + | + | −459 ∼ −99 | ||
| mod028503 | + | + | + | +1059 ∼ +1161 | ||||||||
| mod028502 | + | + | + | + | + | +2209 ∼ +2350 | ||||||
| HNF3G | HNF3GJ | + | + | + | + | + | + | +9430 ∼ +9808 | ||||
| HNF3G 2 | + | + | + | + | + | + | + | + | + | +17703 ∼ +18018 | ||
| CEBPA | CEBPA_1 | + | + | + | + | + | + | + | + | + | +55013 ∼ +54674 | |
| CEBPA_4 | + | + | + | + | + | + | + | + | + | + | +26338 ∼ +26111 | |
| mod054825 | + | + | + | + | +20066 ∼ +19896 | |||||||
| mod054829 | + | + | + | + | + | +2666 ∼ +1978 | ||||||
| mod054831 | + | + | + | −1230 ∼ −1983 |
Both of PReMod and rVISTA were used to predict the HNF1A, FOXA and HNF4A binding sites in each of the evolutionary conserved regions. + in the predicted TRF columns indicates that the potential TFBS was detected, – indicates no potential TFBS. + in the validated TRF and cofactor columns indicates that chromatin binding was demonstrated, – indicates no significant binding detected. Location relative to the TSS is indicated in base pair.
Although PReMod did not detect any conserved region containing potential sites for binding of the TRFs in and around the HNF3G gene, rVISTA predicted a conserved 3′-downstream region containing potential HNF1A and HNF3 binding sites.
Several HNFs bind to the predicted same genomic regions
Next, we experimentally probed the combinatorial binding of the HNF proteins to the conserved regions detected by PReMod or rVISTA. For this purpose, we selected predicted regions containing at least two kinds of matrix for the HNFs (HNF1A, HNF3s and HNF4A). Their bindings to the predicted DNA regions were investigated by X-ChIP/qPCR with specific antibodies against respective HNFs. Because HNF3s share their binding sequence, we used specific antibodies to discriminate chromatin bindings among HNF3A, HNF3B and HNF3G. Target-specific primer sets, each of which was able to amplify the DNA region covering the TFBSs, were used for qPCR. We also made primer sets to amplify the DNA regions located ∼500 bp downstream or upstream of the target regions to evaluate the levels of nonspecific immunoprecipitation as the backgrounds.
All of the three conserved sites predicted by PReMod overlapped with three out of four sites predicted by rVISTA in the HNF1A proximal promoter and intronic regions (Figure 2). Because all of these three overlapping sites are located in an ∼3.9- kb DNA region, we examined the HNF occupancy in this region with eight primer sets, four of which were designed to cover each of the overlapping sites. X-ChIP/qPCR analysis revealed that all of the HNFs (HNF1A, HNF3A, HNF3B, HNF3G and HNF4A) were bound to the predicted site (mod028501 and HNF1A_1) in the proximal promoter of the HNF1A gene (Figure 2). In addition, binding of all HNFs, except HNF4A, to the farthest predicted site (mod028502 and HNF1A_4) in the first intron was also detected (Figure 2). All of the binding data were consistent with the predictions of the TFBSs made by both rVISTA and PReMod. However, we did not detect significant binding of any of the HNFs to two other intronic sites.
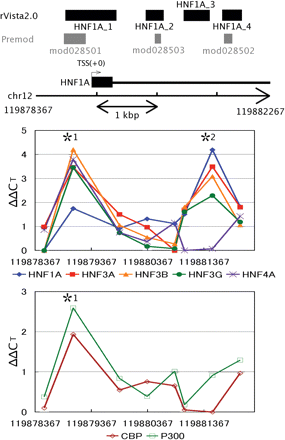
HNFs (HNF1A, HNF3A, HNF3B, HNF3G and HNF4A) occupancy analysis in proximal upstream region of and within the human HNF1A gene. X-ChIP/qPCR analysis was carried out with specific primer sets covering each of the potential HNF binding sites detected in the conserved regions. The enrichment of the target sites was evaluated as the differences between ΔCTsample and ΔCTcalibrator (ΔΔCT) (see ‘Materials and methods’ section for the details). SD values of the differences were calculated and the paired Student's t-tests were made to calculate P-values. The normalized mean ΔΔCT value is plotted against the nucleotide position at the center of each of the corresponding DNA segments amplified (see Supplementary Table 1 for details). The lowest ΔΔCT was normalized to 0 in the region of each TRF. Coincidental enrichment of the target sites with antibodies specific to cofactors, p300 and CBP, was also assessed with the same chromatin samples. In vivo bindings of HNF1A, HNF3B, HNF3G, HNF4A, CBP and p300 to the *1 region and HNF1A, HNF3B and HNF3G to the *2 region cleared the following criteria: mean ΔΔCT ≥ 1.0, mean ΔΔCT ≥ 2× SD, and P < 0.01. Base positions are indicated at both sides of the chromosome fragment and beneath the x-axes.
