





Abstract
The Drosophila species comparative genome database DroSpeGe (Author Webpage) provides genome researchers with rapid, usable access to 12 new and old Drosophila genomes, since its inception in 2004. Scientists can use, with minimal computing expertise, the wealth of new genome information for developing new insights into insect evolution. New genome assemblies provided by several sequencing centers have been annotated with known model organism gene homologies and gene predictions to provided basic comparative data. TeraGrid supplies the shared cyberinfrastructure for the primary computations. This genome database includes homologies to Drosophila melanogaster and eight other eukaryote model genomes, and gene predictions from several groups. BLAST searches of the newest assemblies are integrated with genome maps. GBrowse maps provide detailed views of cross-species aligned genomes. BioMart provides for data mining of annotations and sequences. Common chromosome maps identify major synteny among species. Potential gain and loss of genes is suggested by Gene Ontology groupings for genes of the new species. Summaries of essential genome statistics include sizes, genes found and predicted, homology among genomes, phylogenetic trees of species and comparisons of several gene predictions for sensitivity and specificity in finding new and known genes.
INTRODUCTION
Many new genomes are becoming available this decade. Current contents of public genome archives exceed 1 billion sequence traces from >1000 organisms [Author Webpage; (1)]. This number will increase rapidly as costs drop and scientific uses for comparing many genomes increases (2). Biologists should have rapid access to these new genomes, including basic annotations from well-studied model organisms and predictions to locate potential new genes, to make sense of them. Genome annotation and database management can be streamlined now using generic tools, shared computing resources and common genome database techniques to provide useful access to biologists in weeks instead of several months.
New genome sequencing projects and communities are facing large informatics tasks for incorporating, curating and annotating and disseminating sequence and annotation data. Effective genome studies need an informatics infrastructure that moves beyond individual organism projects to a cost-effective use of common tools. Expertise from existing genome projects should be leveraged into building such tools. The Generic Model Organism Database [GMOD (3)] project has this goal, to fully develop and extend a genome database tool set to the level of quality needed to create and maintain new genome databases. GMOD and related genome database tools now support a portion of the basic tasks for such. Two needs in development for GMOD are the creation of new databases for emerging model organisms, and tools for comparative genome databases that integrate data from many sources.
A common, ongoing task for research that uses genome databases is to compare an organism's genome and proteome with related organisms, and other sequence datasets (ESTs, SNPs, transposable elements). This task requires significant computational infrastructure, one where reusable tools, protocols and resources will be valuable and significantly reduce duplicative infrastructure and maintenance effort. Software tools to fully assembly, analyze and compare these genomes are available to bioscientists. The ability to employ these tools on genome datasets is limited to those with extensive computational resources and engineering talent. Effective use of shared cyberinfrastructure in bioinformatics is a problem today. Cluster and Grid computing in bioinformatics have followed other disciplines in parallelizing applications, but this is costly and limited to a subset of bioinformatics applications. This database enables bioscientists to have usable access to new genomes shortly after sequencing centers make them available, facilitating new science discoveries and understanding of the evolution, comparative biology and genomics of these model organisms.
GENOME INFORMATICS METHODS
Common components
DroSpeGe has been built with common GMOD database components and open source software shared with other genome databases. Use of common components facilitates rapid construction and interoperability. The GMOD ARGOS replicable genome database template (Author Webpage) provides a tested set of integrated components. The genome access tools of GMOD GBrowse [Author Webpage;(3)], BioMart [Author Webpage; (4)] and BLAST (5) are available for the Drosophila species genomes. The GMOD Chado relational database schema (Author Webpage) is used for managing an extensible range of genome information. Middleware in Perl and Java are added to bring together BLAST, BioMart, sequence reports, searches and other bioinformatics programs for public access. Another aid to integrating and mining these data is GMOD Lucegene (Author Webpage), that forms a core component for rapid data retrieval by attributes, GBrowse data retrieval and databank partitioning for Grid analyses. DroSpeGe operates on several Unix computers; the primary server is a SunFire V20z from Sun Microsystems. Genome maps include Drosophila melanogaster DNA and protein homology, homologies to nine eukaryote proteomes, marker gene locations, gene predictions using 15 methods produced by several contributing groups. The assemblies and predicted genes can be BLASTed, with links to genome maps. BioMart provides searches of the full genome annotation sets, allowing selections of genome regions with and without specific features.
New species genomes
Twelve Drosophila genomes, 10 recently sequenced, contain over 2 billion nt, with sizes ranging from a small 133 Mb of D.melanogaster to >230 Mb in Drosophila willistoni (Table 1). The model organism D.melanogaster is approaching its fifth major assembly release, and continues to see significant improvements in genes and genome features. It has a known, located complement of ∼14 000 protein genes. One main impetus for undertaking the sequencing of 11 additional related species is to improve via comparative analyses the knowledge of this major research organism. Drosophila pseudoobscura, the second related genome, is in its second major release. The additional species are at their first major assembly stage, requiring automated annotation, quality assessment and cross-species comparisons. Four of these new genomes (Dsim, Dsec, Dyak and Dere) are close relatives of the model in the melanogaster subgroup. The remainder range through five other taxonomic groups with an estimated divergence time of 40 million years, with the cactus breeder Drosophila mojavensis, widely distributed Drosophila virilis and Hawaiian picture-wing Drosophila grimshawi most distant from Dmel. Assembly sequences of the Drosophila species comparative annotation freeze 1 (CAF1) are distributed at Author Webpage, as listed in Table 1. These form the primary source data for this database. Over the course of two years, this database has provided rapid access to several assembly releases per species, including annotation, searching and viewing services for each release.
Drosophila species genomes, abbreviation, sequencing centers and genome size of CAF1 assemblies used at DroSpeGe
| Abbreviation | Species | Size (Mb) | Sequencing center |
|---|---|---|---|
| Dmel | Drosophila melanogaster | 133 | Berkeley Drosophila Genome Project/Celera |
| Dsim | Drosophila simulans | 142 | Genome Sequencing Center, Washington University |
| Dsec | Drosophila sechellia | 167 | Broad Institute |
| Dyak | Drosophila yakuba | 160 | Genome Sequencing Center, Washington University |
| Dere | Drosophila erecta | 153 | Agencourt Bioscience Corporation |
| Dana | Drosophila ananassae | 231 | Agencourt Bioscience Corporation |
| Dper | Drosophila persimilis | 188 | Broad Institute |
| Dpse | Drosophila pseudoobscura | 153 | Human Genome Sequencing Center, Baylor College of Medicine |
| Dwil | Drosophila willistoni | 237 | J. Craig Venter Institute |
| Dmoj | Drosophila mojavensis | 194 | Agencourt Bioscience Corporation |
| Dvir | Drosophila virilis | 206 | Agencourt Bioscience Corporation |
| Dgri | Drosophila grimshawi | 200 | Agencourt Bioscience Corporation |
| Abbreviation | Species | Size (Mb) | Sequencing center |
|---|---|---|---|
| Dmel | Drosophila melanogaster | 133 | Berkeley Drosophila Genome Project/Celera |
| Dsim | Drosophila simulans | 142 | Genome Sequencing Center, Washington University |
| Dsec | Drosophila sechellia | 167 | Broad Institute |
| Dyak | Drosophila yakuba | 160 | Genome Sequencing Center, Washington University |
| Dere | Drosophila erecta | 153 | Agencourt Bioscience Corporation |
| Dana | Drosophila ananassae | 231 | Agencourt Bioscience Corporation |
| Dper | Drosophila persimilis | 188 | Broad Institute |
| Dpse | Drosophila pseudoobscura | 153 | Human Genome Sequencing Center, Baylor College of Medicine |
| Dwil | Drosophila willistoni | 237 | J. Craig Venter Institute |
| Dmoj | Drosophila mojavensis | 194 | Agencourt Bioscience Corporation |
| Dvir | Drosophila virilis | 206 | Agencourt Bioscience Corporation |
| Dgri | Drosophila grimshawi | 200 | Agencourt Bioscience Corporation |
Annotations produced by several groups collaboratively are provided for map viewing and data mining. Protein coding gene predictions viewable at this resource include contributions listed in Table 2.
Drosophila species genomes, abbreviation, sequencing centers and genome size of CAF1 assemblies used at DroSpeGe
| Abbreviation | Species | Size (Mb) | Sequencing center |
|---|---|---|---|
| Dmel | Drosophila melanogaster | 133 | Berkeley Drosophila Genome Project/Celera |
| Dsim | Drosophila simulans | 142 | Genome Sequencing Center, Washington University |
| Dsec | Drosophila sechellia | 167 | Broad Institute |
| Dyak | Drosophila yakuba | 160 | Genome Sequencing Center, Washington University |
| Dere | Drosophila erecta | 153 | Agencourt Bioscience Corporation |
| Dana | Drosophila ananassae | 231 | Agencourt Bioscience Corporation |
| Dper | Drosophila persimilis | 188 | Broad Institute |
| Dpse | Drosophila pseudoobscura | 153 | Human Genome Sequencing Center, Baylor College of Medicine |
| Dwil | Drosophila willistoni | 237 | J. Craig Venter Institute |
| Dmoj | Drosophila mojavensis | 194 | Agencourt Bioscience Corporation |
| Dvir | Drosophila virilis | 206 | Agencourt Bioscience Corporation |
| Dgri | Drosophila grimshawi | 200 | Agencourt Bioscience Corporation |
| Abbreviation | Species | Size (Mb) | Sequencing center |
|---|---|---|---|
| Dmel | Drosophila melanogaster | 133 | Berkeley Drosophila Genome Project/Celera |
| Dsim | Drosophila simulans | 142 | Genome Sequencing Center, Washington University |
| Dsec | Drosophila sechellia | 167 | Broad Institute |
| Dyak | Drosophila yakuba | 160 | Genome Sequencing Center, Washington University |
| Dere | Drosophila erecta | 153 | Agencourt Bioscience Corporation |
| Dana | Drosophila ananassae | 231 | Agencourt Bioscience Corporation |
| Dper | Drosophila persimilis | 188 | Broad Institute |
| Dpse | Drosophila pseudoobscura | 153 | Human Genome Sequencing Center, Baylor College of Medicine |
| Dwil | Drosophila willistoni | 237 | J. Craig Venter Institute |
| Dmoj | Drosophila mojavensis | 194 | Agencourt Bioscience Corporation |
| Dvir | Drosophila virilis | 206 | Agencourt Bioscience Corporation |
| Dgri | Drosophila grimshawi | 200 | Agencourt Bioscience Corporation |
Annotations produced by several groups collaboratively are provided for map viewing and data mining. Protein coding gene predictions viewable at this resource include contributions listed in Table 2.
Drosophila species genome annotations (partial list) included at DroSpeGe, contributed at Author Webpage
| Contributor | Annotation description |
|---|---|
| S. Batzoglou Lab, Stanford | Contrast [Author Webpage; (6)] predictions |
| M. Brent Lab, Washington University, St Louis | N-SCAN [Author Webpage; 7)] predictions with melanogaster alignments |
| D. Gilbert Lab, Indiana University | SNAP [Author Webpage; (8)] predictions, model organism gene homologies |
| M. Eisen Lab, UC Berkeley/LBNL | GeneWise (9), GeneMapper [Author Webpage; (10)], Exonerate (11) annotations |
| R. Guigó Genome Bioinformatics Lab, Barcelona | Geneid [Author Webpage; (12)] predictions |
| NCBI, Bethesda | Gnomon [Author Webpage; (13)] predictions |
| B. Oliver Lab, LCDB, NIDDK, NIH | Gene expression evidence from microarray [Author Webpage; (14)] |
| L. Pachter Lab, UC Berkeley | GeneMapper (10) annotations |
| C. Ponting Lab, MRC FGU Oxford | Gene prediction pipeline [Author Webpage; (15)] with Exonerate |
| Contributor | Annotation description |
|---|---|
| S. Batzoglou Lab, Stanford | Contrast [Author Webpage; (6)] predictions |
| M. Brent Lab, Washington University, St Louis | N-SCAN [Author Webpage; 7)] predictions with melanogaster alignments |
| D. Gilbert Lab, Indiana University | SNAP [Author Webpage; (8)] predictions, model organism gene homologies |
| M. Eisen Lab, UC Berkeley/LBNL | GeneWise (9), GeneMapper [Author Webpage; (10)], Exonerate (11) annotations |
| R. Guigó Genome Bioinformatics Lab, Barcelona | Geneid [Author Webpage; (12)] predictions |
| NCBI, Bethesda | Gnomon [Author Webpage; (13)] predictions |
| B. Oliver Lab, LCDB, NIDDK, NIH | Gene expression evidence from microarray [Author Webpage; (14)] |
| L. Pachter Lab, UC Berkeley | GeneMapper (10) annotations |
| C. Ponting Lab, MRC FGU Oxford | Gene prediction pipeline [Author Webpage; (15)] with Exonerate |
Drosophila species genome annotations (partial list) included at DroSpeGe, contributed at Author Webpage
| Contributor | Annotation description |
|---|---|
| S. Batzoglou Lab, Stanford | Contrast [Author Webpage; (6)] predictions |
| M. Brent Lab, Washington University, St Louis | N-SCAN [Author Webpage; 7)] predictions with melanogaster alignments |
| D. Gilbert Lab, Indiana University | SNAP [Author Webpage; (8)] predictions, model organism gene homologies |
| M. Eisen Lab, UC Berkeley/LBNL | GeneWise (9), GeneMapper [Author Webpage; (10)], Exonerate (11) annotations |
| R. Guigó Genome Bioinformatics Lab, Barcelona | Geneid [Author Webpage; (12)] predictions |
| NCBI, Bethesda | Gnomon [Author Webpage; (13)] predictions |
| B. Oliver Lab, LCDB, NIDDK, NIH | Gene expression evidence from microarray [Author Webpage; (14)] |
| L. Pachter Lab, UC Berkeley | GeneMapper (10) annotations |
| C. Ponting Lab, MRC FGU Oxford | Gene prediction pipeline [Author Webpage; (15)] with Exonerate |
| Contributor | Annotation description |
|---|---|
| S. Batzoglou Lab, Stanford | Contrast [Author Webpage; (6)] predictions |
| M. Brent Lab, Washington University, St Louis | N-SCAN [Author Webpage; 7)] predictions with melanogaster alignments |
| D. Gilbert Lab, Indiana University | SNAP [Author Webpage; (8)] predictions, model organism gene homologies |
| M. Eisen Lab, UC Berkeley/LBNL | GeneWise (9), GeneMapper [Author Webpage; (10)], Exonerate (11) annotations |
| R. Guigó Genome Bioinformatics Lab, Barcelona | Geneid [Author Webpage; (12)] predictions |
| NCBI, Bethesda | Gnomon [Author Webpage; (13)] predictions |
| B. Oliver Lab, LCDB, NIDDK, NIH | Gene expression evidence from microarray [Author Webpage; (14)] |
| L. Pachter Lab, UC Berkeley | GeneMapper (10) annotations |
| C. Ponting Lab, MRC FGU Oxford | Gene prediction pipeline [Author Webpage; (15)] with Exonerate |
TeraGrid genome analyses
The TeraGrid project (Author Webpage) is part of a shared cyberinfrastructure for sciences, funded primarily by NSF. TeraGrid provides collaborative, cost-effective scientific computing infrastructure much in the same way the GMOD initiative is building common tools for genome databases. The TeraGrid system is particularly suitable for genome assembly, annotation, gene finding and phylogenetic analyses. TeraGrid computers have been employed to analyze the 12 Drosophila genomes, providing the major contents of DroSpeGe database. This has enabled rapid analyses without the expense of obtaining and maintaining a local computer cluster. This experience forms a basis for other genome projects to use TeraGrid. Scripts used for this analysis are available at the GMOD repository (Author Webpage). Genome database tools from GMOD project are used to organize the computations for public access. Results include D.melanogaster genome homology, homologies to nine eukaryote proteomes, gene predictions, marker gene locations and Drosophila microsatellites. For each of 12 Drosophila genomes, a comparison is made to a set of nine proteomes, with 217 000 proteins, drawn from source genome databases, Ensembl and NCBI. The reference proteomes are human, mouse, zebrafish, fruitfly (Dmel), mosquito, bee, worm (Caenorhabditis elegans), mustard weed (Arabidopsis thaliana) and yeast. Sizes of the new genomes are in the 150–250 Mb range. Protein–genome DNA alignment is done using tBLASTn, with a Grid-aware version of NCBI software. The TeraGrid run for each genome took 12–18 h using 64 processors. Whole genome DNA–DNA alignments were performed for a subset of new genomes. Gene predictions with SNAP (8) have been generated. Over the course of 6 months, with 2–3 genome assembly updates each per species, and error corrections, the total TeraGrid 64 cpu usage per genome has been ∼4 days, excluding queue-waiting times.
DATABASE USES
DroSpeGe provides a resource to biologists interested in comparing species differences and similarities, including novel and known genes, genome structure and evolution, gene function associations. Known genes from model organisms are found in the new genomes at expected rates, allowing for variations due to assembly quality. The most divergent species (Dmoj, Dvir, Dgri), with 40 million years divergence from Dmel, match ∼90% of the model species genes. These known genes provide useful access to the new genomes for many researchers interested in locating a particular gene or gene family. The known gene matches also offer searches and cataloging gene contents by known functions. Figure 1 shows the size and similarity to the Dmel model of these genomes. Genome annotations and analyses produced for this database is available at DroSpeGe/data/and in bulk form at , including annotations of CAF1 and prior assembly releases. Related genome projects also provide Drosophila genome data and complementary services (see Related Work).
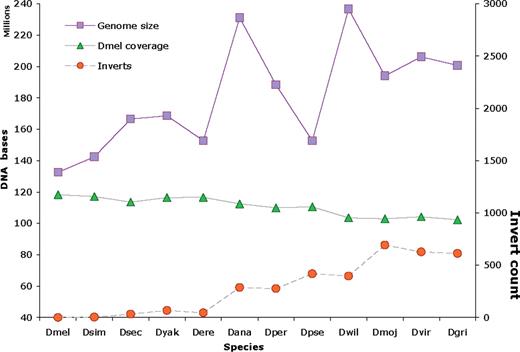
Drosophila species assemblies, showing assembly sizes and coverage of these by D.melanogaster genome DNA (top and middle lines, in megabases, left ordinate), and counts of chromosome segments inverted relative to Dmel (bottom line, right ordinate). Species on abscissa are taxonomically ordered with Dgri most distant from Dmel. This is summarized from Author Webpage.
Genome data mining
An emerging trend among bioscientists and bioinformaticians is to use data mining of large subsets of genome data, often focused on summary information for a range of common attributes. These data are used in spreadsheets and simple databases or analyses. Genomics web databases often lack methods for effectively mining large subsets of genomes, or are limited in the questions one can pose to the underlying complex data (16). The Ensembl project with its off-shoot BioMart (4) is an example of integrated software and data that bridge the gap in biology data access between bulk files and web portals. A tool for creating BioMart-compliant transaction databases, gff2biomart, is a recent addition by the author to GMOD tools collection (Author Webpage). It has been used for DroSpeGe and other genome datasets. BioMart with annotations of 12 Drosophila genomes has provided numerous bioscientists with a unique data mining access to these new genomes.
With BioMart, one can select genome regions with the available annotations, and exclude others and download tables or sequences of the selection set. For instance, select the regions with mosquito gene homologs, but lacking D.melanogaster homologs. Or select regions with gene predictions but no known homology. A major reason to undertake the genome sequencing of 12 Drosophila species is to improve genome knowledge of the widely used D.melanogaster model organism. A significant application for BioMart has been to identify gene predictions in D.melanogaster that do not match known genes. Further phylogenetic analysis of these new gene predictions has identified a subset with cross-species homology and high synonymous substitution rates, validating these as likely new genes and coding exons with phylogenetic evidence. Another application of BioMart has been to compare the qualities of gene predictor methods, identifying predicted exons that coincide with known gene homology, and with gene expression datasets to measure sensitivity at predicting new and known genes.
Genome maps
Maps of the 12 genomes form the core, with BLAST searches, of discovery tools for bioscientists. Maps including all available annotations from several groups are provided using GBrowse (3). The BLAST result reports include hyperlinks from each alignment match to the respective genome map, as well as to sequence and GFF annotation results. As species comparisons are of much interest, BLAST results also link to a comparative map display of the matches. A recent addition to the genome maps is an aligned comparative map set for any group of the 12 species. As seen in Figure 2, this allows one to view phylogenetic evidence of common gene predictions and features in homologous regions. In this example, genes that are predicted in the model Dmel, but previously not located, are found to be orthologously located across eight species (Dmel through Dmoj). This capability of full comparative annotation maps may be unique to DroSpeGe. Other genome maps offer either a single species map with tracks that summarize homology, or a syntenic view of two species. An overview of all species chromosome maps is provided in the DroSpeGe/maps/section. These overviews link to detailed genome maps, with known gene homology locations, gene expression evidence and predictions.
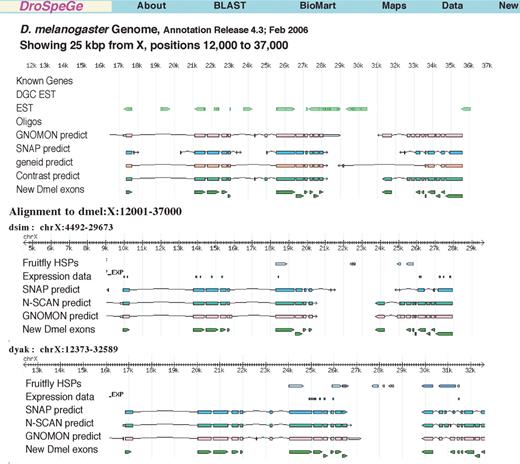
Aligned genomes view of new D.melanogaster gene locations on X chromosome, on D.melanogaster, Drosophila simulans and Drosophila yakuba, identified with cross-species comparison of coding exons, from DroSpeGe/data/dmel-dspp/newgenes. Several gene predictors match these common coding exons. Additional evidence from EST, protein HSP matches and gene expression data corroborate the new genes. Genomes with orthologous gene predictions not shown, but viewable at DroSpeGe maps, include Dsec, Dere, Dana, Dpse and Dmoj.
Common chromosomes in Drosophila
A series of maps show large-scale synteny between genome assembly units (scaffolds or chromosomes), as determined from genome × genome DNA BLAST matches, identified as common Muller elements. These are found in DroSpeGe/maps/muller-elements. Muller's elements are the names A, B, C, D, E, F for six chromosome arms common among Drosophila as coined by Hermann Müller. Chromosome names and centromeric joins differ among the species; Muller elements identify the common units. These synteny maps provide scientists with quick access to common genome regions among the 12 species. The melanogaster group species (Dmel, Dsim, Dsec, Dyak and Dere) have close matching, with large-scale inversions evident in these maps. Among the more distantly related species, the new genome assembly of D.mojavensis has proved most complete, with four Muller elements nearly fully assembled, the autosomes B to E, and the sex chromosome assembled into four major scaffolds.
Gene variation by gene ontology group
To provide an assessment of possible gene gain and loss among Drosophila, gene matches to Gene Ontology categories by species were tabulated, and provided at section DroSpeGe/news/genome-summaries/gene-GO-function-association. These may indicate species differences in functional categories. Statistically significant deviations are indicated. While low counts, suggestive missing genes, may be due to divergence of genes, extra gene matches more strongly suggest categories where species differ. Among the interesting differences, transport genes (GO:0006810) may show a phylogenetic cline with more in the non-melanogaster group (Dana to Dgri); protein binding genes (GO:00055515) may be more common in the Dmel–Dsim–Dsec siblings; protein biosynthesis (GO:0006412) is higher in the Dpse–Dper sibling species. Individual species peaks such as Dwil for catalytic activity genes (GO:0003824) or signal transduction (GO:0007165) in Dgri, suggest species-specific adaptations. The gene matches are high-scoring segment pair (HSP) groupings, and include various events: gene duplications, alternate splice exons within genes, new genes that appear composed of exons from other genes, as well as computational artifacts. Detailed evidence pages provide links to GBrowse genome map views showing all secondary HSPs. Proteome sources in this analysis are those organism with extensive GO annotations: Dmel fruitfly, mouse, C.elegans worm and yeast. GO-Slim groupings are used for Biological Process, Molecular Function, Cell Location (125 categories). A table provides the correspondence between MOD gene ID, GO primary ID and GO-slim groupings. Chris Mungall's GO map2slim software is employed for this, along with current GO gene associations.
RELATED WORK
Drosophila species assemblies, analyses and annotations have been coordinated at Michael Eisen's community Wiki (Author Webpage), in an open way that serves as a model for future genome collaborations. Contributors have here submitted data, genome analyses, summaries and discussion for the benefit of the research community. In conjunction with this, the Eisen Lab provides annotations, analyses and GBrowse maps of Drosophila species. The FlyBase project (Author Webpage) has benefited from these community efforts, recently adding a subset of annotations for 12 species to its map and search services. The most comparable effort to DroSpeGe in approach, if not species, is the Fungal Comparative Genomics resource [Author Webpage; Author Webpage; (17)] that catalogs 56 genomes including the model Saccharomyces cerevisiae and related yeasts and fungi. Fungal Genomics offers GBrowse maps, BLAST, gene predictions and phylogenetic comparisons. Comprehensive genome resources with Drosophila include UCSC Genome Bioinformatics (Author Webpage), Ensembl (Author Webpage), Entrez Genomes (Author Webpage). The latter two currently show only D.melanogaster. UCSC Genomes has most of the insect genomes, but as of October 2006 has yet to update to current Drosophila assemblies and annotations. UCSC provides a very useful set of comparative homology analyses for each species genome.
The DroSpeGe comparative genome database has provided bioscientists with rapid access to new genomes in a usable way, with new annotations, browsing, search and summary services not available elsewhere. Future plans for this database focus on enhancing genome comparison functions, with improvements to category overviews for gene functions, pathways and orthology evidence. Additional insect genomes and the arthropod Daphnia pulex [Author Webpage; (18)] may be integrated to extend the comparative range.
Reviewers who suggested improvements to this paper are thanked for their efforts. This project is supported in part by grants from the National Human Genome Research Institute of the National Institutes of Health, and from Sun Microsystems, to D.G. National Science Foundation TeraGrid access grant provided computing resources. The Open Access publication charges for this article were waived by Oxford University Press.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
Comments