





Abstract
Sequence-specific interactions between aminoacyl-tRNA synthetases and their cognate tRNAs ensure both accurate RNA recognition and the efficient catalysis of aminoacylation. The effects of tRNATrP variants on the aminoacylation reaction catalyzed by wild-type Escherichia coli tryptophanyl-tRNA synthe-tase (TrpRS) have now been investigated by stopped-flow fluorimetry, which allowed a pre-steady-state analysis to be undertaken. This showed that tRNATrP identity has some effect on the ability of tRNA to bind the reaction intermediate TrpRS-tryptophanyl-adenylate, but predominantly affects the rate at which trypto-phan is transferred from TrpRS-tryptophanyl ade-nylate to tRNA. Use of the binding (KtRNA) and rate constants (k4) to determine the energetic levels of the various species in the aminoacylation reaction showed a difference of ∼2 kcal mol-1 in the barrier to transition state formation compared to wild-type for both tRNATrP A→C73 and
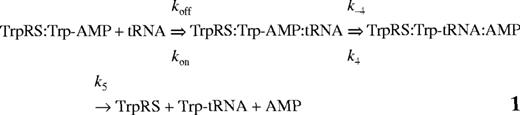
Introduction
The accurate transfer of genetic information from the nucleo-tide sequence of a gene to the amino acid sequence of a protein is dependent on the high fidelity of several molecular processes. Firstly, an RNA message (mRNA) complementary to the DNA sequence containing the gene is transcribed (for some viruses this is preceded by reverse transcription of RNA-encoded genes). This mRNA, or a processed form of it, is then used as a template for protein synthesis on ribosomes. This second process, by which the correct sequence of amino acids is assembled to form a protein, encompasses two distinct molecular recognition events. The first of these is the pairing of codons in mRNA with the corresponding anticodons in aminoacylated transfer RNA (tRNA). The other is the aminoacylation of these tRNA molecules, whereby an amino acid is esterified to the 3′-end of a tRNA containing the corresponding anticodon. It is this pairing of amino acids and tRNAs which defines the genetic code. The aminoacylation of tRNAs with the corresponding amino acid is catalyzed by the aminoacyl-tRNA synthetases (AARSs) and in a few cases the aminoacyl-tRNA is further processed before its use in translation (1). Thus, one of the main criteria for accurate translation of genetic information is that the AARSs should display an extremely high level of substrate specificity.
The processes of molecular recognition which allow only the appropriate amino acid and tRNAs to be selected from the cellular pool have been studied in great detail. Differences in the side chains of amino acids are often sufficient to allow their specific selection. In those cases where errors do occur, such as during the discrimination of isoleucine from valine, structurally distinct AARS editing mechanisms prevent the synthesis or release of incorrectly aminoacylated tRNAs (2). The editing of erroneously activated amino acids has been found in some cases to depend upon the cognate tRNA substrate, which sequence-specifically enables the enzymatic hydrolysis of the non-cognate aminoacyl-adenylate (3).