No other transcript has been detected in the 130-kb region encompassing the human CEBPA gene that has no intron and the DNA region covering all the predicted regulatory sites was extended more than 56 kb. Thus, we designed five groups of primer sets for X-ChIP/qPCR analysis: each of the groups consisted of 3–4 primer sets to amplify a 1.5–2.5-kb region containing any of the potential regulatory sites. Only two (CEBPA_1 and _4, predicted by rVISTA) out of the five CEBPA potential regulatory sites predicted were found to be actually bound by all of the HNFs examined (Figure 3A). These sites are located ∼26- and 55-kb downstream of the CEBPA TSS. The rVISTA prediction and the X-ChIP/qPCR result at the +26-kb site completely coincided with each other. Enrichment by X-ChIP of the DNA fragments corresponding to this site was also confirmed by comparative PCR analysis (Figure 3B). Figure 4 shows the nucleotide sequences of HNF1A_1 and CEBPA_4, for both of which the rVISTA prediction and experimental data are perfectly consistent with each other.
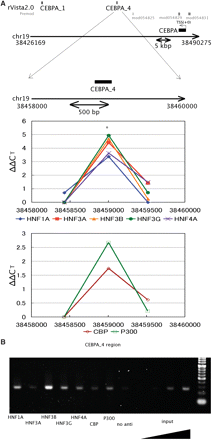
HNFs occupancy analysis in upstream and downstream regions of the human CEBPA gene. (A) The experimental setting is the same as that in Figure 2. In the region with an asterisk, in vivo bindings of HNF1A, HNF3A, HNF3B, HNF3G, HNF4A, CBP and p300 cleared the criteria for enrichment as described in the footnote of Figure 2. (B) DNA samples recovered from the X-ChIP as in (A) were amplified by PCR and electrophoresed in a 3% agarose gel. The precipitates obtained without antibodies (no anti) and genome DNA (input) were also used for PCR amplification as the controls.
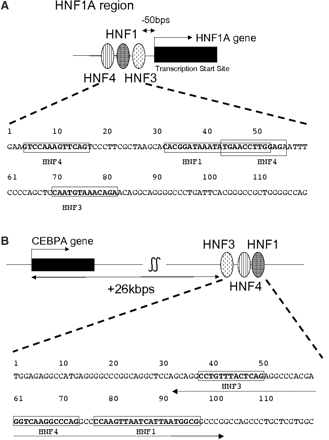
Nucleotide sequences of some of the putative combinatorial regulatory regions identified. Sequence corresponding to the TRANSFAC matrix for each TRF binding is indicated by boldface in a rectangle. (A) HNF1A_1 (nucleotide number 1 corresponds to −267 relative to the TSS); (B) CEBPA_4 (nucleotide number 1 corresponds to +26278 relative to the TSS). The dotted line with arrows at both sides shows the region used as the 70-bp probe in EMSA.
The X-ChIP/qPCR assay also showed the bindings of all of the HNFs tested in a region about 17 kb downstream (HNF3G_2 in rVISTA analysis) from the TSS of HNF3G gene (Figure 5). The DNA stretch corresponding to the HNF3G_2 was a syntenic region that was previously identified as an enhancer of the mouse HNF3G gene by reporter assay in rat cultured cells (34).
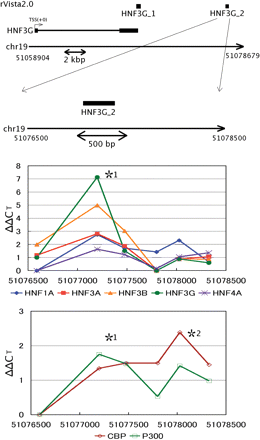
HNFs occupancy analysis in the 3′-flanking and far downstream regions of the human HNF3G gene. The experimental setting is the same as that in Figure 2. In vivo bindings of HNF1A, HNF3A, HNF3B, HNF3G, HNF4A, CBP and p300 to the *1 region and the only cofactors (CBP and p300) to the *2 region cleared the enrichment criteria as described in the footnote of Figure 2.
CBP and p300 coincidentally bind to almost all the predicted conserved regions
Transcriptional regulation is known to be coupled with TRF–cofactor interactions and cofactors can be immunoprecipitated with DNA fragments derived from transcriptional regulatory regions by their specific antibodies (35). Thus, we next examined the coincidental immunoprecipitation of the DNA fragments, which were derived from the sites that were strongly suggested by RNAi knockdown, rVISTA prediction and X-ChIP–qPCR analysis to be involved in the transcriptional regulation of the three TRF genes, with well-known cofactors CBP and p300. X-ChIP/qPCR assays clearly showed the concomitant recovery of two cofactors with almost all of these sites (five in six sites; Figures 2, 3 and 5; Table 1). Based on the high probability of coincidental immunoprecipitation of the cofactor- and HNF-bound DNA fragments, their common binding sites can be expected to correspond to the potential combinatorial regulatory sites.
Overall, we identified four potential combinatorial regulatory sites that had binding matrices for all of the three TRFs and were actually bound by these transcriptional regulators (summarized in Table 1). TRF binding to all but one of the 14 binding sites predicted by rVISTA were experimentally confirmed and three additional sites that were not predicted were also detected. On the other hand, 7 out of the 15 TFBSs predicted by PReMod were bound by the cognate TRFs and two additional sites were detected. Based on the superior prediction, rVISTA is conclusively considered as the tool of choice for probing the potential combinatorial regulatory sites.
Preferential TRF binding to DNA regions with clustered binding sites
We focused on the DNA regions where multiple (clustered) HNF binding sites resided and identified their involvement in combinatorial transcriptional regulation. On the other hand, a number of potential HNF binding sites that are not clustered are found in the noncoding regions in and around the HNF1A, HNF3G and CEBPA genes. The question arises of whether these sites are bound by each of the corresponding HNFs and involved in transcriptional regulation. If this is the case, the DNA fragments containing each of the single TFBSs should be recovered with efficiencies comparable to those observed for multiple TFBSs. We then examined the enrichment of DNA fragments derived from the CEBPA gene in which we identified many single TFBSs as well as multiple TFBSs focusing on HNF3s and HNF4A bindings.
rVISTA detected 18 sites containing at least one HNF1, HNF3 or HNF4 matrix in the 130-kb CEBPA gene locus: 14 sites having only a single HNF3 consensus sequence, two sites having an isolated HNF4-recognizable sequence and two sites having HNF3 and HNF4A recognizable sequences (Table 2). We evaluated the enrichment of the specific DNA fragments containing a multiple TRF site or an isolated TRF site by calculating the ΔΔCT values (see ‘Materials and methods’ section). As a result, the isolated binding sites for any of the TRFs examined (HNF3A, HNF3B, HNF3G and HNF4A) exhibited less enrichment of the corresponding DNA fragments than those of multiple TFBSs (Figure 6). This indicates that these TRFs are bound to the potential combinatorial regulatory sites more frequently than to the isolated TRF-recognizable sites.
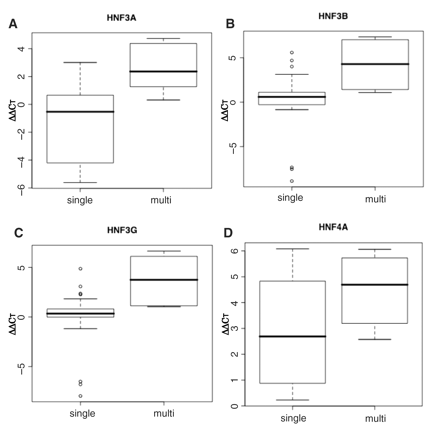
Preferential HNF binding to the putative combinatorial regulatory sites of the human CEBPA gene. The putative combinatorial regulatory sites (multi) correspond to the composite TFBSs 1 and 4 and the remaining 16 sites (single) to the isolated sites in Table 2. The enrichment of each target site was assessed by X-ChIP/qPCR with specific primer sets measuring ΔΔCT, the difference between the threshold cycles observed with input chromatin DNA (1 ng) and with ChIP sample recovered by using antibody raised against HNF3A, HNF3B, HNF3G or HNF4A. The median, upper and lower quartiles and minimum and maximum values of ΔΔCT are shown in the boxplot. Double-sided P-values for the Student's paired t-tests were 0.0007, 0.001, 0.0009 and 0.18 for the bindings of HNF3A, HNF3B, HNF3G and HNF4A, respectively.
Prediction of composite and isolated potential HNF binding sites in the DNA region surrounding the human CEBPA gene and experimental validation of their interactions with the corresponding HNF proteins
| Predicted | Validated | Predicted | Validated | Validated | Validated | Predicted | Validated | Validated | Validated | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Site | HNF1 | HNF1A | HNF3 | HNF3A | HNF3B | HNF3G | HNF4 | HNF4A | CBP | P300 |
| CEBPA | CEBPA_1 | + | + | + | + | + | + | + | |||
| CEBPA_2 | + | ||||||||||
| CEBPA_3 | + | + | + | ||||||||
| CEBPA_4 | + | + | + | + | + | + | + | + | + | + | |
| CEBPA_5 | + | + | + | + | + | ||||||
| CEBPA_6 | + | + | |||||||||
| CEBPA_7 | + | + | + | ||||||||
| CEBPA_8 | + | + | + | + | |||||||
| CEBPA_9 | + | + | + | + | + | + | + | + | |||
| CEBPA_10 | + | ||||||||||
| CEBPA_11 | + | + | |||||||||
| CEBPA_12 | + | ||||||||||
| CEBPA_13 | + | ||||||||||
| CEBPA_14 | + | ||||||||||
| CEBPA_15 | + | + | |||||||||
| CEBPA_16 | + | ||||||||||
| CEBPA_17 | + | ||||||||||
| CEBPA_18 | + |
| Predicted | Validated | Predicted | Validated | Validated | Validated | Predicted | Validated | Validated | Validated | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Site | HNF1 | HNF1A | HNF3 | HNF3A | HNF3B | HNF3G | HNF4 | HNF4A | CBP | P300 |
| CEBPA | CEBPA_1 | + | + | + | + | + | + | + | |||
| CEBPA_2 | + | ||||||||||
| CEBPA_3 | + | + | + | ||||||||
| CEBPA_4 | + | + | + | + | + | + | + | + | + | + | |
| CEBPA_5 | + | + | + | + | + | ||||||
| CEBPA_6 | + | + | |||||||||
| CEBPA_7 | + | + | + | ||||||||
| CEBPA_8 | + | + | + | + | |||||||
| CEBPA_9 | + | + | + | + | + | + | + | + | |||
| CEBPA_10 | + | ||||||||||
| CEBPA_11 | + | + | |||||||||
| CEBPA_12 | + | ||||||||||
| CEBPA_13 | + | ||||||||||
| CEBPA_14 | + | ||||||||||
| CEBPA_15 | + | + | |||||||||
| CEBPA_16 | + | ||||||||||
| CEBPA_17 | + | ||||||||||
| CEBPA_18 | + |
All of these HNF binding sites were predicted by rVISTA. CEBPA_1 and _4 were predicted to be composite TFBSs and the remaining sites were predicted to be isolated ones. + indicates that the TFBSs were predicted or the TRF bindings were demonstrated.
Prediction of composite and isolated potential HNF binding sites in the DNA region surrounding the human CEBPA gene and experimental validation of their interactions with the corresponding HNF proteins
| Predicted | Validated | Predicted | Validated | Validated | Validated | Predicted | Validated | Validated | Validated | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Site | HNF1 | HNF1A | HNF3 | HNF3A | HNF3B | HNF3G | HNF4 | HNF4A | CBP | P300 |
| CEBPA | CEBPA_1 | + | + | + | + | + | + | + | |||
| CEBPA_2 | + | ||||||||||
| CEBPA_3 | + | + | + | ||||||||
| CEBPA_4 | + | + | + | + | + | + | + | + | + | + | |
| CEBPA_5 | + | + | + | + | + | ||||||
| CEBPA_6 | + | + | |||||||||
| CEBPA_7 | + | + | + | ||||||||
| CEBPA_8 | + | + | + | + | |||||||
| CEBPA_9 | + | + | + | + | + | + | + | + | |||
| CEBPA_10 | + | ||||||||||
| CEBPA_11 | + | + | |||||||||
| CEBPA_12 | + | ||||||||||
| CEBPA_13 | + | ||||||||||
| CEBPA_14 | + | ||||||||||
| CEBPA_15 | + | + | |||||||||
| CEBPA_16 | + | ||||||||||
| CEBPA_17 | + | ||||||||||
| CEBPA_18 | + |
| Predicted | Validated | Predicted | Validated | Validated | Validated | Predicted | Validated | Validated | Validated | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Site | HNF1 | HNF1A | HNF3 | HNF3A | HNF3B | HNF3G | HNF4 | HNF4A | CBP | P300 |
| CEBPA | CEBPA_1 | + | + | + | + | + | + | + | |||
| CEBPA_2 | + | ||||||||||
| CEBPA_3 | + | + | + | ||||||||
| CEBPA_4 | + | + | + | + | + | + | + | + | + | + | |
| CEBPA_5 | + | + | + | + | + | ||||||
| CEBPA_6 | + | + | |||||||||
| CEBPA_7 | + | + | + | ||||||||
| CEBPA_8 | + | + | + | + | |||||||
| CEBPA_9 | + | + | + | + | + | + | + | + | |||
| CEBPA_10 | + | ||||||||||
| CEBPA_11 | + | + | |||||||||
| CEBPA_12 | + | ||||||||||
| CEBPA_13 | + | ||||||||||
| CEBPA_14 | + | ||||||||||
| CEBPA_15 | + | + | |||||||||
| CEBPA_16 | + | ||||||||||
| CEBPA_17 | + | ||||||||||
| CEBPA_18 | + |
All of these HNF binding sites were predicted by rVISTA. CEBPA_1 and _4 were predicted to be composite TFBSs and the remaining sites were predicted to be isolated ones. + indicates that the TFBSs were predicted or the TRF bindings were demonstrated.
We also compared the occupancy of CBP and p300 at the isolated and multiple TRF sites and again found the preferential binding of these cofactors at multiple sites over the isolated TRF sites. More significantly, the multiple TRF bindings at sites CEBPA_5, 8 and 9 were not computationally predicted but the X-ChIP/qPCR assay with a combination of multiple TRFs and cofactor antibodies clearly detected their bindings. These results indicate much higher commitment of the regions with clustered TFBSs to transcriptional regulation of mammalian genes.
The novel putative combinatorial regulatory region of the CEBPA gene has a strong transcription-enhancing activity
To examine the stimulatory activity of the putative combinatorial regulatory region deduced by DNA conservation, RNAi knockdown and X-ChIP experimental data, we performed the luciferase reporter assay in HepG2 cells. In the assay, the 433-bp DNA fragment corresponding to the human CEBPA proximal promoter (–426 to +7) were inserted in front of the firefly luciferase reporter gene in the pGL4.12 reporter vector to construct a ‘promoter vector’. We also made a ‘promoter-enhancer vector’ in which the deduced regulatory region (CEBPA_4 and its flanking regions) was inserted at the end of the luciferase reporter gene in the CEBPA promoter vector by In-fusion cloning system (see ‘Materials and methods’ section for the details). We used the pGL4.12 reporter vector without any additional insertion as a negative control. Each of these vectors was introduced into HepG2 cells and the luciferase activity in the cell extracts was measured 48 h after transfection. As shown in Figure 7A, insertion of the DNA fragment derived from the putative combinatorial regulatory region in the CEBPA promoter vector induced a dramatic increase in the luciferase gene expression.
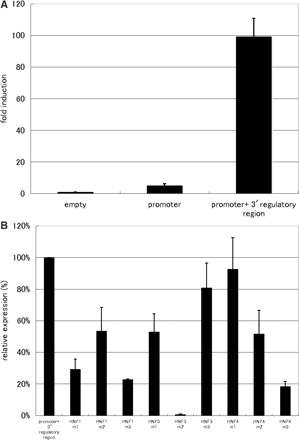
Transcription-enhancing activity of the CEBPA_4 regulatory region. Three independent experiments were performed in triplicate (n = 9). (A) The empty pGL4.12 vector was used as the control. The ‘promoter vector’ designated as ‘Promoter’ was the construct having an insertion of the CEBPA proximal promoter region in front of the luciferase gene. The ‘promoter-enhancer vector’ designated as ‘Promoter + 3′ regulatory region’ has an insertion of the deduced regulatory region (CEBPA_4 region) located downstream of the CEBPA gene and its surrounding region at the end of the luciferase gene. Fold induction indicates the level of luciferase expression (mean with a standard deviation) relative to that (= 1.0) detected with only the ‘empty’ vector. (B) The luciferase gene expressions by the constructs containing a different mutated sequence (m1–m3) for each of the three TFBSs (HNF1, HNF3 and HNF4) were comparatively examined. Expression level is indicated (mean with a standard deviation) relative to that caused by the ‘promoter-enhancer vector’ (taken as 100% here for comparative purposes).
The putative combinatorial regulatory region contains the binding sites for three TRFs (HNF1, HNF3 and HNF4). To evaluate the involvements of these TRFs in the regulation of transcription through the CEBPA proximal promoter, we generated various mutant vectors in which the TFBS for any one of the three TRFs had a set of nucleotide substitutions as follows: TGGCCAA (m1), TGGCGAA (m2) and TGCCCAA (m3) instead of TGATTAA in the HNF1 binding site; ATTGA (m1), ATGGA (m2) and ACTCA (m3) for AAACA in the HNF3 binding site; GATAATA (m1), GACAATA (m2) and GAGAATA (m3) for GCCTTGA in the HNF4 binding site.
Most of the mutant sequences caused a significant decrease in luciferase gene expression (Figure 7B). In particular, several mutants such as m1 and m3 for HNF1, m2 for HNF3 and m3 for HNF4 resulted in a dramatic reduction in the reporter gene expression by more than 70% as compared with the regulatory region having the wild-type nucleotide sequence. The lower but significant level of transcription-enhancing activity was observed when the 70-bp DNA fragment containing the binding sites for all of the three TRFs was inserted in the same location as where the CEBPA_4 region was, indicating the involvement of this narrow region in transcriptional enhancing. This result also implies the additive stimulatory effects of the surrounding region(s). Overall, the reporter assays demonstrate that all of these three TRFs and their binding sites are needed for the full transcription-enhancing activity of the regulatory region, consistent with the data obtained by the RNAi knockdown and X-ChIP/qPCR analysis.
EMSA supports specific bindings of the HNFs to the putative combinatorial regulatory region
We then investigated protein binding to their cognate recognition sites within the combinatorial regulatory region of the human CEBPA gene by EMSA. Several shifted bands were detected by gel electrophoresis and imaging after incubation of the radiolabled 70-bp oligonucleotide probe, which was derived from CEBPA_4 and covered all of the potential HNF1, HNF4 and HNF3 recognition sequences that were located in this order, with the nuclear extracts of HepG2 cells (Figure 8A). The addition of the unlabeled 70 mer markedly decreased the intensities of most of the bands. Short oligonucleotides corresponding to HNF 1 or 3 consensus recognition sequence also reduced the intensities of the cognate shifted bands (Figure 8A). Moreover, the addition of the specific antibody against either HNF1A or HNF3B formed a super-shifted band. These results indicate the specific bindings of HNF1A and HNF3B to their target sites. Contrary to this, neither clear competition by a ds-oligonucleotide competitor specific for HNF4A nor a supershift by anti-HNF4A antibody was detected. However, two distinct shifted bands were detected when a smaller probe (21-mer) derived from CEBPA_4 and containing a HNF4 core recognition sequence was used as the probe (Figure 8B). The intensities of these two bands were severely diminished in the presence of 100-fold excess amount of the unlabeled probe with the same sequence. No supershift by anti-HNF4 antibody with this short probe was again observed as with the 70-mer probe. Any of three kinds of competitor with different mutations in the HNF4 core sequence failed to diminish the intensities of the two retarded bands in contrast to the competitor with the wild-type sequence.
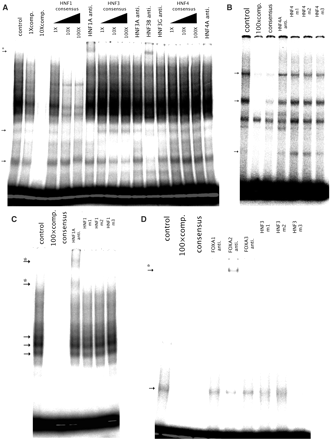
In vitro binding of HNFs to the regulatory region located downstream of the CEBPA gene. HepG2 nuclear extracts were incubated with the 70-mer probe containing the recognition sequences for HNF1, HNF3 and HNF4 or each of the TRF-specific probes (control). Competition assays were carried out with the equimolar (1×), 10-fold (10×) or 100-fold (100×) molar ratio of unlabeled probes (comp.), each of the TRF-specific consensus sequences (consensus) or mutated sequences of each of the TRF recognition sites (m1, 2 and 3). Specific antibody raised against each TRF (HNF1A, HNF3A, HNF3B, HNF3G and HNF4A anti.) was used for supershift assay. (A) 70-mer probe, (B) HNF4-specific probe, (C) HNF1-specific probe, (D) HNF3-specific probe. TRF-specific complexes are showed by arrows (→) and supershifted complexes are showed by arrows with an asterisk (*→).
When the smaller (21 bp) oligonucleotides containing a HNF1 or HNF3 recognition sequence derived from CEBPA_4 were used as the probes, supershifted bands (two for HNF1 and one for HNF3B) appeared by the incubation with HNF1A- or HNF3B-specific antibody as were the case for EMSA with the longer (70-bp) probe (Figure 8C and D). All of the three mutant sequences did not affect the intensities of the distinct retarded bands in the EMSA of HNF1. The competitor containing only the HNF3 consensus sequence also reduced markedly a shifted band. On the other hand, the mutant competitors differently affected the intensity of the shifted band. The mutant m1 that reduced moderately the luciferase gene expression showed a modest competition. The m3 competitor that slightly reduced the enhancing activity showed a clear competition, whereas the shifted band was scarcely competed by the m2 mutant sequence that caused a dramatic reduction in luciferase gene expression.
Co-transfection experiments demonstrate the combinatorial regulation of the CEBPA gene by a set of the HNFs
Based on the findings that the three TRFs (HNF1A, HNF3B and HNF4A) bind to the novel putative regulatory region in the downstream of the CEBPA gene and RNAi knockdown of any of these TRFs results in the downregulation of the gene, we expected that the region may be involved in a combinatorial transcriptional regulation. To demonstrate the combinatorial regulation of the CEBPA gene transcription by these three TRFs, carrying a luciferase reporter gene into HeLa cells. The TRF expression vectors were exogenously introduced into HeLa cells in a combinatorial manner along with the ‘promoter-enhancer vector’, a firefly luciferase reporter vector and the luciferase activities were measured at 48 h after co-transfection. HeLa cells were chosen as the hosts suitable for the reporter assays because the three TRFs were much less expressed in HeLa cells compared with HepG2 cells (Genome Network Platform Viewer URL://genomenetwork.nig.ac.jp/public/sys/gnppub/portal.do) and the combinatorial effects of the TRF genes, which were exogenously expressed, were expected to be clearer. We tested all combinations of the three TRFs for their combinatorial contributions to the transcriptional regulation of CEBPA gene. As shown in Figure 9, double transfections gave a modest or poor level of transcriptional enhancement. On the other hand, the highest reporter gene activation was occurred by the triple transfection with all of the three TRF expression vectors (statistically significant on paired t-test with P-value of 0.007). These data demonstrate that these three TRFs (HNF1A, HNF3B and HNF4A) act in a combinatorial manner for transcriptional regulation of the CEBPA gene.
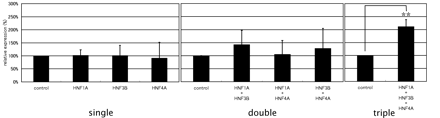
Combinatorial regulation of the CEBPA gene by a set of the HNFs. Three independent experiments were performed in triplicate (n = 9). The ‘promoter-enhancer vector’, a luciferase reporter vector, and various combinations of HNF (HNF1A, HNF3B and HNF4A) expression vectors or the control vectors were transfected into HeLa cells. Expression levels are indicated (mean with a standard deviation) relative to those achieved by co-transfection with the control vector (taken as 100% here for comparative purposes). single, co-transfection with the reporter vector and expression vectors for a single TRF gene; double, co-transfection with expression vectors for each of the two TRF genes; triple, co-transfection with all of the three TRFs. As a control, we used the same amount of the control vectors for each co-transfection experiment. Asterisks (**) mean a statistically significant combination compared with the control data set. P-value was calculated by Student's t-test (P-value <0.01).
DISCUSSION
To identify DNA regions and a set of TRFs that are involved in combinatorial regulation, we used a strategy including several criteria closely connected to functional definition of transcriptional regulation in mammalian systems. The combination of these criteria was found to be effective for identifying the functional combinatorial regulatory regions and regulators. Involvement of the TRFs identified in combinatorial transcriptional regulation may be also supported by the fact that the TRFs lie very close to each other. For example, HNFs bound to HNF1A_1 or CEBPA_4 should be located in their immediate vicinity (Figure 4). These TRFs may interact directly with each other or communicate through interaction with cofactors such as CBP and p300 or mediator(s). Physical association of CBP with HNF1A has been reported (36). In addition of CBP, HNF1A also interacts with HNF4A (37), supporting that the combinatorial regulation by HNF1A and HNF4A takes place via the physical interaction between these TRFs.
The HNF1A gene is known to be regulated by binding to its proximal promoter region of HNF3B (38), HNF4A (24) and HNF1A itself (39,40) in mammalian cultured cells and by HNF3G in salmon (41). We found significant perturbation of HNF1A gene expression by RNAi knockdown of HNF3A, HNF3B, HNF3G and HNF4A in HepG2 cells (submitted). Binding of almost all of these TRFs as well as CBP and p300 at the proximal upstream region (HNF1A_1 and mod028501) were clearly observed, indicating the validity of the present approach for characterizing combinatorial transcriptional regulation. Because regulation of the HNF1A gene by a multiple binding of HNF3s has not yet been reported, our findings add new insights into the mechanism of regulation of the mammalian HNF1A gene, whose heterozygous germ-line mutations are linked to the occurrence of maturity-onset diabetes of the young (MODY3) in humans (42). The reason why only anti-HNF3B antibody caused the super shift in contrast with the successful ChIP with all of the antibodies against the three HNF3s is unknown. Another region (HNF1A_4 and mod028502) showed clear bindings of HNF1A, HNF3A, HNF3B and HNF3G, but its involvement in the combinatorial transcriptional regulation of HNF1A gene is not conclusive because no clear bindings of HNF4A, CBP and p300 were detected.
CEBPA-mediated transcriptional regulation has been extensively studied (43–45). However, the mechanism of transcriptional regulation of the CEBPA gene itself remains to be elucidated. We identified two regulatory sites in the far downstream region of the CEBPA gene and demonstrated that the coactivators and HNFs examined bind to that region. Especially, one of the regulatory sites, CEBPA_4 that was bound by almost all of the HNFs and coactivators in X-ChIP assays showed a strong transcription-enhancing activity in the luciferase reporter assays. Most of the mutations at the recognition sites of HNF1, HNF3 and HNF4 in this region caused significant reduction in transcription-enhancing activities, supporting the involvement of these TRFs in combinatorial transcriptional regulation through the bindings to this enhancing region. EMSA also supported specific bindings of all of HNF1, HNF3 (mainly HNF3B based on the supershift) and HNF4 to this region. Although no supershift by the HNF4-specific antibody was observed with either the 70-mer or smaller probe, competition by an unlabeled competitor and the supershift by the same HNF4-specific antibody were clearly shown when the HNF4 consensus sequence derived from human prothrombin enhancer was used as the probe in EMSA with the same nuclear extracts (data not shown). Therefore, the extracts contained active HNF4 proteins and the HNF4-specific antibody really had the TRF-binding potential as demonstrated in X-ChIP assays. Based on the findings that the mutant sequences cannot inhibit the band shifts as competitors but can induce a marked (m3) and a pronounced (m2) reduction in enhancing the luciferase gene expression in the reporter assays, it is reasonable to think that HNF4 does not bind efficiently to these mutant sequences but does to the wild-type sequence in the form in which its epitope may be masked by some other protein(s), which interact with HNF4 to prevent it from binding of the HNF4-specific antibody. Interactions of HNF4A with other transcription factors including HNF1A and coactivators including CBP have been well documented (37,46–49).
Finally, co-transfection assays with the expression vectors for HNF1A, HNF3B and HNF4A genes along with the luciferase reporter vector containing the CEBPA promoter and novel regulatory region provided strong evidence of their combinatorial transcriptional regulation of the CEBPA gene. It should be noted that double transfection of HNF3B and either HNF1A or HNF4A exhibited a modest transcriptional stimulation and the combination of HNF1A and HNF4A was scarcely effective to enhance the CEBPA gene transcription. These data suggest that the three TRFs are needed to act in a combinatorial manner for the attainment of full transcriptional stimulation of their common target CEBPA gene. This is consistent with the finding that RNAi knockdown of any of these TRF genes resulted in a significant reduction of the levels of the CEBPA gene expression.
The transcriptional regulation of CEBPA gene is critical for various biological events such as inhibition of mitotic growth and tumor suppression (43–45). Information about its distant regulatory sites and the TRFs involved in the combinatorial regulation should provide insights into the mechanism of its functioning in mammalian cells.
HNF3G is an essential TRF for the maintenance of specific liver functions (50–52), but the mechanism of regulation of HNF3G gene expression remains poorly understood. We found that HNF3G_2 site may be engaged in the transcriptional regulation. Binding to this site by all the HNFs and two coactivators examined was observed. Although rVISTA could not locate any consensus sequence for HNF4A recognition, a manual search led us to detection of two possible HNF4A binding sequences lying ∼300 bp apart from each other within this site. A portion of the HNF3G_2 is homologous to the enhancer identified at +16 kb of mouse HNF3G gene (34), also supporting the validity of using the present approach to identify distantly located mammalian enhancers.
In general, individual TFBSs are more conserved than is their surrounding DNA, and genes having high sequence conservation in their upstream regions are predominantly TRF genes (8). If this is the case, the present approach based on sequence conservation analysis can be effectively applied to the characterization of combinatorial regulation of mammalian TRF genes. In conclusion, the present approach could be effectively used to identify TRFs and their cognate cis-regulatory elements and to investigate their involvements in combinatorial transcriptional regulation leading to understanding of their regulatory mechanisms.
FUNDING
A research grant for the RIKEN Genome Exploration Research Project from the Ministry of Education, Culture, Sports, Science and Technology of Japan; and a grant of the Genome Network Project from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to Y.H.) (http://genomenetwork.nig.ac.jp/index_e.html). Funding for open access charges: the grant to the Genome Network Project.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank Alistair Forrest for his critical reading of the manuscript and comments.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments