





Abstract
A major challenge to successful antiviral therapy is the emergence of drug-resistant viruses. Recent studies have developed several automated analyses of HIV sequence polymorphism based on calculations of selection pressure (Ka/Ks) to predict drug resistance mutations. Similar resistance analysis programs for HCV inhibitors are not currently available. Taking advantage of the recently available sequence data of patient HCV samples from a Phase II clinical study of protease inhibitor boceprevir, we calculated the selection pressure for all codons in the HCV protease region (amino acid 1–181) to identify potential resistance mutations. The correlation between mutations was also calculated to evaluate linkage between any two mutations. Using this approach, we identified previously known major resistant mutations, including a recently reported mutation V55A. In addition, a novel mutation V158I was identified, and we further confirmed its resistance to boceprevir in protease enzyme and replicon assay. We also extended the approach to analyze potential interactions between individual mutations and identified three pairs of correlated changes. Our data suggests that selection pressure-based analysis and correlation mapping could provide useful tools to analyze large amount of sequencing data from clinical samples and to identify new drug resistance mutations as well as their linkage and correlations.
INTRODUCTION
More than 170 million people worldwide are chronically infected with the hepatitis C virus (HCV). Combination therapy with pegylated interferon-alpha plus ribavirin achieves sustained virologic response rates of only ∼50% in HCV genotype 1 infected patients (1–3), which emphasizes the need for new antiviral drugs. The viral serine protease NS3-4A is responsible for the cleavage of the viral polyprotein into functional individual proteins (4), and has become a promising drug target for specific antiviral treatment. Data for clinical efficacy drug resistance have been published for several protease inhibitors BILN-2061 (ciluprevir) (5,6), VX-950 (telaprevir) (7,8) and SCH 503034 (boceprevir) (9). A major challenge to successful antiviral therapy is the emergence of drug-resistant viruses. Due to the rapid viral dynamics and high mutation rate of HCV polymerase, large pools of genetically distinct but closely related variants are generated in infected patients (10). During treatment, the strong selective pressure allows the outgrowth of variants resistant to therapy which can result in viral load rebound and reduced clinical efficacy.
The protease inhibitor boceprevir (SCH 503034) binds to the enzyme active site and inhibits cleavage of the viral polyprotein (11). It has been under clinical investigation in mono- and combination therapy regimens. Resistance mutations to boceprevir have been identified in the HCV replicon system as well as in clinical trials. The resistance loci are V36, Q41, F43, T54, R155, A156 and V170, which are located near the inhibitor binding site (12–15).
Although the replicon system has proven to be a valuable tool to identify resistance mutations to protease inhibitors, comprehensive evaluation of clinical sequencing data may provide further insight into the development of resistance and evolution of viral population under clinical settings. Several automated analyses of HIV sequence polymorphism to predict drug resistance mutations were published in the past few years (16,17). One very widely used metric of selection pressure that has been adapted in these studies is known as Ka/Ks (18,19) which measures the ratio of observed non-synonymous mutations over observed synonymous mutations, normalized by the ratio expected under a neutral model. Methods used for inferring Ka/Ks ratios are constantly being developed (20–23). A ratio around 1 indicates either neutral evolution or an averaging of positive and negative selective pressures. A ratio less than 1 indicates negative selection, with most amino-acid mutations being deleterious. A ratio significantly greater than 1 indicates positive selective pressure, which is less common and suggests that amino acid changes are favored, i.e. they reflect a change in the function of a gene or a change in environment conditions that forces the organism to adapt, thus increase organism's fitness.
In silico analysis of drug resistant mutation using a selection pressure-based method in these studies has provided a valuable resource for HIV research community. Similar studies for HCV are not currently available. In this study, taking advantage of the recently available sequence data of patient HCV samples from a boceprevir Phase II clinical study, we calculated the selection pressure for all the codons on the HCV protease region and by combining with frequency based filtering approach, we identified all major known resistant mutations as well as novel non-synonymous mutations that confer resistance to HCV protease inhibitor treatment.
MATERIALS AND METHODS
Sample selections
Samples used in this study were obtained from a Phase II clinical study of HCV protease inhibitor boceprevir (SCH 503034) in genotype 1 non-responders. Patient blood samples at time points specified by study protocol were collected and HCV RNA was extracted, amplified and sequenced. Additional samples were selected by study clinicians based on virological response of individual patients. Because samples and protease sequences were not available from all subjects at all time points, it was not possible to select a single on-treatment time point for analysis. Therefore, the last on-treatment time point was used for each subject. Samples collected from subjects prior to boceprevir treatment were used as baseline controls.
Amplification of HCV NS3 protease region and DNA sequencing
Viral RNA was extracted from human plasma samples using a commercially available silica-gel membrane based kit (QIAamp Virus BioRobot 9604 Kit, Qiagen, Valencia, CA) and processed on an automated BioRobot 9604 system (Qiagen). Reverse transcription of RNA was performed using a SuperScript III First Strand Synthesis Supermix kit (Invitrogen, Carlsbad, CA), with random hexamers according to manufacturer's instructions. PCR was conducted with a Platinum PCR SuperMix kit (Invitrogen), using 3 µl cDNA, 200 nM NS3 protease gene-specific primers (forward primer: GTAGAGCCCGTCGTCTTCTC; reverse primer: GTGCTCTTGCCGCTGCCAGT), and 45 µl of Platinum PCR Supermix (proprietary mix contains anti-Taq DNA polymerase antibody, Mg2+, dNTPs and recombinant Taq DNA polymerase). Cycle sequencing reactions were performed using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA), gene-specific primer and 5–10 ng of purified DNA according to manufacturer's instructions. Reaction products were purified on a Biomek FX system (Beckman Coulter, Fullerton, CA) using a magnetic bead kit (Agencourt CleanSEQ Kit, Agencourt Bioscience Corporation, Beverly, MA). DNA sequencing of purified material was conducted on a 3730xl DNA Analyzer (Applied Biosystems).
Clonal sequencing was carried out on a subset of patient samples. Purified RT-PCR products were cloned using the TOPO TA Cloning Kit (pCR 2.1TOPO vector, Invitrogen). For each serum sample, 96 bacteria colonies were sent to Qiagen or Genewiz for sequencing, using M13 forward and reverse primers as well as two protease specific primers (56f, GACATCATCTTGGGTCTGCCCGTCTC, 65r, GTGGGAGCGTGTAGGTGGGC). Sequence reads were aligned with HCV template sequence D90208 and mutations were analyzed.
Sequencing data analysis
The sequenced region included codons 1–181 of the HCV protease NS3 region. Base calling was done using PHRED (24). Quality scores from PHRED output were extracted and used to select chromatograms with good quality for subsequent analysis. For mixture positions where the chromatogram indicated that a mixture of more than one nucleotide was present, only the major peak was called. For each sample, at least four sequencing reads with good quality were required to cover each nucleotide position and at least one of them was required to come from a different sequencing direction. ClustalW (25) was used to align sequences to a template HCV sequence (D90208). Consensus NS3 region sequences from both before- and after-treatment samples were generated for each subject from the ClustalW alignments and were compared at each nucleotide position for each subject. Nucleotide changes were recorded and mutation type (transition or transversion) were determined. Amino-acid changes before and after treatment for each codon position were also recorded. HCV NS3 region sequences from before and after treatment used in this study can be obtained from GenBank (FJ830936 to FJ831439).
Ka/Ks calculation and selection pressure analysis

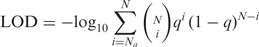
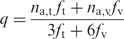
Confirmation of boceprevir resistance of novel mutations
To generate mutant proteases carrying resistance mutations, the nucleotide changes were introduced using the QuikChange mutagenesis kit (Stratagene). The parental plasmid expressing His-tagged single chain NS4A-NS3 protease domain, NS4A21–32-GSGS-NS33–181, was described by Taremi et al. (26). The expression and purification protocol was described in detail by Taremi et al. (26). Recombinant proteases were tested using a chromogenic assay as described by Zhang et al. (27). The overall inhibition constant Ki* (where vs = VmaxS/(Km(1 + 1/Ki*))) was used to measure inhibitor potency.
The construction and sensitivity testing of mutant replicons have been described previously (12). The QuikChange (Stratagene) template was plasmid pUC18 1–5, which contained the 5′ half of the subgenomic replicon sequence (T7 promoter to EcoRI site in NS5A). The PmeI-EcoRI fragment, which contained the coding sequence for NS3 to the N-terminal of NS5A, was sequenced to confirm the engineered mutations and used to replace the same region in pUC18 Bart, which contained the complete subgenomic replicon sequence (28). To measure anti-replicon activity of compounds, replicon cells (in triplicates) were dosed with protease inhibitors for 3 days. The replicon RNA level was measured using real-time PCR (Taqman assay) on the ABI PRISMS 7900HT Sequence Detection System. The dCT values (CT5B − CTGAPDH) were plotted against drug concentration and fitted to the sigmoid dose response model using Graphpad PRISM software (Graphpad Software Inc.). IC50 was the drug dose necessary to achieve dCT = 1 over the projected baseline. IC90 was the drug dose necessary to achieve dCT = 3.2 over the baseline.
Corrleation map of selection pressure

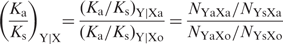
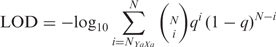
Structural modeling
The crystal structure of boceprevir (SCH 503034) complexed with the NS3/4A protease was solved in house [PDB code 2OBO (29)]. To model the binding mode of telaprevir (VX-950) to the NS3/4A protease, the crystal structure of boceprevir-NS3/4A protease was used as a template. The backbone atoms and the ketoamide positions of boceprevir served as initial coordinates upon which the P1–P4 side chains and P1 cap of telaprevir were built. The P4 cap was built based on the binding mode of another ketoamide inhibitor which has the same N-terminal cap (PDB code 1RGQ) (30). The inhibitor molecule was then energy minimized in the protein environment. In addition, point mutations were also modeled into the three-dimensional structure of the protein. InsightII molecular modeling program (Accelrys Software Inc., San Diego, CA) was used in the modeling.
RESULTS AND DISCUSSION
During HCV antiviral therapy, viruses with drug-resistant mutations are expected to be positively selected. To identify such positively selected amino acids, the Ka/Ks values were calculated for each position of the HCV NS3 protease region (amino acid 1–181). A total of 252 subjects from this HCV protease inhibitor clinical study with sequence data from before and on treatment samples were included in this analysis. Because the collecting schedule of serum samples varied with individual patients, it was difficult to choose a single fixed time point for all patients. Instead, the last on-treatment time was analyzed to maximize the time under selection pressure. For ‘mixture positions’ where the chromatogram indicated more than one nucleotide was present, only the major peak was analyzed. Out of the Ka/Ks values for 746 different non-synonymous mutations in the NS3 protease, 198 amino-acid changes have Ka/Ks value >1. In order to focus on major mutations which may have more impact on clinical outcome and to differentiate mutations selected specifically for drug resistance from ‘neutral drifts’, the following criteria were built into the mutation analysis process: (i) Low frequency non-synonymous mutations (in <1% treated subjects) were excluded from further analysis. (ii) Frequency differences of mutations in before and after treatment samples were compared using Fisher's exact test. Mutations whose prevalence did not increase significantly during therapy (P > 0.1) were excluded. Table 1 listed the resulting resistant mutations with Ka/Ks value >1 and meeting the criteria described above. As shown in Table 1, the major known boceprevir-resistant mutations (V36M, T54A/S, R155K/T, A156S, V170A) (12,13) were correctly identified by this analysis. V55A is a resistance mutation identified recently by clonal sequencing analysis of a phase I clinical trial of boceprevir, and has been shown to confer resistance to boceprevir and telaprevir in replicons (31). Structural analysis of V55 supports observed phenotypic resistance (31). In our study, two mutations at V55 (V55A and V55I) were positively selected based on Ka/Ks values, although V55A appears to be the more favored amino-acid change based on calculated P-values. In addition, our analysis identified a novel mutation V158I, which fit all the filtering criteria. Another mutation V163L was positively selected based on Ka/Ks value; although the P-value was greater than 0.1, it was also included in subsequent confirmation studies to evaluate the stringency of filtering criteria. The patient population of this trial consists of approximately equal mixture of genotype 1a and 1b subjects. Mutation V55A/I was found in five 1a subjects, seven 1b subjects and two subjects with unknown subtypes. Mutation V158I was found in five 1b subjects, and mutation V163L in two 1b subjects and one 1a subject. The number of subjects is too small to show any statistically significant difference in mutation distribution in genotype 1a and 1b subjects. Mutations V55A/I, V158I and V163L mutations are very rare among natural isolates based on the amino-acid frequency database constructed from GenBank data as described previously (32).
Positively selected mutations in HCV protease region (after filtering as described in the text)
| Position | AA_SCRN | AA_Therapy | Ka/Ks | LOD | Number of Mutation_Therapy | Number of Mutation_SCRN | P-value | Mutation% in Genbank |
|---|---|---|---|---|---|---|---|---|
| 36 | V | M | 18.27 | 90.47 | 48 | 0 | 6.0e–16 | 0 |
| 54 | T | A | 4.87 | 30.20 | 15 | 0 | 4.9e–5 | 0 |
| 54 | T | S | 47.15 | 160.07 | 66 | 1 | <2.2e–16 | 0 |
| 155 | R | K | 80.80 | 227.17 | 113 | 1 | <2.2e–16 | 0 |
| 156 | A | S | 19.30 | 49.00 | 15 | 0 | 4.9e-5 | 0 |
| 170 | V | A | 40.92 | 11.95 | 6 | 0 | 0.03 | 0.39 |
| 55 | V | A | 5.11 | 13.71 | 10 | 3 | 0.09 | 0 |
| 55 | V | I | 2.19 | 6.76 | 4 | 1 | 0.37 | 0 |
| 158 | V | I | 4.38 | 18.22 | 5 | 0 | 0.06 | 0.39 |
| 163 | V | L | 2.24 | 8.76 | 3 | 0 | 0.25 | 0 |
| Position | AA_SCRN | AA_Therapy | Ka/Ks | LOD | Number of Mutation_Therapy | Number of Mutation_SCRN | P-value | Mutation% in Genbank |
|---|---|---|---|---|---|---|---|---|
| 36 | V | M | 18.27 | 90.47 | 48 | 0 | 6.0e–16 | 0 |
| 54 | T | A | 4.87 | 30.20 | 15 | 0 | 4.9e–5 | 0 |
| 54 | T | S | 47.15 | 160.07 | 66 | 1 | <2.2e–16 | 0 |
| 155 | R | K | 80.80 | 227.17 | 113 | 1 | <2.2e–16 | 0 |
| 156 | A | S | 19.30 | 49.00 | 15 | 0 | 4.9e-5 | 0 |
| 170 | V | A | 40.92 | 11.95 | 6 | 0 | 0.03 | 0.39 |
| 55 | V | A | 5.11 | 13.71 | 10 | 3 | 0.09 | 0 |
| 55 | V | I | 2.19 | 6.76 | 4 | 1 | 0.37 | 0 |
| 158 | V | I | 4.38 | 18.22 | 5 | 0 | 0.06 | 0.39 |
| 163 | V | L | 2.24 | 8.76 | 3 | 0 | 0.25 | 0 |
P-values are the mutation frequency differences in before and after treatment samples. Mutation % in GenBank are largely based on 1b (64%) and 1a (27%) sequences.
Positively selected mutations in HCV protease region (after filtering as described in the text)
| Position | AA_SCRN | AA_Therapy | Ka/Ks | LOD | Number of Mutation_Therapy | Number of Mutation_SCRN | P-value | Mutation% in Genbank |
|---|---|---|---|---|---|---|---|---|
| 36 | V | M | 18.27 | 90.47 | 48 | 0 | 6.0e–16 | 0 |
| 54 | T | A | 4.87 | 30.20 | 15 | 0 | 4.9e–5 | 0 |
| 54 | T | S | 47.15 | 160.07 | 66 | 1 | <2.2e–16 | 0 |
| 155 | R | K | 80.80 | 227.17 | 113 | 1 | <2.2e–16 | 0 |
| 156 | A | S | 19.30 | 49.00 | 15 | 0 | 4.9e-5 | 0 |
| 170 | V | A | 40.92 | 11.95 | 6 | 0 | 0.03 | 0.39 |
| 55 | V | A | 5.11 | 13.71 | 10 | 3 | 0.09 | 0 |
| 55 | V | I | 2.19 | 6.76 | 4 | 1 | 0.37 | 0 |
| 158 | V | I | 4.38 | 18.22 | 5 | 0 | 0.06 | 0.39 |
| 163 | V | L | 2.24 | 8.76 | 3 | 0 | 0.25 | 0 |
| Position | AA_SCRN | AA_Therapy | Ka/Ks | LOD | Number of Mutation_Therapy | Number of Mutation_SCRN | P-value | Mutation% in Genbank |
|---|---|---|---|---|---|---|---|---|
| 36 | V | M | 18.27 | 90.47 | 48 | 0 | 6.0e–16 | 0 |
| 54 | T | A | 4.87 | 30.20 | 15 | 0 | 4.9e–5 | 0 |
| 54 | T | S | 47.15 | 160.07 | 66 | 1 | <2.2e–16 | 0 |
| 155 | R | K | 80.80 | 227.17 | 113 | 1 | <2.2e–16 | 0 |
| 156 | A | S | 19.30 | 49.00 | 15 | 0 | 4.9e-5 | 0 |
| 170 | V | A | 40.92 | 11.95 | 6 | 0 | 0.03 | 0.39 |
| 55 | V | A | 5.11 | 13.71 | 10 | 3 | 0.09 | 0 |
| 55 | V | I | 2.19 | 6.76 | 4 | 1 | 0.37 | 0 |
| 158 | V | I | 4.38 | 18.22 | 5 | 0 | 0.06 | 0.39 |
| 163 | V | L | 2.24 | 8.76 | 3 | 0 | 0.25 | 0 |
P-values are the mutation frequency differences in before and after treatment samples. Mutation % in GenBank are largely based on 1b (64%) and 1a (27%) sequences.
To test whether V158I and V163L confer resistance to boceprevir, each mutation was introduced into the single-chain genotype 1b protease (26). As shown in Table 2, V158I conferred low level resistance (as shown by a 2.5-fold increase in inhibition constant Ki*) to boceprevir but not to telaprevir, another ketoamide inhibitor under clinical evaluation. To confirm in vitro enzyme results, mutation V158I was cloned into a genotype 1b replicon and shown to decrease replicon sensitivity (as shown by a ∼3-fold increase in replicon EC50 value) to boceprevir but not to telaprevir (Figure 1 and Table 2). In contrast, mutation V163L did not affect boceprevir inhibition of the protease in biochemical assays (Table 2). Structural analysis (Figure 2) of the NS3 protease active site showed that V158 is part of the substrate binding pocket and is in direct contact with the terbutyl P3 capping group of boceprevir. Change of this residue to a slightly bulkier Ile could interfere with boceprevir binding. Telaprevir has a more extended interaction with the active site via its P4 group which could compensate the potency loss caused by the V158I mutation. V163 is about 9 Å away from the terbutyl P3 capping group of boceprevir and is not expected to be involved in inhibitor binding.
Enzyme and replicon resistance of newly identified resistance mutations
| Mutations | Fold change over WT ± SD | ||
|---|---|---|---|
| Enzyme (Ki*) | Replicon (EC50) | ||
| Boceprevir | V158l | 2.5 ± 1.1 | 3.3 ± 0.5 |
| V163L | 1 ± 0.3 | not done | |
| Telaprevir | V158l | 0.5 ± 0.2 | 0.9 ± 0.1 |
| V163L | 1 ± 0.3 | not done | |
| Mutations | Fold change over WT ± SD | ||
|---|---|---|---|
| Enzyme (Ki*) | Replicon (EC50) | ||
| Boceprevir | V158l | 2.5 ± 1.1 | 3.3 ± 0.5 |
| V163L | 1 ± 0.3 | not done | |
| Telaprevir | V158l | 0.5 ± 0.2 | 0.9 ± 0.1 |
| V163L | 1 ± 0.3 | not done | |
Averages and SD of two to four experiments are reported.
Enzyme and replicon resistance of newly identified resistance mutations
| Mutations | Fold change over WT ± SD | ||
|---|---|---|---|
| Enzyme (Ki*) | Replicon (EC50) | ||
| Boceprevir | V158l | 2.5 ± 1.1 | 3.3 ± 0.5 |
| V163L | 1 ± 0.3 | not done | |
| Telaprevir | V158l | 0.5 ± 0.2 | 0.9 ± 0.1 |
| V163L | 1 ± 0.3 | not done | |
| Mutations | Fold change over WT ± SD | ||
|---|---|---|---|
| Enzyme (Ki*) | Replicon (EC50) | ||
| Boceprevir | V158l | 2.5 ± 1.1 | 3.3 ± 0.5 |
| V163L | 1 ± 0.3 | not done | |
| Telaprevir | V158l | 0.5 ± 0.2 | 0.9 ± 0.1 |
| V163L | 1 ± 0.3 | not done | |
Averages and SD of two to four experiments are reported.
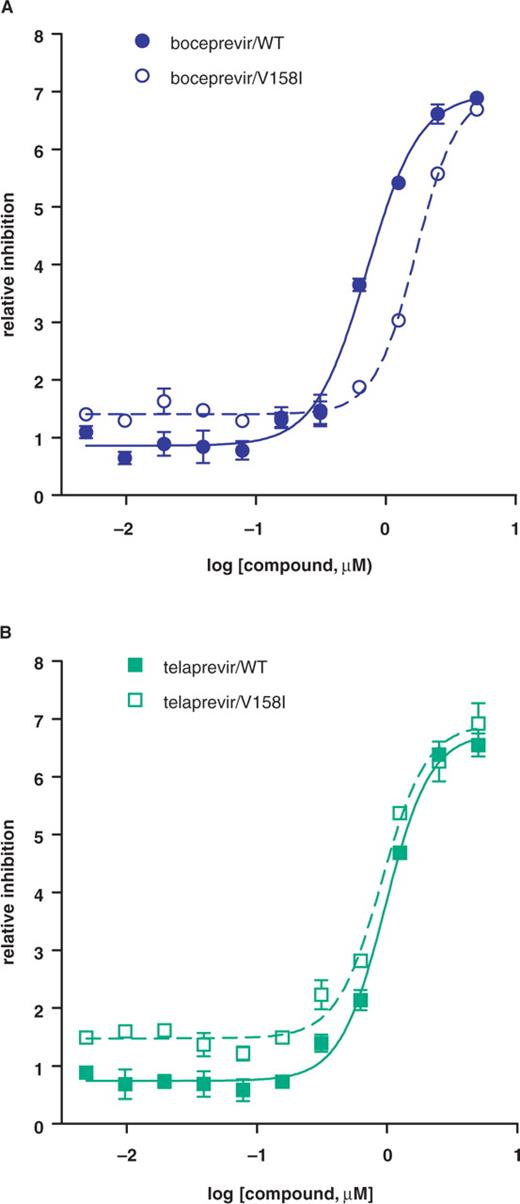
Inhibition of mutant replicons by protease inhibitors. Replicon cells carrying wild-type or mutant V158I subgenomes were treated with boceprevir (A) or telaprevir (B) for 3 days, and replicon RNA levels were measured by Taqman assay. The relative reduction in replicon RNA level (dCT) was plotted against compound concentration.
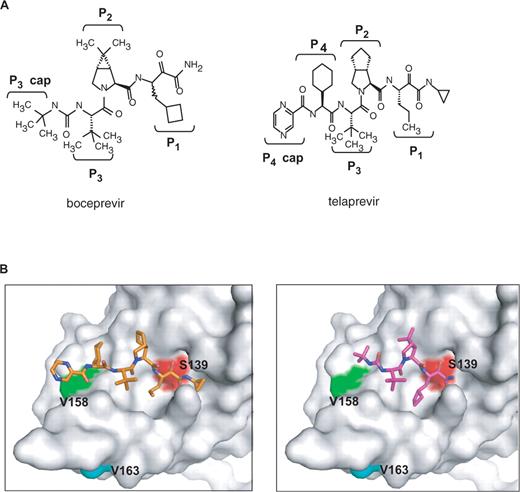
(A) Structures of boceprevir and telaprevir. (B) Three-dimensional structure of NS3 protease-inhibitor complex. The structures of boceprevir (purple, right panel) and telaprevir (gold, left panel) are presented as stick models. Side chains of the active site Ser (Ser139) and two mutation sites (V158 and V163) are depicted on the Connolly surface of the NS3 protease.
Current research has largely focused on the impact of single mutations on drug resistance, and the potential of interactions among mutations has received little attention. The impact of an individual mutation may vary depending on other mutations present. The interactions could be positive (synergistic), leading to higher levels of drug resistance than would be expected from the individual mutations alone; or negative (antagonistic), with some mutations effectively reducing the effects of others. Compensatory mutations also can increase the replication fitness of the mutant virus, increasing the apparent effects of resistance mutations. The lack of understanding of coordinations between mutations is at least partly due to the limitation of commonly used sequencing methods. Clonal sequencing can provide linkage information for multiple mutations, but the method is labor intensive and can only be applied to relatively small number of samples. Population sequencing is more cost effective in evaluating large number of samples, but can not evaluate directly the linkage of mutations at different positions. The multiple mutations observed in a patient by population sequencing could be linked in a single genome or could arise from two or more individual virus strains.
Correlated mutations may occur at both short range positions (33) and distant positions (34). Several bioinformatics methods for detecting correlated mutations have been developed over the years (35–37). One of the key problems that all the methods share is distinguishing signal from noise, such as those arise from evolutionary processes related to common ancestry. In this study, to extract correlated mutation information from population sequencing, we calculated Ka/Ks values for mutation at one position (Y) conditioned on the mutation on another position (X), a method reported by Pan et al. (17). A total of 627 pairs of amino-acid mutations show conditional Ka/Ks values >1. Similar filters as described for single mutations were used to reduce false positives and the resulting amino-acid pairs were shown in Table 3. All three pairs of double mutations have been identified by clonal sequencing of samples from 19 patients from this clinical study. V36M+R155K was observed in 15/19 patients, T54S+R155K in 10/19 patients (a related mutation T54A+R155K in 1/19 patient), T54S+A156S in 1/19 patient (a related mutation T54A+A156S in 1/19 patient). Double mutations (e.g. V36M+R155K) have also been observed in patient treated with telaprevir (8). The level of resistance conferred by double mutations is generally multiplicative or greater (13,38). For example, double mutant V36M+155K confers 14-fold resistance to boceprevir, while single mutants V36M and R155K confer only 2- and 3-fold resistance, respectively (13). The mechanism of interaction between those mutations on drug resistance remains to be analyzed.
Interaction of mutations in HCV protease region (after filtering as described in text)
| Position 1 | AA1 | Position 2 | AA2 | Number of double mutationa | Ka/Ks | LOD | P-value |
|---|---|---|---|---|---|---|---|
| 36 | M | 155 | K | 33 | 4.44 | 57.84 | 7.6e–10 |
| 54 | S | 156 | S | 12 | 2.69 | 28.25 | 4.3e–4 |
| 155 | K | 36 | M | 33 | 2.79 | 57.11 | 7.6e–10 |
| 155 | K | 54 | S | 31 | 3.49 | 53.64 | 3.5e–10 |
| 156 | S | 54 | S | 12 | 10.67 | 36.04 | 4.3e–4 |
| Position 1 | AA1 | Position 2 | AA2 | Number of double mutationa | Ka/Ks | LOD | P-value |
|---|---|---|---|---|---|---|---|
| 36 | M | 155 | K | 33 | 4.44 | 57.84 | 7.6e–10 |
| 54 | S | 156 | S | 12 | 2.69 | 28.25 | 4.3e–4 |
| 155 | K | 36 | M | 33 | 2.79 | 57.11 | 7.6e–10 |
| 155 | K | 54 | S | 31 | 3.49 | 53.64 | 3.5e–10 |
| 156 | S | 54 | S | 12 | 10.67 | 36.04 | 4.3e–4 |
P-values are the double mutation frequency differences in before and after treatment samples.
aThe number of double mutation observed on therapy, no double mutations were detected in SCRN samples.
Interaction of mutations in HCV protease region (after filtering as described in text)
| Position 1 | AA1 | Position 2 | AA2 | Number of double mutationa | Ka/Ks | LOD | P-value |
|---|---|---|---|---|---|---|---|
| 36 | M | 155 | K | 33 | 4.44 | 57.84 | 7.6e–10 |
| 54 | S | 156 | S | 12 | 2.69 | 28.25 | 4.3e–4 |
| 155 | K | 36 | M | 33 | 2.79 | 57.11 | 7.6e–10 |
| 155 | K | 54 | S | 31 | 3.49 | 53.64 | 3.5e–10 |
| 156 | S | 54 | S | 12 | 10.67 | 36.04 | 4.3e–4 |
| Position 1 | AA1 | Position 2 | AA2 | Number of double mutationa | Ka/Ks | LOD | P-value |
|---|---|---|---|---|---|---|---|
| 36 | M | 155 | K | 33 | 4.44 | 57.84 | 7.6e–10 |
| 54 | S | 156 | S | 12 | 2.69 | 28.25 | 4.3e–4 |
| 155 | K | 36 | M | 33 | 2.79 | 57.11 | 7.6e–10 |
| 155 | K | 54 | S | 31 | 3.49 | 53.64 | 3.5e–10 |
| 156 | S | 54 | S | 12 | 10.67 | 36.04 | 4.3e–4 |
P-values are the double mutation frequency differences in before and after treatment samples.
aThe number of double mutation observed on therapy, no double mutations were detected in SCRN samples.
In summary, five major known HCV boceprevir drug resistance mutations were successfully identified using the selection pressure-based method described here, two additional mutations V55A and V158I were identified that were previously not revealed in subjects from this clinical study. Use of more sensitive sequencing method and inclusion of more data set from future studies will increase the likelihood of identifying low prevalence mutations. Although the data set included in this study is relatively small compared to datasets used in similar HIV studies and other frequency based mutation detection methods have been reported for HCV clonal sequence analysis (8,31,39), the high success rate of our method suggests that Ka/Ks analysis and correlated mutation mapping combined with proper filtering mechanism described above could provide useful tools to identify new drug resistant mutations in HCV and shed light on potential interactions between mutations at different sites.
FUNDING
Funding for open access charge: Schering-Plough Research Institute.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
Comments