During substrate selection tRNA first binds the aminoacyl-tRNA synthetase (either as a free enzyme or as an enzyme-aminoacyl-adenylate complex) with recognition dependent on sequence-specific protein-RNA interactions. Then, this transient protein-RNA complex catalyzes attachment of the amino acid to the 3′-terminal adenosine of the tRNA. Selection of the correct tRNA is assumed to occur as a result of preferential reaction kinetics for cognate protein-RNA complexes (4). Investigation of numerous experimental systems has indicated that the second process is predominantly responsible for the discrimination of cognate from non-cognate tRNAs by aminoacyl-tRNA synthetases. Much of this work has been based upon the use of mutant tRNAs in steady-state kinetic studies to determine Michaelis-Menten parameters. In the majority of cases, the use of mutant or non-cognate tRNAs predominantly affects the reaction rate (kcat) while having a far less pronounced effect on the apparent substrate dissociation constant (Km). Since the aminoacylation reaction proceeds via two transition states and, therefore, does not conform to the simple Michaelis-Menten mechanism, kcat is a complex term which reflects enzyme-substrate, enzyme-intermediate and enzyme-product complexes (5). Thus, while substantial changes in kcat directly result from disruption of critical protein-RNA interactions, the inherent limitations of steady-state kinetics mean that such changes cannot be unequivocally assigned to a single point in the aminoacylation reaction. Stopped-flow fluorimetry has previously been used to study the attachment of cognate and non-cognate amino acids to tRNA by wild-type and mutant forms of the tyrosyl- and phenyl-alanyl-tRNA synthetases, as well as by bovine tryptophanyl-tRNA synthetase (TrpRS) (6–8). Pre-steady-state analyses arising from such studies allowed the calculation of microscopic kinetic parameters for aminoacylation. This methodology has now been extended to compare the aminoacylation of cognate and non-cognate tRNAs by Escherichia coli TrpRS. Substrate recognition by TrpRS has previously been extensively investigated using a combination of biochemical, genetic and structural approaches (9–13) and the ability of TrpRS to aminoacylate non-cognate tRNAs is well documented (14). It has also been shown that specific interactions between TrpRS and tRNATrP determine the apparent affinity for tryptophan of the enzyme, providing additional evidence for a role of differential reaction kinetics in tRNA selection (15). We therefore used a combination of steady-state and pre-steady-state kinetics to investigate the ability of wild-type E.coli TrpRS to aminoacylate a range of cognate and non-cognate tRNAs.
Materials and Methods
General
The overexpression and purification of E.coli TrpRS (12) and T7 RNA polymerase have been described (16). In vitro transcription of tRNA genes and subsequent purification and refolding of transcripts were performed as previously detailed (16). In vivo E.coli tRNATrP was purchased from Subriden RNA (Rolling Bay, WA). Plasmid preparation for transcription of tRNA genes was performed by standard techniques using precipitation with polyethylene glycol (17) and the tRNA gene sequences confirmed by sequencing prior to transcription. Template DNA for transcription by T7 RNA polymerase was digested with BstNI for both tRNATrP and tRNAGln templates. L-[5-3H]Tryptophan (27 Ci/mmol) and L-[3-14C]tryptophan (53.8 mCi/mmol) were purchased from NEN (Boston, MA). Media for bacterial growth and molecular biology protocols were standard unless otherwise noted (17).
Strains and plasmids
Construction of plasmids for use as templates during the in vitro transcription of E.coli tRNATrPΔAl, (Gl) and (Gl) have been previously described (14). The tRNATrP variants Al (wild-type), G36 (Al) and C73 (Al) were constructed by mutagenesis of the plasmid containing the gene encoding E.coli tRNATrPΔAl. The Bacillus stearothermophilus tRNATrP gene was also subcloned into a suitable vector for in vitro transcription (M.Prætorius-Ibba, unpublished results).

Aminoacylation assays
Aminoacylation assays were performed as described (12). For all kinetic assays the concentrations of Trp and tRNATrP varied over the range 0.3-5 times the Km, with all other substrates at saturating concentrations. TrpRS concentrations, as determined by active site titration, were between 0.3 and 30 nM. The discs were then washed and radioactivity was measured by scintillation counting (18). Kinetic parameters were calculated by fitting data to the Michaelis-Menten equation using the program Kaleidagraph (Abelbeck Software).
Synthesis of tryptophanyl-adenylate
The aminoacyl-adenylate complex was prepared by incubating the TrpRS enzyme (10 (AM) in 50 mM sodium cacodylate buffer (pH 7.0), 10 mM magnesium acetate and 4 mM DTT with 2 mM ATP and 0.1 mM [14C]tryptophan (sp. act. ∼36 c.p.m./pmol) and 1 U of inorganic pyrophosphatase at 37°C for 10 min. The complex was then purified by gel filtration over PD10 gel filtration columns (Pharmacia) in 10 mM Bis Tris (pH 6.0), 10 mM MgCl2 and 4 mM DTT. Fractions containing complex were determined by spotting 20 μl of the 0.5 ml fractions to the nitrocellulose filters (BA85) already pre-soaked in buffer containing 50 mM sodium cacodylate buffer, pH 7.0, 10 mM MgCl2 and 10 mM tryptophan. Filters were dried for 25 min at 65°C and radioactivity was measured by scintillation counting. Correlation of tryptophanyl-adenylate (Trp-AMP) concentration with total protein concentration determined by standard methods indicated a stoichiometry of approximately 1 molecule of Trp-AMP per molecule of TrpRS, as expected in the presence of excess tryptophan (11).
Use of a stopped-flow apparatus to observe the reaction of TrpRS:Trp-AMP with tRNA
In order to measure the rate constants for the formation of the complex between TrpRS:Trp-AMP and tRNA and the subsequent transition state of Trp transfer, it is necessary to measure the rate of approach to the steady-state. Therefore, the absolute rate constants were obtained by monitoring changes in intrinsic fluorescence using a stopped-flow spectrophotometer (Applied Photophysics model SF 17 MV). Excitation was at 285 nm and emission was measured at wavelengths >320 nm using a cut-off filter. The concentration of TrpRS:Trp-AMP complex in one syringe was 0.5 μM in 10 mM Bis Tris (pH 6.0). In the other syringe, concentrations for tRNATrP variants ranged between 0.5 and 12 μM in 50 mM sodium cacodylate (pH 7.0), 10 mM MgCl2 and for between 4 and 40 μM. The stopped block of the apparatus was positioned such that 50 μl volumes from each syringe were mixed. All reactions were repeated at least five times and the data collected used to produce average traces. These were fitted to exponential equations using the KaleidaGraph software package. The rates of the fluorescence changes observed were plotted against tRNA concentrations as described above.
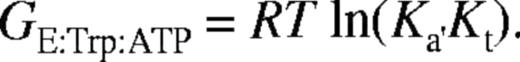
Results
Steady-state aminoacylation of tRNATrP and tRNAGln variants by TrpRS
It has previously been shown for E.coli TrpRS that interactions between tRNA identity nucleotides and their recognition sites determine the steady-state kinetic parameters of the enzyme for both tryptophan and tRNATrP. To further investigate this process in TrpRS, the steady-state kinetic parameters in the presence of cognate and non-cognate tRNA molecules were determined. The non-cognate tRNAs selected were , which has a tryptophan anticodon and is aminoacylated by TrpRS with a relative kcat/Km ratio only 10 times lower than in vitro transcribed tRNATrPΔAl, and , which contains the anticodon corresponding to the opal stop codon and is also aminoacylated by TrpRS (14). tRNATrP identity element mutations [A→G36 (14) and G→C73 (10,13)] and the heterologous tRNATrP from B.stearothermophilus were also used (Fig. 1). The steady-state kinetic parameters are shown in Table 1. In vitro transcribed tRNATrP shows an ∼2-fold reduction in catalytic efficiency (kcat/Km) compared to the in vivo expressed tRNATrP, consistent with the proposed role of certain base or nucleotide modifications in stabilizing tRNA tertiary structure. When non-cognate tRNA molecules were used as substrates, a further increase in the Km for Trp was observed, accompanied by a decrease in kcat. Trp kinetics with in vitro transcribed show a 5-fold increase in the Km for Trp and a 3-fold decrease in kcat when compared with the kinetic parameters of the in vitro transcribed tRNATrP. The replacement of C with U at position 34 in also led to a 5-fold increase in the Km for Trp and an 800-fold decrease in kcat. Of all the tRNAs tested, only showed a significant change in Km for tRNA compared to wild-type.
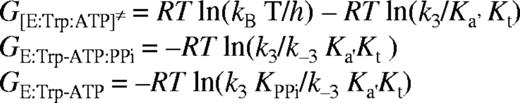
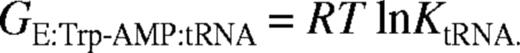
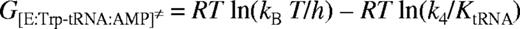
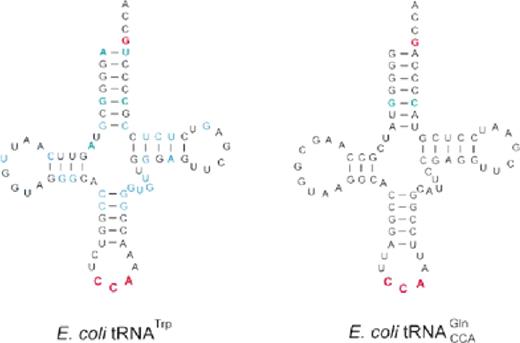
The predicted secondary structures of unmodified E.coli tRNATrP and tRNAGlnCCA Gl shown in cloverleaf form. Nucleotides which act as strong identity elements for tRNATrP are shown in red, weak identity elements in green. Positions which differ in B.stearothermophilus tRNATrP are shown in blue.
Pre-steady-state analysis of Trp-tRNATrP synthesis
The application of pre-steady-state kinetic techniques is necessary to complete the kinetic analysis of the overall pathway of Trp-tRNATrP synthesis and to elucidate the underlying mechanisms which contribute to specificity during catalysis. In order to investigate the second step of the aminoacylation reaction, transfer of the tryptophanyl moiety of Trp-AMP to the 3′-end of tRNA, preformed isolated TrpRS:Trp-AMP was mixed with tRNA and the resulting spectral change monitored by stopped-flow fluorimetry. Figure 2 shows a typical fluorescence trace observed upon mixing tRNATrPΔAl and the wild-type TrpRS:Trp-AMP complex. The observed fluorescence change could be due either to a conformational change associated with the transfer of tryptophan to the tRNA or to product release, since no fluorescence changes are seen upon mixing tRNATrPΔAl with enzyme alone (data not shown). Since previous studies have indicated that the release of Trp-tRNA by E.coli TrpRS is not rate limiting (19), the observed rate of the exponential change (k2obs) can be related to the rate of transfer of the tryptophanyl moiety to tRNA. The accuracy of the fit is indicated by the residual (Fobs-Fcalc, Fig. 2).
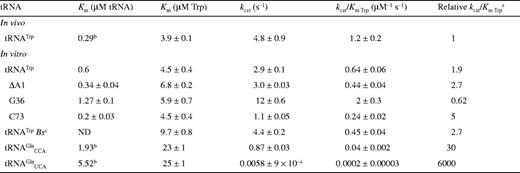
Aminoacylation kinetics for Trp in the presence of cognate and non-cognate tRNAs
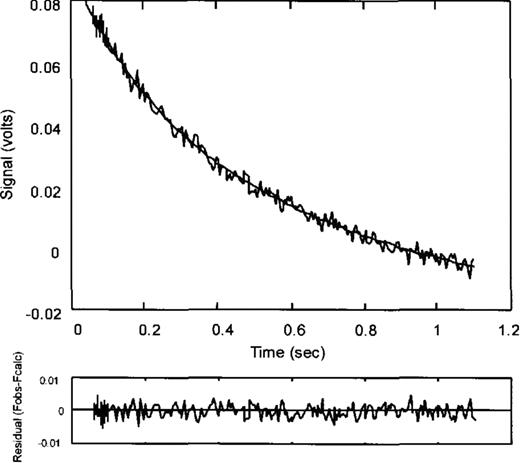
Typical fluorescence change (recorded as a signal voltage, an increase in which is equivalent to a decrease in fluorescence) observed upon mixing equal volumes of tRNATrPAAl with TrpRS. The trace is fitted to an exponential equation and the accuracy of the fit is indicated by the residual (Fobs-Fcalc).
Assuming that the transfer of tryptophan to the tRNA molecule proceeds according to equation 1 (below) the observed rate constant for the fluorescence change, k2obs, can be related to the concentration of tRNA and used to estimate k4 (the rate constant for tryptophan transfer) and KtRNA (dissociation constant of tRNA from the E:Trp-AMP complex), as previously described for tyrosyl-tRNA synthetase (20).
Mutation of nucleotides in tRNATrP predicted to be involved in complex formation were found to reduce k4 but to have a less pronounced effect on KtRNA (Table 2). The most significant change was seen for the discriminator base mutant C73, which showed a 20-fold fall in k4. Transfer of tryptophan to the non-cognate occurred at a significantly lower rate than the transfer to the cognate tRNATrP, as shown in a 30-fold lower k4 value. In addition, there is a 4-fold increase in the dissociation constant, KtRNA. Since contains both the CCA anticodon and G73, which are major identity elements for tRNATrP, the higher KtRNA and lower k4 indicate that additional nucleotides in tRNATrP contribute to tryptophan identity. Pre-steady-state analysis was also attempted using , however, no significant change in fluorescence was observed, presumably as a result of an extremely low k2obs for this mutant.
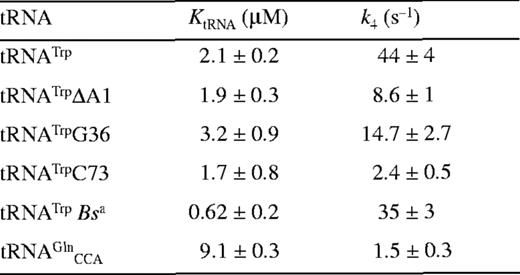
Pre-steady-state kinetic constants for reaction of tRNATrP variants and tRNAGlnCCA with wild-type TrpRS-tryptophanyl adenylate complexes
Calculation of the energy levels of enzyme-bound species during Trp-tRNA synthesis
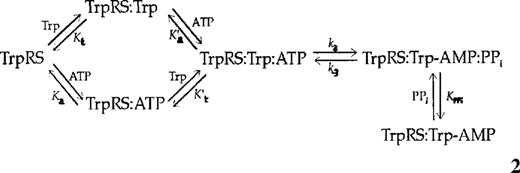
By substituting pre-steady-state dissociation and rate constants into thermodynamic equations it is possible to calculate the energy level of an enzyme during each step of a reaction. Since pre-steady-state kinetic analyses have previously been performed for the formation of tryptophanyl-adenylate by E.coli TrpRS (11), it is possible to use these data to calculate free energies for the first step of the aminoacylation reaction using procedures previously described for TyrRS (21). The results described here can then be used to extend the free energy profile to the second step of the aminoacylation reaction (6,20). The scheme for aminoacyl-adenylate synthesis (equation 2) assumes half-of-the-sites reactivity as previously described in the presence of excess tryptophan (11). The energy of the free enzyme (GE) was set to 0 and the values of the reaction intermediates were calculated relative to this value.
For binding of tryptophan:
For binding of ATP:
Substrate binding is assumed to be random order and KaKt = KaKt,
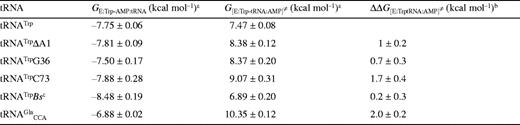
Gibbs' free energies of enzyme species in the tRNA charging reaction, calculated relative to the enzyme-aminoacyl-adenylate complex (E:Trp-AMP)
For formation of tryptophanyl-adenylate:
For the complex with tRNA:
For formation of the transition state of the tryptophan transfer step (from transition state theory and ΔGT≠ = G≠- ΔGS):
where R is the gas constant, T is the absolute temperature, kB is Boltzmann's constant and ≠ indicates the transition state. All calculated energies are presented in Table 3.
The free energy of the TrpRS:Trp-AMP complex relative to free enzyme, calculated from previously published data (11), is -5.8 kcal mol-1. The Gibbs' free energies calculated for two further species on the TrpRS reaction pathway when cognate tRNATrP was used, relative to free enzyme (taking GE = 0), are -13.5 kcal mol-1 for GE:Trp-AMP.tRNA and 1.7 kcal mol-1 for GE:Trp-tRNA:AMP≠. The TrpRS:Trp-AMP:tRNA complex is stabilized by 7.7 kcal mol-1 relative to the preceding enzyme-bound reaction species, which is TrpRS:Trp-AMP (Table 3), and by 13.5 kcal mol-1 relative to free enzyme. The free energies of activation are 14.5 kcal mol-1 for the first transition state and 15.2 kcal mol-1 for the second. The free energies for the ground state species TrpRS:Trp-AMP:tRNA and for the transition state species [TrpRS:Trp-tRNA:AMP]≠ were calculated for non-cognate tRNA transcripts and compared to the value for the wild-type tRNATrP transcript (Table 3). Little difference is seen between the various substrates with respect to the ground state, in agreement with the proposal that tRNA binding provides little scope for the discrimination of cognate from non-cognate tRNAs. Significant differences are seen in the transition state energies, most notably for tRNATrPC73 and , which show increases of 1.6 and 2.9 kcal mol-1, respectively, compared to wild-type. These values for the ground state and transition state were then used to calculate the differences in the size of the barrier to transition state formation compared to wild-type (ΔΔG[TrpRS:Trp-tRNA:AMP]≠). The largest changes were seen with tRNATrPC73 (1.7 kcal mol-1) and (2 kcal mol-1), both of which showed significant decreases in their ability to stabilize the transition state of tryptophan transfer.
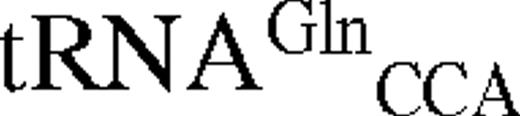
Discussion
tRNA identity as a determinant of substrate recognition and catalysis
It has been extensively demonstrated that single nucleotide substitutions in numerous tRNAs can reduce or abolish the ability of these molecules to function in aminoacylation (reviewed in 22). As expected from earlier studies with TrpRS (10,13,14), the series of tRNA substrates selected only showed moderate reductions in the efficiency of tryptophanylation and were therefore suitable for both steady-state and pre-steady-state kinetic analysis. The greatest reductions in steady-state catalytic efficiency were observed with the tRNAGln derivatives which contain C35, A36 and G73, reaffirming previous proposals that nucleotides outside the anticodon and discriminator base are important for tRNATrP function (10,13). Comparison of the conserved nucleotides in tRNATrP from B.stearothermophilus and E.coli, which show almost no difference in their kinetic parameters (Tables 1 and 2), with corresponding positions in (Fig. 1) point to C6:G67, A9, the D stem and loop and U31:A39 as possible additional identity elements. Two of these predictions are in agreement with previous studies with Bacillus subtilis tRNATrP (which only differs from B.stearothermophilus tRNATrP at nucleotide 16), which showed A9 and suggested U31:A39 as identity elements (13).
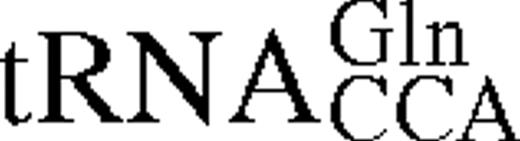
The function of the discriminator base (G73), the A1-U72 base pair and the first anticodon base (A36) as identity elements in a cognate tRNA context were only apparent from pre-steady-state kinetics, where mutations of these positions led to a reduction in the rate of tryptophan transfer (k4, Table 2). This indicates that pre-steady-state analyses allow a more sensitive means to investígate tRNA identity with this particular set of tRNA mutations.
To further analyze the role of tRNA in amino acid recognition, steady-state kinetics with respect to tryptophan were also analyzed. The only tRNAs which gave rise to significant changes in the apparent affinity for tryptophan were the two tRNAGln derivatives. In these cases, the decrease in apparent affinity towards tryptophan did not depend solely on tRNA identity, since both non-cognate tRNAs having different anticodons exhibited the same increase in Km for Trp but 150-fold different kcat values. Comparable effects have previously been observed with E.coli GlnRS, where glutaminyl-adenylate synthesis is strictly tRNA dependent (15). However, the reason for such tRNA-dependent changes in tryptophan binding are unclear since tRNA is not required for Trp-AMP synthesis by TrpRS. Recent suggestions that tRNA may influence the entire aminoacylation reaction even for certain aminoacyl-tRNA synthetases that do not require tRNA during aminoacyl-adenylate synthesis may partially explain these results (23). The presence of tRNATrP throughout the aminoacylation reaction may also explain the low overall reaction rate (2.9 s-1) compared to the individual rate constants for tryptophanyl-adenylate synthesis (146 s-1) and tryptophan transfer (44 s-1). In addition, the ability of E.coli TrpRS to synthesize a second tryptophanyl-adenylate per protein dimer under certain conditions, together with the cooperativity of tryptophan binding (11), further hinder direct correlation of steady-state and pre-steady-state kinetic parameters.
tRNA-dependent transition state stabilization
A considerable body of experimental data has accumulated on the structural and biochemical aspects of the tRNA aminoacylation reaction (4,21,24). Particularly in recent years, the majority of these studies have investigated the key structural requirements for efficient aminoacylation of specific tRNA substrates. This has allowed the identification and characterization of the key tRNA-synthetase interactions (22), most of which appear to be important during catalysis rather than recognition (‘kinetic specificity’; 25). The direct determination of free energy changes during the transfer of tryptophan to the cognate and non-cognate tRNAs provides new insights into the exact mechanism by which tRNA identity is manifested. In agreement with previous studies (26), changes in tRNA identity have little effect on the ability of aminoacyl-tRNA synthetases to bind tRNA. Instead, tRNA identity changes, as initially determined by steady-state kinetics, mainly correlate with differences in the energy for formation of the transition state for tryptophan transfer. The net result is that complexes formed between TrpRS and non-cognate tRNAs have to overcome a larger free energy barrier (ΔG≠) than cognate complexes for tryptophan transfer to take place; for the tRNATrPC73 mutant and the differences in ΔG≠ are ∼2 kcal mol-1. From transition state theory (see for example 5,27) it can be deduced that these changes in ΔG≠ reflect differences in the complementarity of the active site to the transition state. Thus, tRNATrP identity contributes to the degree of transition state complementarity in the active site of tRNATrP:TrpRS complexes. This represents, to the best of our knowledge, the first direct demonstration of an exact point at which tRNA identity can influence the efficiency of the aminoacylation reaction.
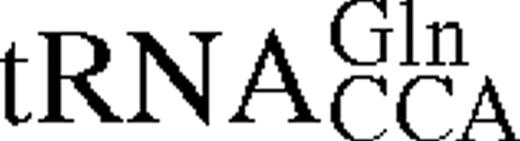
The determination of kinetic parameters for tryptophan transfer also allowed the extension of the free energy profile to the second step of the reaction catalyzed by E.coli TrpRS (Fig. 3). The extended free energy profile shows that the energetic barriers to the two transition states are similar in size, optimizing the overall rate of the reaction (20). The values, 14.5 kcal mol-1 for the formation of tryptophanyl adenylate and 15.2 kcal mol-1 for the transfer of tryptophan to tRNATrP, are comparable to those previously determined for B.stearo-thermophilus TyrRS and E.coli PheRS (6,7).
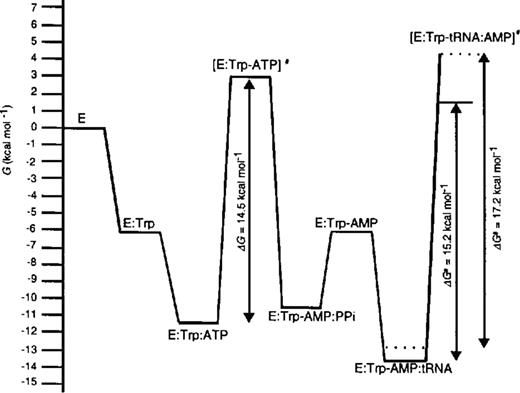
Gibbs' free energy profile for the aminoacylation of tRNATrP (solid line) and
Acknowledgement
This work was supported by a grant from the National Institute of General Medical Sciences to D.S. (GM 22854).
References
Author notes
Present addresses: Sanja Sever, The Scripps Research Institute, 10550 North Torrey Pines Road, IMM, Room 211, La Jolla, CA 92037, USA Mette Praetorius-Ibba, Carlsberg Laboratory, Department of Chemistry, Gamle Carlsberg Vej 10, DK-2500 Valby, Copenhagen, Denmark
The authors wish it to be known that, in their opinion, the first two authors should be considered as joint First Authors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments