





Abstract
Eukaryotic gene expression depends on a complex interplay between the transcriptional apparatus and chromatin structure. We report here a yeast model system for investigating the functional interaction between the human estrogen receptor (hER) and CTF1, a member of the CTF/NFI transcription factor family. We show that a CTF1-fusion protein and the hER transactivate a synthetic promoter in yeast in a synergistic manner. This interaction requires the proline-rich transactivation domain of CTF1. When the natural estrogen-dependent vitellogenin B1 promoter is tested in yeast, CTF1 and CTF1-fusion proteins are unable to activate transcription, and no synergy is observed between hER, which activates the B1 promoter, and these factors. Chromatin structure analysis on this promoter reveals positioned nucleosomes at −430 to −270 (±20 bp) and at −270 to− 100 (±20 bp) relative to the start site of transcription. The positions of the nucleosomes remain unchanged upon hormone-dependent transcriptional activation of the promoter, and the more proximal nucleosome appears to mask the CTF/NFI site located at −101 to −114. We conclude that a functional interaction of hER with the estrogen response element located upstream of a basal promoter occurs in yeast despite the nucleosomal organization of this promoter, whereas the interaction of CTF1 with its target site is apparently precluded by a nucleosome.
INTRODUCTION
Regulation of gene expression in eukaryotic cells is influenced by the packaging of DNA into nucleosomes as well as by other higher order chromosomal structures (1–5). In vitro experiments using reconstituted chromatin templates for transcriptional analyses have attempted to reconstruct some of the constraints imposed on transcription factors for binding to nucleosome-compacted DNA (see Ref. 1 for reviews). Depending on the relative positions of factor-binding sites and nucleosomes in chromatin, nucleosomes can either inhibit or facilitate the binding of transcription factors. It is now well established that multiple mechanisms exist to modulate the accessibility of key DNA sequences within or between nucleosomes, allowing transcription factors to function correctly in a chromosomal environment (4, 5).
Regulatory regions of genes are often maintained free of nucleosomes, as indicated by a high degree of nuclease sensitivity. Although it is generally accepted that sequence-specific factors are involved in the generation of these nuclease-hypersensitive sites, the mechanism by which this is achieved is still unclear. Possibly, the regulatory regions become accessible during DNA replication to sequence-specific factors produced at specific stages of development and tissue differentiation, such that these factors can compete with histones for DNA binding. Alternatively, transcription factors that bind to DNA between or within a nucleosome may themselves promote nucleosome remodeling or attract the appropriate factors to allow the assembly of a functional transcription machinery (Ref. 3 and references therein).
Although nucleosomes are generally thought to play an inhibitory role, DNA sequence-dependent constraints could position a nucleosome in such a way that it facilitates transcription. For instance, a static loop in the DNA may be generated by a positioned nucleosome, which juxtaposes otherwise distant cis-acting elements and favors the interaction and clustering of transcription factors to achieve stimulation of gene activity (1, 6).
The induction of gene expression by nuclear hormone receptors associated with chromatin has served as a model to analyze the interplay between inducible transcription factors and nucleosomal arrangement. A classic example is the opening of the mouse mammary tumor virus (MMTV) promoter by the glucocorticoid receptor (GR) (7). A well positioned nucleosome restricts the access of CTF/NFI to DNA. However, the activated GR can bind to glucocorticoid response elements at the surface of a positioned nucleosome leading to its perturbation in vivo, which enables CTF/NFI to bind and stimulate transcription (Refs. 8, 9 and references therein). A similar mechanism of nucleosome invasion by an activator that is not a nuclear receptor occurs during PHO5 induction in yeast (10). Others have extended such studies with different experimental systems showing disruption of chromatin structures after hormonal stimulation (11–13), confirming the prevailing view that nucleosomes are inhibitory and need to be structurally altered during the transcription activation process.
Another classic model system for investigating hormone-induced gene expression is provided by the vitellogenin genes (14). A role for several transcription factors, including the estrogen receptor (ER), liver-enriched and ubiquitous factors, has been suggested in the control for the liver- specific expression of these genes (Ref. 15 and references therein; Refs. 16–18). In vivo experiments have revealed alterations in chromatin structure in the promoter region of these genes after estrogen stimulation (19–21). Unexpectedly, in vitro transcription experiments showed that the combination of chromatin assembly and transcription factor binding involving a NFI-like factor strongly enhanced transcription from the Xenopus vitellogenin gene B1 promoter (22). Moreover, ER-dependent stimulation of transcription in nuclear extracts suggested that a positioned nucleosome, which wrapped DNA around the histone octamer core to create a loop, bringing distal ER and other binding sites close to proximal promoter elements, potentiates transcriptional activation (6). Although it was suggested that this nucleosome-mediated clustering of transcription factors close to the initiation site was responsible for transcriptional potentiation, it remained to be determined whether a nucleosome is positioned similarly in vivo.
The demonstration that the ER functions in yeast (23) makes it possible to study changes in chromatin structure that are dependent on the receptor and its activation by hormone. In this work, we have reconstituted an estrogen-responsive model system in Saccharomyces cerevisiae, to test whether the nucleosome positioning detected by in vitro chromatin assembly of the vitellogenin gene B1 promoter also occurs in vivo. We have screened for a functional interaction between the human estrogen receptor (hER) and CTF/NF1 (hereafter referred to as CTF1) similar to that observed in vitro, and we have examined how the nucleosomal organization of the vitellogenin gene B1 promoter in yeast affects its estrogen-dependent regulation.
The results from the yeast system demonstrate functional cooperation between hER and CTF1 fusion proteins on a synthetic promoter and indicate that the proline-rich transactivation domain of CTF1 is responsible for this interaction. Chromatin structure analysis in vivo of the natural vitellogenin B1 promoter in yeast cells reveals a nucleosome with multiple positions in the same region as that identified in in vitro assembly experiments. However, in yeast this nucleosome assumes a favored position more promoter proximal, such that the CTF1-binding site is included within the nucleosomal DNA. The location of this nucleosome is not altered by hormone-induced transcription nor by the presence of CTF1 during chromatin assembly. Thus, although hER does bind and activate the B1 promoter when organized into chromatin in yeast, CTF1 is unable to bind its consensus within the B1 promoter chromatin, and consequently fails to activate either alone or in concert with hER. Differences between yeast and mammalian nucleosomes, promoter flanking sequences, and/or the absence of accessory factors may be responsible for this discrepancy.
RESULTS
Establishment of an Estrogen-Responsive Yeast System
To create an inducible model system in which to study interactions between the ER and CTF1 on the natural vitellogenin promoter assembled into chromatin, we first subcloned the human ER [hERval 400-variant (24)] under the control of the galactose-inducible yeast GAL1 promoter on a 2μ plasmid (high-copy number episomal plasmid) (Fig. 1A). The correct expression of hER in yeast was analyzed by Western blot (Fig. 2A), and the in vitro DNA-binding activity in the presence or absence of hormone and antagonists was analyzed by a gel retardation assay (Fig. 2B). In agreement with previous finding (23), no DNA-protein complex is formed when estradiol is omitted (Fig. 2B, lane 2), even though the hER is highly expressed (see Western blot, Fig. 2A). In contrast, increasing the amounts of estradiol added to galactose-induced cultures enhances the levels of specific DNA-protein complex formed in vitro. In extracts of cells grown in presence of 5μ m of the antiestrogens, tamoxifen, nafoxidine, and ICI 164,384 (hereafter called ICI), alone, no complexes were detected although hER levels were similar to those found in extracts of cells grown in absence or presence of estrogen (data not shown). Furthermore, the levels of complexes were reduced in protein extracts from cells grown with both 10 nm estradiol and 5 μm antagonist compared with extracts of cells grown in the presence of estradiol alone (Fig. 2B), even though the hER was expressed in similar amounts (Fig. 2A). Together, these results demonstrate that the hER produced in yeast cells is strictly dependent on estradiol for binding to DNA in vitro. Moreover, anti-estrogens do not enhance binding, but rather reduce it, when they are added to the culture medium together with estrogen.
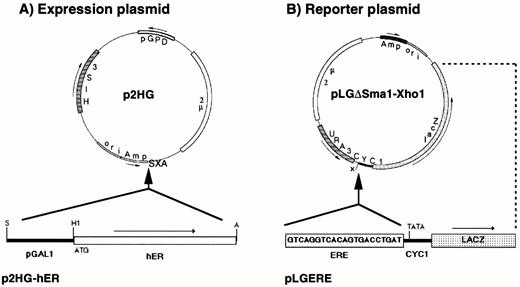
Plasmids Used for the Expression and Functional Analysis of hER in Yeast A, The hER cDNA was subcloned under the control of the inducible yeast GAL1 promoter (bottom scheme) into the SalI and ApaI sites (SXA) of the 2μ plasmid, p2HG. Arrows show orientation of transcription. The site of translation initiation, ATG, is positioned five nucleotides downstream from the BamHI site. Restriction enzyme sites used for the subcloning are indicated: A, ApaI; H1, BamHI; S, SalI; X, XhoI; pGPD indicates the promoter of the yeast glyceraldehyde-3-phosphate dehydrogenase gene. B, The reporter plasmid pLGΔSmaI-XhoI, in which all upstream activation sequences from the CYC1 promoter were removed by a religation of SmaI and filled XhoI sites, has only the sequences for basal transcription of the CYC1 promoter and a restored XhoI site. One copy of an ERE was cloned into the XhoI site at nucleotide −247 from the first ATG of the yeast gene CYC1. The E. coli β-galactosidase gene lacZ is the reporter gene used for the transcriptional analysis of the hybrid promoter in yeast.
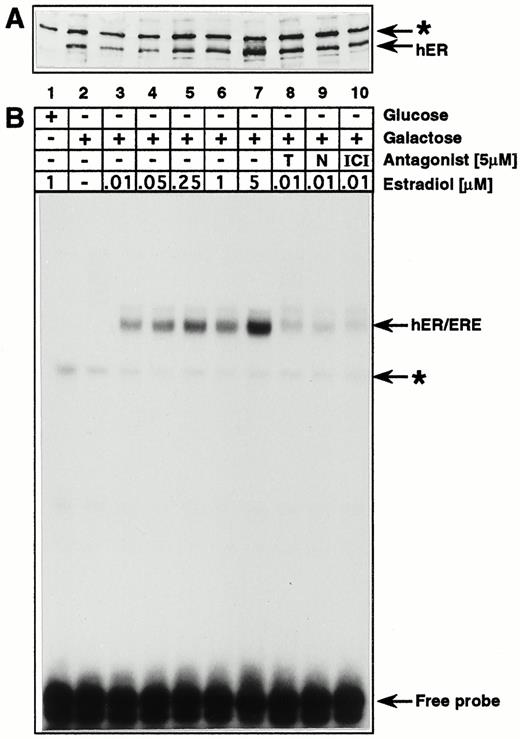
Expression and Estrogen-Dependent DNA-Binding Activity of hER in Yeast A, Western blot analysis of protein extracts from p2HG-hER-transformed yeast cells. Cells were grown in minimal medium in the presence of lactate and glycerol without glucose to A600 = 0.7. Then galactose or glucose and estradiol or estradiol antagonists were added as indicated in panel B, and the growth was continued for 8 h before protein extracts were prepared. Thirty micrograms of extract were loaded for each slot. A specific polyclonal antisera against the ER from X. laevis was used to detect the hER in yeast extracts. The arrows indicate the specific band for the hER and a nonspecific band for a cross-reacting yeast protein (*). B, Bandshift assay to analyze the DNA binding activity of the hER produced in yeast. Binding reactions were performed in buffer A supplemented with 0.1 m KCl as described in Materials and Methods, using 30 μg each of the same protein extracts as in panel A with 4 pmol specific DNA probe. A 32P-end-labeled oligomer, ds(GATCCGGGGTCACAGTGACCTA), containing one specific ERE with Sau3A ends, was used as a specific DNA probe. The abbreviations T, N, and ICI stand for the estradiol antagonists, tamoxifen, nafoxidine, and ICI, respectively. The arrows indicate the hER-specific DNA-protein complex and a nonspecific yeast protein-DNA complex (*).
We next analyzed the ability of the hER to stimulate transcription of a target gene in yeast, which consists of a consensus estrogen response element [ERE (25, 26)] inserted upstream of the minimal yeast CYC1 promoter driving transcription of lacZ coding sequences (Fig. 1B). β-Galactosidase activity was determined in protein extracts from transformed yeast cells grown in the absence or presence of increasing amounts of estradiol (Table 1). In the absence of hormone, a basalβ -galactosidase activity was detected, which corresponds to the amount found in yeast cells transformed with the control p2HG expression plasmid that does not carry hER sequences. Depending on the copy number of the hER expression plasmid, maximal stimulation is reached with 1 μm estradiol for the CEN-based plasmid (single-copy number centromere-containing plasmid), and with 50 nm estradiol for the 2μ-based hER expression plasmid (Table 1). To analyze the effects of antiestrogens on the transcriptional activity of the hER in yeast on a CEN-based (single-copy) expression plasmid, β-galactosidase activity was determined in protein extracts isolated from galactose-induced cells grown in the presence of 5 μm antagonist, with or without the addition of 60 nm estradiol. None of the tested antagonists, tamoxifen, nafoxidine, or ICI, showed any agonist activity in the absence of estrogen (Table 2). In the presence of 60 nm estradiol and 5 μm of either tamoxifen, nafoxidine, or ICI, the β-galactosidase levels drop by 40–55% compared with those with 60 nm estrogen alone (Table 2). The effect of antiestrogens on the transcriptional activity of hER is less pronounced than that observed on in vitro DNA binding (Fig. 2B). This discrepancy, however, reflects the nature of the two different methods used: β-galactosidase accumulates in the yeast cell during the growth in the presence of overexpressed hER, whereas the gel retardation assay represents the DNA binding activity of hER at a given timepoint. Our results allow us to conclude that the expressed hER mediates a strictly estrogen-dependent and an antagonist-sensitive activation of transcription in yeast.
Estrogen-Responsive Transcriptional Activity of the hER in Yeast
| Transcriptional Activationa | ||
|---|---|---|
| Estradiol | Expression plasmid | |
| CEN | 2μ | |
| None | 5 ± 1 | 6 ± 1 |
| 10 nm | 413 ± 119 | 1373 ± 339 |
| 50 nm | 1484 ± 117 | 2052 ± 351 |
| 1 μm | 1836 ± 104 | 2098 ± 795 |
| 5 μm | 2156 ± 506 | 2115 ± 522 |
| Transcriptional Activationa | ||
|---|---|---|
| Estradiol | Expression plasmid | |
| CEN | 2μ | |
| None | 5 ± 1 | 6 ± 1 |
| 10 nm | 413 ± 119 | 1373 ± 339 |
| 50 nm | 1484 ± 117 | 2052 ± 351 |
| 1 μm | 1836 ± 104 | 2098 ± 795 |
| 5 μm | 2156 ± 506 | 2115 ± 522 |
β-Galactosidase activities from extracts of yeast cells transformed with the ERE-containing reporter plasmid pLGERE and a CEN-based (single-copy) or a 2μ-based (multi-copy) hER expression plasmid p2HG-hER (see Fig. 1 for plasmid structure). Transformed yeast cells were grown as indicated in Fig. 2 in the presence of galactose, and estradiol was added as shown. Protein extracts were prepared and β-galactosidase activities were determined as described in Materials and Methods. Two independent transformants were analyzed.
Estrogen-Responsive Transcriptional Activity of the hER in Yeast
| Transcriptional Activationa | ||
|---|---|---|
| Estradiol | Expression plasmid | |
| CEN | 2μ | |
| None | 5 ± 1 | 6 ± 1 |
| 10 nm | 413 ± 119 | 1373 ± 339 |
| 50 nm | 1484 ± 117 | 2052 ± 351 |
| 1 μm | 1836 ± 104 | 2098 ± 795 |
| 5 μm | 2156 ± 506 | 2115 ± 522 |
| Transcriptional Activationa | ||
|---|---|---|
| Estradiol | Expression plasmid | |
| CEN | 2μ | |
| None | 5 ± 1 | 6 ± 1 |
| 10 nm | 413 ± 119 | 1373 ± 339 |
| 50 nm | 1484 ± 117 | 2052 ± 351 |
| 1 μm | 1836 ± 104 | 2098 ± 795 |
| 5 μm | 2156 ± 506 | 2115 ± 522 |
β-Galactosidase activities from extracts of yeast cells transformed with the ERE-containing reporter plasmid pLGERE and a CEN-based (single-copy) or a 2μ-based (multi-copy) hER expression plasmid p2HG-hER (see Fig. 1 for plasmid structure). Transformed yeast cells were grown as indicated in Fig. 2 in the presence of galactose, and estradiol was added as shown. Protein extracts were prepared and β-galactosidase activities were determined as described in Materials and Methods. Two independent transformants were analyzed.
Effects of Estradiol Antagonists onβ -Galactosidase Expression
| Transcriptional Activationa | ||
|---|---|---|
| Antagonist | Estradiol | |
| None | 60 nm | |
| Without | 6 ± 1 | 1658 ± 163 |
| Tamoxifen | 8 ± 1 | 736 ± 46 |
| Nafoxidine | 7 ± 1 | 861 ± 52 |
| ICI | 6 ± 1 | 987 ± 35 |
| Transcriptional Activationa | ||
|---|---|---|
| Antagonist | Estradiol | |
| None | 60 nm | |
| Without | 6 ± 1 | 1658 ± 163 |
| Tamoxifen | 8 ± 1 | 736 ± 46 |
| Nafoxidine | 7 ± 1 | 861 ± 52 |
| ICI | 6 ± 1 | 987 ± 35 |
Cells containing the CEN-based expression plasmid p2HG-hER and the reporter plasmid pLGERE were grown as in Table 1 in the presence of galactose. In addition, estradiol and 5 μm of the antagonists, tamoxifen, nafoxidine, or ICI, were included as indicated. Two independent transformants were analyzed.
Effects of Estradiol Antagonists onβ -Galactosidase Expression
| Transcriptional Activationa | ||
|---|---|---|
| Antagonist | Estradiol | |
| None | 60 nm | |
| Without | 6 ± 1 | 1658 ± 163 |
| Tamoxifen | 8 ± 1 | 736 ± 46 |
| Nafoxidine | 7 ± 1 | 861 ± 52 |
| ICI | 6 ± 1 | 987 ± 35 |
| Transcriptional Activationa | ||
|---|---|---|
| Antagonist | Estradiol | |
| None | 60 nm | |
| Without | 6 ± 1 | 1658 ± 163 |
| Tamoxifen | 8 ± 1 | 736 ± 46 |
| Nafoxidine | 7 ± 1 | 861 ± 52 |
| ICI | 6 ± 1 | 987 ± 35 |
Cells containing the CEN-based expression plasmid p2HG-hER and the reporter plasmid pLGERE were grown as in Table 1 in the presence of galactose. In addition, estradiol and 5 μm of the antagonists, tamoxifen, nafoxidine, or ICI, were included as indicated. Two independent transformants were analyzed.
Expression of Functional CTF1 in Yeast
We have previously shown that a Gal4-CTF1 fusion protein can synergize with hER in HeLa cells to activate the transcription of a synthetic promoter composed of the adenovirus E1b TATA box, an ERE, and a Gal4-binding site (27). Furthermore, in liver nuclear extracts isolated from Xenopus laevis, CTF1 cooperates with ER to stimulate in vitro the natural vitellogenin B1 promoter on which the ERE is relatively far upstream of the transcription initiation site (28). Therefore, it was of interest to test the functional interaction between these two transcription factors in yeast. To this end, CTF1 was expressed under the control of the galactose-inducible yeast GAL1 promoter (Fig. 3A). First, whole-cell extracts were tested for CTF1 DNA-binding activity by gel retardation assay (Fig. 3C). Only extracts isolated from galactose-induced cells show a specific DNA-protein complex (lane 2), in contrast to extracts isolated from cells grown in the presence of glucose, in which no complexes are formed (lane 1).
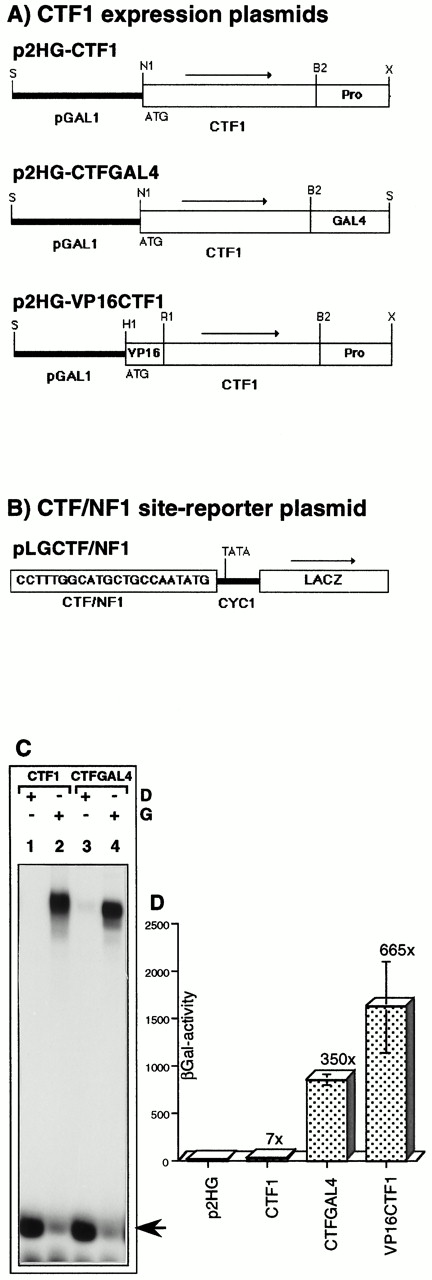
Expression, DNA Binding Activity, and Transcriptional Activity of the Human Transcription Factor, CTF1/NFI, in Yeast A, Diagrams of the inserts used for expressing CTF1 and CTF1 derivatives in yeast, all under the control of the inducible yeast GAL1 promoter into the 2 μ plasmid, p2HG. The names of the plasmids are indicated above each diagram. Arrows show the orientation of transcription. The site of translation initiation, ATG, is positioned two nucleotides downstream from the NcoI site (N1 in p2HG-CTF1 and p2HG-CTFGAL4), respectively, nine nucleotides from the filled BamHI site (H1 in p2HG-VP16CTF1). In p2HG-CTFGAL4, the C-terminal proline-rich transcription activation domain of CTF1 was replaced by the acidic transcription activation domain of the yeast GAL4 protein. In p2HG-VP16CTF1, the acidic transcription activation domain of the viral VP16 protein was fused in frame to the N terminus of CTF1. p2HG-VP16CTF1 is a 2μ-based derivative of p2HG-VP16CTF1/hER from Fig. 4. Restriction enzyme sites used for the subcloning are indicated: B2, BglII; H1, BamHI; N1, NcoI; R1, EcoRI; S, SalI; X, XhoI. B, Reporter plasmid used for CTF1-dependent transcription activation analysis. One copy of a CTF/NF1 DNA-binding site was inserted into the XhoI site of pLGΔSmaI-XhoI as in Fig. 1. C, Bandshift assay to analyze the DNA-binding activity of CTF1 derivatives expressed in yeast. Cells transformed either with the expression plasmid, p2HG-CTF1 or p2HG-CTFGAL4, were grown as described in Fig. 2. Binding reactions were performed as described in Materials and Methods, using 50 μg protein extract each with 0.2 pmol specific DNA probe. A 32P-end-labeled oligomer, ds(CCTTTGGCATGCTGCCAATATG), containing one specific CTF/NFI DNA-binding site, was used as a specific DNA probe, and glucose (D) or galactose (G) were added as indicated. The arrow indicates the migration of the free probe. D, Transcriptional activity of CTF1 and CTF1-derivatives. β-Galactosidase activities were measured in extracts isolated from yeast cells transformed with p2HG-based CTF1 expression- and pLGCTF/NFI reporter plasmids. Cells were grown and the GAL1 promoter was induced by galactose as in panel C. Protein extracts were prepared and β-galactosidase activities were determined as described in Materials and Methods. The error bars indicate sd values from measurements of three independent transformants each. The fold stimulation of β-galactosidase activity is given with respect to the basal activity (2.5 ± 1.3) determined in the p2HG transformants.
Second, to analyze the transactivation capacity of CTF1 in yeast, a reporter plasmid was constructed (Fig. 3B) containing one copy of a consensus CTF/NFI DNA-binding site (29) upstream of the minimal yeast CYC1 promoter that drives expression of lacZ coding sequences. β-Galactosidase activity was determined in protein extracts from yeast cells transformed with both the CTF1 expression plasmid and the CTF/NFI-dependent reporter plasmid. CTF1 induces a weak 7-fold stimulation of β-galactosidase activity as compared with the basal activity of the reporter gene cotransformed with the control p2GH expression plasmid lacking CTF1-coding sequences (Fig. 3D). To test whether this low transcription activation mediated by CTF1 reflects the weak activity in yeast of proline-rich transactivation domains, the latter was replaced by the C-terminal 129 amino acids (aa) of yeast Gal4, which is an acidic transcription activation domain (see Materials and Methods and Fig. 3A). The expression and DNA-binding activity of this fusion protein, CTF1-Gal4, was tested by gel retardation assay (Fig. 3C). An abundant and specific DNA-protein complex is only visible in extracts isolated from galactose-induced cells (lane 4). Furthermore, the hybrid CTF1-Gal4 factor is 50 times more active in the transcription induction assay than wild-type CTF1 (Fig. 3D). Similarly, a fusion protein comprising the full-length CTF1 and the C-terminal 80 aa of the herpes simplex virus protein VP16 is 95 times stronger than CTF1 (Fig. 3, A and D). These results indicate that the proline-rich transactivation domain of human CTF1 functions poorly in yeast, although it does not negatively affect the function of an acidic domain when associated with it in a fusion protein (VP16-CTF1). Finally, full substitution of the proline-rich domain by the GAL4 acidic domain activates strongly in vivo.
Functional Interaction between hER and CTF1 in Yeast
Having shown that hER as well as CTF1 and CTF1 derivatives can be expressed in yeast and that they transactivate their corresponding reporter genes, we tested the ability of these transcription factors to function additively or synergistically on a promoter containing both binding sites. To this end, an ER- and CTF1-dependent reporter plasmid was constructed by inserting two consensus EREs [(ERE)2] upstream of the CTF/NFI element in pLGCTF/NFI to give pLGERE-CTF/NFI (Fig. 4B). In addition, expression plasmids were constructed that coexpress the hER and CTF1 derivatives under the control of the yeast GAL1 promoter (Fig. 4A). To overcome saturation of induction, which would mask synergism due to a high overexpression of hER and CTF1-derivatives with 2μ-based plasmids, a CEN-based plasmid was used for hER and CTF1 derivative coexpression. This reduces the level of expression of hER and CTF1 derivatives by about 50-fold, although their transcriptional activity remains similar (data not shown).β -Galactosidase activity was determined in protein extracts from yeast cells transformed with plasmids expressing either hER, CTF1-GAL4 and hER, VP16-CTF1 and hER, or VP16-CTFΔ and hER (VP16-CTFΔ is a VP16-CTF1 derivative, in which the C-terminal 100 aa proline-rich domain of CTF1 was deleted). Since the activity of hER is strictly dependent on estrogen (see above), the stimulation mediated by CTF1 or the CTF1 derivatives can be determined in coexpression experiments in the absence of hormone. When CTF1 is coexpressed with hER, the hormone-dependent stimulation is similar to that obtained with hER alone, indicating that there is no synergism between the two factors (data not shown). The highest transcriptional stimulation of the reporter gene is obtained when the hER and VP16-CTF1 fusion protein are coexpressed in the presence of estrogen. The induced level is 16,000-fold over that of basal promoter activity (Fig. 4C). This strong estrogen-dependent activation of the reporter promoter represents an almost 3-fold higher activity than the sum of the transcription activities obtained with each activator tested separately (Fig. 4C). In contrast, the stimulation obtained with CTF1-Gal4 and hER is additive; stimulation obtained with VP16-CTFΔ and hER is slightly more than additive, suggesting the absence of functional interaction and a weak synergism, respectively.
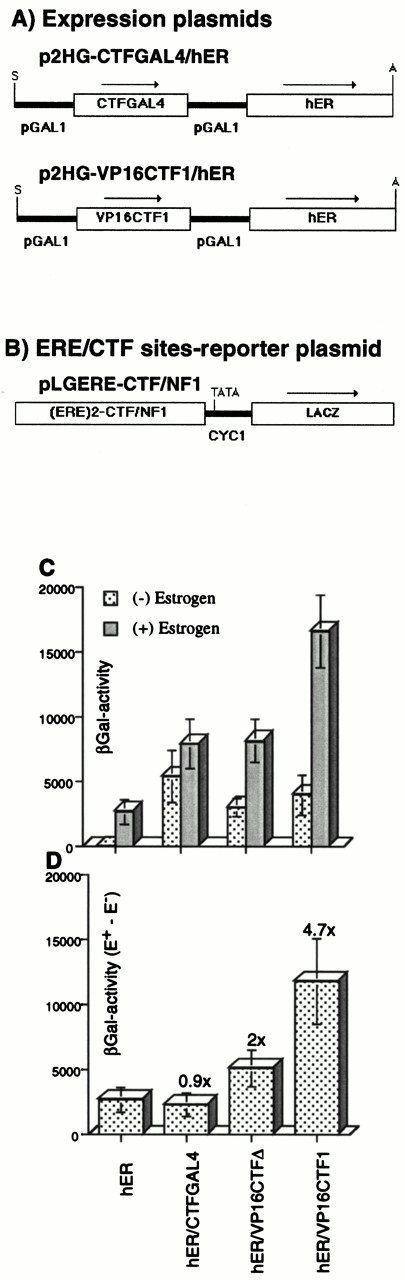
Synergistic Transcriptional Activity of Coexpressed hER and CTF1 A, Diagram of the CEN-based yeast expression vectors used. Both the hER and CTF1 derivatives were subcloned under the control of the yeast GAL1 promoter into a CEN-based p2HG-derivative, in which the 2.2-kb yeast 2 μ DNA element was replaced by a 2.5-kb DNA element, containing the yeast ARS1-CEN4 sequence. Arrows show the orientation of transcription. Restriction enzyme sites used for the subcloning are indicated: A, ApaI; S, SalI. Panel B shows the reporter plasmid used. Two copies of an estrogen response element (ERE)2 and one copy of a CTF/NFI-specific DNA element were fused at nucleotide position −247 from the first ATG of the yeast CYC1 gene into the XhoI site of pLGΔSmaI-XhoI as in (Fig. 1). C, Transcriptional analysis of coexpressed ER and CTF1 derivatives in yeast cells. β-Galactosidase activities were measured in extracts isolated from yeast cells transformed with either p2HG-hER, p2HG-CTFGAL4/hER, p2HG-VP16CTFΔ/hER, or p2HG-VP16CTF1/hER CEN-based expression plasmids and the pLGERE-CTF/NFI 2μ -based reporter plasmid. p2HG-VP16CTFΔ/hER is identical to p2HG-VP16CTF1/hER, except it has the 100 aa long proline-rich domain of CTF1 deleted. Cells were grown, protein extracts were prepared, and β-galactosidase activities were determined as indicated in Table 1. D, Potentiation of the hER activity by CTF1-derivatives. β-Galactosidase activities from panel C are shown as the differences between activities of extracts grown in the presence of estrogen minus the activities from extracts grown in the absence of estrogen (Δ (E+ − E−). The error bars indicate sd from measurements of five independent transformants each. The potentiation factor (fold synergism) is given.
Another way to determine whether the presence of the CTF1 derivatives potentiates hER-induced stimulation, while taking into account varying levels of expression in different transformants, is to represent the difference (Δ) of the β-galactosidase activities from each individual transformants grown in the presence (E+, combined effects of CTF1 derivatives and hER) or absence (E−, effect of CTF1 derivatives only) of estrogen, i.e. Δ [E+ − E−] (Fig. 4D). The results confirm that the CTF1-Gal4 construct that lacks the proline-rich activation domain does not synergize with the hER. Similarly, VP16-CTFΔ, which also lacks the proline-rich domain of CTF1, shows only a 2-fold higher Δ (E+ − E−) activity than seen with hER alone. This suggests that the acidic activation domain of VP16 alone can only weakly synergize with the hER in yeast. However, since the VP16-CTF1 fusion potentiates the action of hER by a factor of 4.7, we conclude that the proline-rich domain of CTF1, in association with the acidic domain of VP16, creates a strong synergistic activation.
Analysis of the X. laevis Vitellogenin Promoter B1 in Yeast
We have demonstrated above that CTF1 derivatives and hER are capable of synergistic activation in yeast of a synthetic promoter composed of two estrogen-responsive elements and one copy of a consensus CTF/NFI site, which thus allow functional cooperativity between factors binding to them. However, it is not clear whether this holds true for a natural estrogen-dependent promoter with its native arrangement of ER and CTF/NFI-binding sites and its propensity for forming nucleosomes. To answer this, we analyzed the promoter of the vitellogenin B1 gene of X. laevis (30), which contains three imperfect copies of the ERE at −314, −334 [forming the estrogen-responsive unit (ERU)], and −555 as well as a strong CTF/NF1 site at −101 (15, 26, 28). Either the full-length vitellogenin B1 promoter (−596 until −42) or a 3′-deleted version of it (−596 until −235) was subcloned upstream of the yeast CYC1 minimal promoter driving lacZ, to create yeast reporter plasmids dependent on the vitellogenin B1 promoter region (Fig. 5). In the latter, the EREs are brought close to the TATA box, and the strong CTF/NFI binding site is deleted. In these constructs, the vitellogenin B1 TATA box region (−41 until+ 1) was replaced by the yeast CYC1 TATA box sequences (−245 until +1), because the former shows a very high basal activity in yeast.
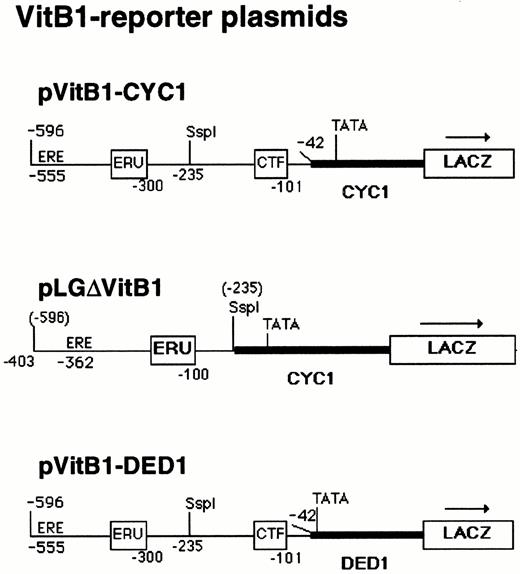
Reporter Plasmids Used for the Transcriptional Analysis of the X. laevis Vitellogenin (VitB1) Promoter in Yeast pVitB1-CYC1 contains 554 nucleotides of vitellogenin B1 promoter sequences including all control elements, except the TATA box, fused to the minimal yeast CYC1 promoter in pLGΔSmaI-XhoI (see Fig. 1). In pLGΔVitB1, 193 nucleotides of promoter-proximal sequences including the CTF/NF1 binding site (CTF) were deleted, locating the vitellogenin B1 upstream promoter sequences (−596 to −235) close to the CYC1 TATA box in pLGΔSmaI-XhoI. pVitB1-DED1 is a derivative of pVitB1-CYC1, in which the CYC1 promoter is replaced by the minimal yeast DED1 promoter. Positions of the EREs (ERE and ERU) and the CTF/NF1-specific DNA element (CTF) are indicated. Numbers correspond to vitellogenin B1 promoter sequences upstream from the site of transcription initiation. Numbers in parentheses above the pLGΔVitB1 diagram correspond to original vitellogenin B1 promoter sequences before the deletion. Arrows show the orientation of transcription from the reporter gene, lacZ.
Transcription activation analyses of these reporter promoters were performed and are summarized in Table 3. In the absence of estrogen, CTF1-Gal4 showed a weak 3-fold stimulation, and VP16-CTF1 was unable to stimulate the full-length vitellogenin B1 promoter (pVitB1-CYC1), although both CTF1 and a CTF1 derivative were able to bind the CTF/NF1 site in gel retardation assays in vitro (data not shown). Control experiments showed that both CTF1-derivatives are fully competent for activation from a synthetic promoter containing a single CTF/NF1-binding site. Furthermore, the integrity of this binding site in the full-length B1 promoter construct was confirmed by sequencing. On the other hand, hER mediates a more than 40-fold estrogen-dependent activation of the full-length promoter and an even stronger activation (∼200-fold) of its 3′-truncated version (pLGΔVitB1). Finally, the VP16-CTF1 fusion fails to synergize with hER on the vitellogenin B1 promoter, although the two proteins activate synergistically on a synthetic promoter, which bears both binding sites immediately upstream of the TATA box (see Fig. 4). Compared with the activation of this latter synthetic promoter, the stimulation by the hER of the full-length vitellogenin B1 promoter and its 3′-deleted version is at least 20 times and 5 times lower, respectively. From these results, we conclude that the EREs in the native vitellogenin B1 promoter are functional in yeast in a distance-dependent manner. In contrast, the CTF/NFI-binding site very weakly promotes transcription by binding CTF1-Gal4 (100–150 times less than on the synthetic promoter) and is unable to do so by binding CTF1 or VP16-CTF1 either on their own or in combination with hER.
Estrogen-Dependent Activities from the Vitellogenin B1 Promoter1
| Transactivator | Replication (origin) | Reporter Plasmid | |||||
|---|---|---|---|---|---|---|---|
| pVitB1-CYC1 | pLGΔVitB1 | pVitB1-DED1 | |||||
| Estrogen | Estrogen | Estrogen | |||||
| None | 1 μm | None | 1 μm | None | 1 μm | ||
| p2HG | (2μ) | 2.0 ± 0.6 | 1.6 ± 0.8 | 1.2 ± 0.2 | 0.8 ± 0.3 | 175 ± 25 | 200 ± 25 |
| p2HG-hER | (2μ) | 1.8 ± 0.5 | 87 ± 10 | 1.0 ± 0.5 | 440 ± 18 | 232 ± 21 | 553 ± 179 |
| p2HG-hER | (CEN) | 0.3 ± 0.1 | 5.4 ± 1.0 | n.d. | n.d. | 6.4 ± 2.5 | 62 ± 13 |
| p2HG-CTFGAL4 | (2μ) | 6.1 ± 2.3 | n.d. | 1.6 ± 0.2 | n.d. | 695 ± 126 | 713 ± 91 |
| p2HG-CTFGAL4/hER | (2μ) | 6.9 ± 3 | 88 ± 6 | 1.8 ± 0.4 | 753 ± 65 | 509 ± 140 | 806 ± 203 |
| p2HG-VP16CTF1/hER | (2μ) | 1.8 ± 0.7 | 103 ± 41 | 1.9 ± 0.1 | 380 ± 65 | 403 ± 39 | 638 ± 42 |
| p2HG-VP16CTF1/hER | (CEN) | 0.7 ± 0.2 | 9.1 ± 2.0 | n.d. | n.d. | 5.3 ± 1.3 | 47 ± 6 |
| Transactivator | Replication (origin) | Reporter Plasmid | |||||
|---|---|---|---|---|---|---|---|
| pVitB1-CYC1 | pLGΔVitB1 | pVitB1-DED1 | |||||
| Estrogen | Estrogen | Estrogen | |||||
| None | 1 μm | None | 1 μm | None | 1 μm | ||
| p2HG | (2μ) | 2.0 ± 0.6 | 1.6 ± 0.8 | 1.2 ± 0.2 | 0.8 ± 0.3 | 175 ± 25 | 200 ± 25 |
| p2HG-hER | (2μ) | 1.8 ± 0.5 | 87 ± 10 | 1.0 ± 0.5 | 440 ± 18 | 232 ± 21 | 553 ± 179 |
| p2HG-hER | (CEN) | 0.3 ± 0.1 | 5.4 ± 1.0 | n.d. | n.d. | 6.4 ± 2.5 | 62 ± 13 |
| p2HG-CTFGAL4 | (2μ) | 6.1 ± 2.3 | n.d. | 1.6 ± 0.2 | n.d. | 695 ± 126 | 713 ± 91 |
| p2HG-CTFGAL4/hER | (2μ) | 6.9 ± 3 | 88 ± 6 | 1.8 ± 0.4 | 753 ± 65 | 509 ± 140 | 806 ± 203 |
| p2HG-VP16CTF1/hER | (2μ) | 1.8 ± 0.7 | 103 ± 41 | 1.9 ± 0.1 | 380 ± 65 | 403 ± 39 | 638 ± 42 |
| p2HG-VP16CTF1/hER | (CEN) | 0.7 ± 0.2 | 9.1 ± 2.0 | n.d. | n.d. | 5.3 ± 1.3 | 47 ± 6 |
β-Galactosidase activities from vitellogenin B1 promoter derivatives were measured in extracts isolated from yeast cells transformed with the 2μ- or CEN-based pVitB1-CYC1, pLGΔVitB1, or pVitB1-DED1 reporter plasmids (see Fig. 5) and one of the following 2μ-based expression plasmids: p2HG, p2HG-hER (see Fig. 1), p2HG-CTFGAL4 (see Fig. 3), p2HG-CTFGAL4/hER, and p2HG-VP16CTF1/hER (see Fig. 4). Cells were grown and β-galactosidase activities were determined as described in Table 1. sd values are from measurements of three independent transformants each. (n.d., not determined).
Estrogen-Dependent Activities from the Vitellogenin B1 Promoter1
| Transactivator | Replication (origin) | Reporter Plasmid | |||||
|---|---|---|---|---|---|---|---|
| pVitB1-CYC1 | pLGΔVitB1 | pVitB1-DED1 | |||||
| Estrogen | Estrogen | Estrogen | |||||
| None | 1 μm | None | 1 μm | None | 1 μm | ||
| p2HG | (2μ) | 2.0 ± 0.6 | 1.6 ± 0.8 | 1.2 ± 0.2 | 0.8 ± 0.3 | 175 ± 25 | 200 ± 25 |
| p2HG-hER | (2μ) | 1.8 ± 0.5 | 87 ± 10 | 1.0 ± 0.5 | 440 ± 18 | 232 ± 21 | 553 ± 179 |
| p2HG-hER | (CEN) | 0.3 ± 0.1 | 5.4 ± 1.0 | n.d. | n.d. | 6.4 ± 2.5 | 62 ± 13 |
| p2HG-CTFGAL4 | (2μ) | 6.1 ± 2.3 | n.d. | 1.6 ± 0.2 | n.d. | 695 ± 126 | 713 ± 91 |
| p2HG-CTFGAL4/hER | (2μ) | 6.9 ± 3 | 88 ± 6 | 1.8 ± 0.4 | 753 ± 65 | 509 ± 140 | 806 ± 203 |
| p2HG-VP16CTF1/hER | (2μ) | 1.8 ± 0.7 | 103 ± 41 | 1.9 ± 0.1 | 380 ± 65 | 403 ± 39 | 638 ± 42 |
| p2HG-VP16CTF1/hER | (CEN) | 0.7 ± 0.2 | 9.1 ± 2.0 | n.d. | n.d. | 5.3 ± 1.3 | 47 ± 6 |
| Transactivator | Replication (origin) | Reporter Plasmid | |||||
|---|---|---|---|---|---|---|---|
| pVitB1-CYC1 | pLGΔVitB1 | pVitB1-DED1 | |||||
| Estrogen | Estrogen | Estrogen | |||||
| None | 1 μm | None | 1 μm | None | 1 μm | ||
| p2HG | (2μ) | 2.0 ± 0.6 | 1.6 ± 0.8 | 1.2 ± 0.2 | 0.8 ± 0.3 | 175 ± 25 | 200 ± 25 |
| p2HG-hER | (2μ) | 1.8 ± 0.5 | 87 ± 10 | 1.0 ± 0.5 | 440 ± 18 | 232 ± 21 | 553 ± 179 |
| p2HG-hER | (CEN) | 0.3 ± 0.1 | 5.4 ± 1.0 | n.d. | n.d. | 6.4 ± 2.5 | 62 ± 13 |
| p2HG-CTFGAL4 | (2μ) | 6.1 ± 2.3 | n.d. | 1.6 ± 0.2 | n.d. | 695 ± 126 | 713 ± 91 |
| p2HG-CTFGAL4/hER | (2μ) | 6.9 ± 3 | 88 ± 6 | 1.8 ± 0.4 | 753 ± 65 | 509 ± 140 | 806 ± 203 |
| p2HG-VP16CTF1/hER | (2μ) | 1.8 ± 0.7 | 103 ± 41 | 1.9 ± 0.1 | 380 ± 65 | 403 ± 39 | 638 ± 42 |
| p2HG-VP16CTF1/hER | (CEN) | 0.7 ± 0.2 | 9.1 ± 2.0 | n.d. | n.d. | 5.3 ± 1.3 | 47 ± 6 |
β-Galactosidase activities from vitellogenin B1 promoter derivatives were measured in extracts isolated from yeast cells transformed with the 2μ- or CEN-based pVitB1-CYC1, pLGΔVitB1, or pVitB1-DED1 reporter plasmids (see Fig. 5) and one of the following 2μ-based expression plasmids: p2HG, p2HG-hER (see Fig. 1), p2HG-CTFGAL4 (see Fig. 3), p2HG-CTFGAL4/hER, and p2HG-VP16CTF1/hER (see Fig. 4). Cells were grown and β-galactosidase activities were determined as described in Table 1. sd values are from measurements of three independent transformants each. (n.d., not determined).
The CTF/NFI Site on the Vitellogenin B1 Promoter Is within a Nucleosome
To better understand why CTF1 and CTF1 fusion proteins do not activate the X. laevis vitellogenin B1 promoter in yeast, we tested the possibility that the chromatin structure of the vitellogenin B1 promoter prevents the binding of CTF1. It has previously been shown on in vitro reconstituted chromatin templates that a nucleosome is positioned between −300 and− 140 on the vitellogenin B1 promoter, immediately upstream of the CTF/NFI-binding site at position −101 to −114 (6, 15). This positioned nucleosome does not prevent CTF1 binding (6). If nucleosome positioning also occurs in yeast in this region, a slight shift in the position of the nucleosome might interfere with CTF1 binding. Therefore, we analyzed the low-resolution chromatin structure of the vitellogenin B1 promoter by mapping double-strand cuts of micrococcal nuclease (MNase)- digested yeast nuclei using the indirect end-labeling method (Fig. 6A). In comparison to the cleavage of naked DNA, three preferred MNase cleavage sites are centered around positions −430, −270, and −100 bp (±20 bp) relative to the start site of transcription on the vitellogenin B1 promoter, with the latter position being slightly diffuse. The pattern is the same in the absence or presence of hER and estradiol, and it is consistent with nucleosomes being positioned between the cleavage sites as the enzyme cuts preferentially in the linker regions between nucleosomes.

MNase Digestion Analysis of the X. laevis Vitellogenin B1 Promoter in Yeast A, Analysis of the vitellogenin B1-CYC1 fusion promoter. Yeast cells were transformed with pVitB1-CYC1 (see Fig. 5) and either p2HG or p2HG-hER (see Fig. 1). Cells were grown in minimal medium in the presence of lactate and glycerol without glucose until A600 = 0.7. Galactose and 1 μm estradiol were added, and growth was continued for 5–6 h. Yeast nuclei were prepared as described in Materials and Methods. MNase digestions were performed with nuclei isolated from p2HG-hER-transformed yeast cells (+hER) and from p2HG-transformed cells (−hER). For the naked DNA control, DNA was isolated from nuclei before MNase digestion. Samples were treated with MNase at 37 C for 10 min with 2.5 U/ml (lanes 1 and 7), 5 U/ml (lanes 2 and 8), 10 U/ml (lanes 3 and 9), 20 U/ml (lanes 4 and 10), 40 U/ml (lanes 5 and 11), 80 U/ml (lanes 6 and 12), 0.8 U/ml (lane 13), 1.6 U/ml (lane 14), 3.2 U/ml (lane 15), 6.5 U/ml (lane 16), and 26 U/ml (lane 17). The MNase cleavage sites were mapped relative to the XbaI site (X), which is located 12 bp upstream from the 5′-end (−596) of the vitellogenin B1 promoter in pVitB1-CYC1. The indirect end-label analysis was performed as described in Materials and Methods using a 158-bp long DNA fragment that overlaps the 5′-end of the VitB1 promoter (−596 to −446). The positions of marker (M) bands are given in base pairs; M1 is a 123-bp DNA ladder. The vitellogenin B1-CYC1 fusion promoter is shown as a diagram along the autoradiogram. Numbers in parentheses correspond to vitellogenin B1 promoter sequences upstream from the site of transcription initiation (−), respectively, to lacZ gene sequences downstream from the site of translation initiation (+). MNase digestion-hypersensitive sites, corresponding to nucleosome linker regions, are indicated by arrows. Lanes 9 and 16 were scanned and plotted as nuclei (9 ) and as Δ9–16, in which the signal from the free DNA (lane 16) was subtracted from the nuclei (lane 9). B, Analysis of the vitellogenin B1-DED1 fusion promoter. Yeast cells were transformed with pVitB1-DED1 (see Fig. 5) and p2HG-hER (see Fig. 1). Samples were digested with MNase as in panel A using 0.3 U/ml (lane 1), 0.63 U/ml (lane 2), 1.25 U/ml (lane 3), 2.5 U/ml (lane 4), 5 U/ml (lane 5), 10 U/ml (lane 6), 20 U/ml (lane 7), and 40 U/ml (lane 8). The MNase cleavage sites were mapped as in panel A. M2 is a 1-kb DNA ladder. A scheme for the vitellogenin B1-DED1 fusion promoter is shown along the autoradiogram as in panel A. Lanes 2 and 4 were scanned and plotted as in panel A.
The CYC1 promoter contains five TATA box sequences (31), and at least two of them are required for transcription at the normal level (32). We could test whether the diffuse positioning of the promoter-proximal nucleosome (−100) is due to different transcription initiation sites of the VitB1-CYC1 fusion promoter, by replacing the CYC1 promoter in pVitB1-CYC1 with the yeast DED1 minimal promoter (33). In pVitB1-DED1 (Fig. 5), all DED1 promoter upstream sequences were removed, except for 13 bp upstream from the unique DED1 TATA box. Analysis of β-galactosidase activities with the VitB1-DED1 fusion promoter showed a 80- to 100-fold higher basal activity as compared with the VitB1-CYC1 fusion promoter (Table 3). The presence of CTF1 derivatives induces β-galactosidase activity 2- to 4-fold over basal level, although the hER-mediated activation was a mere 4-fold in the presence of estrogen (Table 3). Analysis of the VitB1-DED1 fusion promoter on a CEN-based reporter plasmid reduced the basal activity 35-fold as compared with the 2μ-based reporter plasmid, and in turn a 10-fold rather than a 4-fold enhanced transactivation was observed with the estrogen-stimulated hER (Table 3).
Indirect end-labeling analysis of yeast nuclei containing the VitB1-DED1 fusion promoter reveals three preferred MNase cleavages sites around positions −430, −270, and −100 bp (±20 bp, Fig. 6B), as seen with the VitB1-CYC1 promoter, although the most promoter-proximal cleavage at position −100 bp is now precisely positioned. This shows that the surrounding DNA sequences influence the positioning of the promoter proximal nucleosome (34). Consistent with the masking of the CTF/NF1-binding site by the positioned nucleosome, in none of the conditions tested (CEN- vs. 2μ-based vitellogenin B1 reporter- or expression plasmids, with variable estrogen concentrations) did CTF1 and CTF1 derivatives show synergism with hER (Table 3 and data not shown).
We can draw four conclusions from these results. First, the vitellogenin promoter sequences are able to position nucleosomes in yeast as they do in vitro. We note, however, that the nucleosome positioned between −300 and −140 bp after in vitro nucleosome reconstitution (6) shifts slightly more promoter proximal (between −270 and −100 ± 20) in yeast. Interestingly, the nucleosomal DNA encompasses the CTF/NFI-binding site and raises the possibility that the site is inaccessible due to the nucleosome. Second, the unique transcription initiation site in the VitB1-DED1 construct reduces the variation in nucleosome positioning observed on the VitB1-CYC1 promoter, but conserves the nucleosomal phasing over the vitellogenin B1 promoter. Third, the region containing the EREs also falls within a nucleosome, yet these EREs remain accessible to hER. Fourth, in addition to the positioned nucleosomes over the vitellogenin promoter, a 150- to 200-bp periodic MNase cleavage pattern is visible over the lacZ reporter gene, while the CYC1 promoter region and the proximal lacZ gene are either nucleosome free or do not bear positioned nucleosomes (Fig. 6A).
High-Resolution Footprinting Analysis on the Vitellogenin B1 Promoter in Yeast
To analyze the structure of the vitellogenin B1 promoter with high resolution, we used MNase and deoxyribonuclease I (DNase I) digestions on gently lysed spheroplasts, followed by primer extension, which allows base pair resolution of nucleosome locations as well as the detection of other proteins bound to DNA (35, 36). Digestions were performed on estrogen-stimulated nuclei isolated from yeast cells transformed with p2HG-hER or p2HG insert-free control and pVitB1-CYC1. The genomic footprinting analyses showed identical nuclease cleavage patterns both in the presence and absence of hER (activated or silent promoter, data not shown), consistent with the indirect end-labeling analyses above (see Fig. 6A).
The vitellogenin B1 promoter reveals an unexpectedly complex MNase cleavage pattern. Analysis of the promoter proximal MNase cleavage sites in nuclei shows a protection from around −60 (data not shown) until −171 (±2), with slightly higher accessibility at −98 and −135 (±2, Fig. 7; compare lanes 1+2 with 3+4; see asterisks). Between −175 and −300 (±2) we also observe an altered pattern compared with naked DNA with both enhanced cleavage sites (open arrows) and protected sites (not indicated) and with a pronounced MNase-specific hypersensitive region between −225 and −250 (±2; Fig. 7, lane 1). There is also a pronounced site of cleavage near −180. Similar complex patterns have been observed by Tanaka et al. (36) and interpreted as identifying a nucleosome with multiple positions.
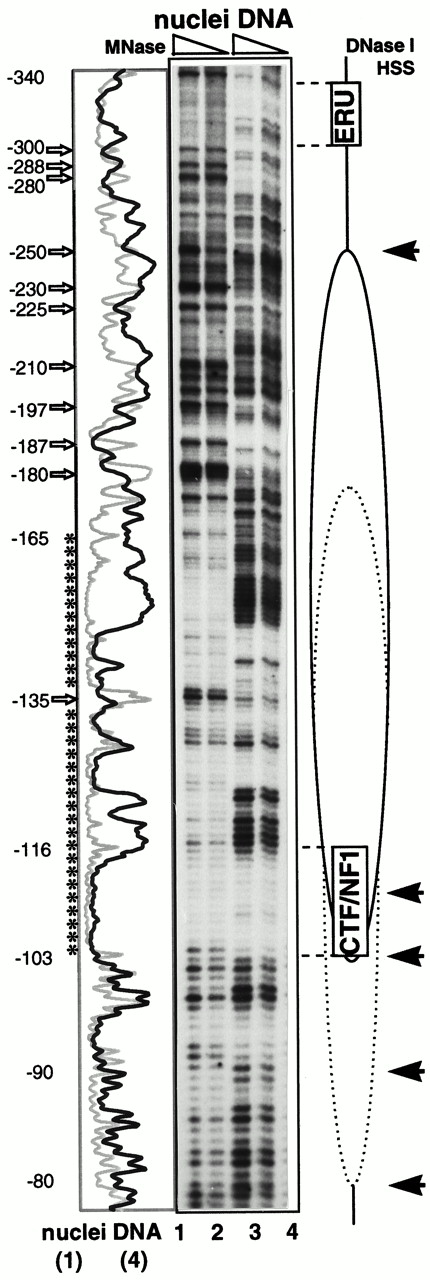
Genomic Footprinting Analysis of the Vitellogenin B1 Promoter in Yeast by Primer Extension Yeast cells were transformed with pVitB1-CYC1 (see Fig. 5) and p2HG-hER. Cells were grown and nuclei isolated as indicated in Fig. 6. Genomic footprinting analysis was performed with the primer SG102. The location of the ERU, the CTF/NFI-specific DNA element, and the nucleosome with multiple positions are shown schematically along the autoradiogram. Numbers correspond to vitellogenin B1 promoter sequences upstream from the site of transcription initiation. Regions of MNase protections are indicated by asterisks, and hypersensitive cleavage sites are shown by open arrows along the scan. DNase I-hypersensitive sites (HSS) are indicated by solid arrows. Nuclei were digested with MNase at room temperature for 5 min with 2500 U/ml (lane 1) and 1300 U/ml (lane 2). Purified genomic DNA was digested with 0.8 U/ml (lane 3) and 0.4 U/ml (lane 4) MNase as for nuclei above. Selected lanes were scanned and plotted along the autoradiogram for MNase-digested nuclei (gray lines) and free DNA (black lines).
To corroborate the MNase digestion pattern, we treated the genomic chromatin with DNase I. There is a strong DNase I hypersensitive cleavage site around −250 that would be consistent with the border of a positioned nucleosome, and specific DNase I cuts with a 10-bp periodicity between −110 and −80 (±2; Fig. 7, solid arrows). Taken with the end-labeling results (Fig. 6A), we interpret the complex digestion pattern over the analyzed promoter region (MNase protection over 100 bp and MNase and DNase I hypersensitive sites up- and downstream of it) to indicate a nucleosome whose position is not strictly fixed, but has a restricted mobility resulting in alternative positions within the region of interest, as suggested by the scheme of Fig. 7. This proposed nucleosome covers the CTF/NFI-binding site at −114 to −101, and there is no indication of a CTF1-specific footprint (data not shown). Thus, the lack of CTF1-dependent transcriptional activation from this promoter is consistent with the idea that this nucleosome masks the CTF/NFI-binding site.
The analysis of the more distal vitellogenin B1 promoter sequence shows that the region between −340 and −300, including two imperfect EREs at −302 to −334, is partially protected from MNase digestion (Fig. 7). These observations hold true whether or not hER and estrogen are present. The fact that hER can stimulate transcription ∼50 fold from this promoter indicates that the hER can bind the EREs even though they might be associated with a nucleosome (see Fig. 6).
DISCUSSION
We have established an experimental system in yeast with the goal of studying the functional interactions between the ER and CTF1 on the vitellogenin gene B1 promoter, which had previously been observed during in vitro transcription of naked DNA templates (28). This system allows us to perform experiments in vivo with a template assembled into chromatin.
hER Stimulates the Native Vitellogenin B1 Promoter in Yeast
We show here that the hER functions in yeast as it does in vertebrate cells, confirming observations first made by Metzger et al. (23). Under our conditions hER requires estrogen to stimulate transcription of a simple responsive promoter that consists of an ERE placed directly upstream of the minimal yeast CYC1 promoter. More importantly, hER is also able to activate the natural vertebrate vitellogenin B1 promoter in yeast cells. In this system, the antiestrogens, tamoxifen, nafoxidin, and ICI, have no agonistic effect, but rather they antagonize estrogen action. Previous analyses of the role of estrogen and antiestrogens in ER DNA binding and transactivation led to conflicting results, most likely due to the experimental conditions, as well as the different sources and forms of ER (see Ref. 37 and references therein). Since the natural vitellogenin gene B1 promoter is readily stimulated in yeast by the liganded ER, we were able to analyze potential interactions of the hER with other transcription factors, notably with CTF1, in a controlled chromatin environment.
The Proline-Rich Transactivation Domain of CTF1 Is Required for a Synergistic Interaction with the hER in Yeast
Our previous demonstration of a functional interaction between members of the CTF/NFI family and hER in vitro suggested that their synergistic interaction results from the stronger tethering of a common factor that participates in the transcriptional machinery, rather than from cooperative DNA binding (28). Furthermore, the activation domains of both hER and CTF1 were required for the observed synergism (27). In yeast we find that the proline-rich activation domain of CTF1 is roughly 50–100 times weaker than the acidic transciptional activation domains of Gal4 or VP16 (see also Refs. 38, 39). However, addition of the strong acidic transcription activation domain of VP16 to the N terminus of CTF1 resulted in a 5-fold synergism between the hER and VP16-CTF1 on a synthetic promoter. Synergism was observed only when VP16-CTF1 and hER were expressed from CEN-based plasmids that avoid high levels of overexpression, suggesting either that the promoter is quickly saturated or that a yeast factor involved in transactivation is limiting and is titrated out by the overexpressed polypeptides. Similar results were found previously in HeLa cells expressing GAL4-CTF1 fusion proteins and hER on a synthetic promoter (27). Interestingly, no synergism could be detected when the proline-rich transcription activation domain of CTF1 was replaced by the acidic transcription activation domain of Gal4, and only a 2-fold synergism was observed when the proline-rich transcription activation domain of CTF1 was deleted in the VP16-CTFΔ chimera. Thus, we conclude that in yeast, as in vertebrate cells, the proline-rich domain of CTF1 participates in the strong synergism between VP16-CTF1 and hER. The proline-rich domain might either make the acidic domain more accessible or with the aid of this latter domain it may itself promote interaction between hER, CTF1, and yeast coactivators. Since the proline domain does not activate on its own, it is unlikely that it interacts with the basal transcription machinery.
A Positioned Nucleosome Masks the CTF/NFI Site on the Vitellogenin B1 Promoter in Yeast
We were unable to detect stimulation of the X. laevis vitellogenin B1 promoter by CTF1 and observed only a weakly transactivating activity of CTF1 derivatives, although all can bind the CTF/NFI site of the vitellogenin B1 promoter in vitro. This result suggests that the CTF/NFI-binding site is masked in yeast cells. The vitellogenin B1 promoter contains a strong nucleosome positioning element that places a nucleosome between −300 and −140, i.e. close to the CTF/NF1 site, during in vitro chromatin assembly (6). Analyses of both MNase- and DNase I-digested yeast nuclei are consistent with the presence of a nucleosome with multiple positions between −250 and −80 on the vitellogenin B1 promoter. The nucleosome appears to be shifted slightly toward the transcription initiation site when compared with that mapped from in vitro chromatin assembly (6). The local heterogeneity revealed by the high-resolution MNase mapping has also been observed in high-resolution chromatin structure analyses of the yeast URA3 gene (36). These authors also detect unexpectedly complex digestion patterns for MNase and DNase I, despite the clear chromatin features observed by low-resolution mapping. From their high-resolution mapping of six nucleosomes on the URA3 gene they could conclude that despite the local heterogeneity revealed by the high-resolution mapping, the low-resolution map is a reasonably accurate representation of the chromatin structure. In view of these results, our low- and high-resolution nuclease-sensitivity patterns are most consistent with a population of overlapping nucleosome positions over the proximal vitellogenin B1 promoter, which cover the CTF/NFI site (−101 to −114). This appears to render the site inaccessible to CTF1 or to derivatives of CTF1 and may explain the lack of transcriptional activation by these factors on the vitellogenin promoter in yeast.
It was recently shown that the proline-rich domain of CTF1 interacts specifically with histone human H3, and full-length CTF1 was shown to alter the interaction of reconstituted nucleosomal cores with DNA (40). Were this true in yeast, we might expect that CTF1 would displace the positioned nucleosome on the B1 promoter. Recent studies suggest, however, that CFT1 interacts with yeast histone H3 with significantly reduced affinity (N. Mermod, personal communication), possibly rendering it unable to remodel yeast chromatin even during assembly after DNA replication. Consistently, using a specific DNA-bending sequence to direct CTF/NFI-binding sites to different positions around an in vitro reconstituted nucleosome, DNA-binding studies using CTF1 showed a 100- to 300-fold reduced binding affinity for all nucleosomal targets as compared with free DNA (41). It was concluded that nucleosomal inhibition of CTF/NFI binding was an inherent characteristic of the factor, which is in excellent agreement with our observations on the vitellogenin promoter. However, there is also some support from in vivo experiments for the idea that CTF might bind to a preexisting nondisplaced nucleosome on the MMTV promoter (42, 43). In vivo, transcriptional activation of this promoter by the GR recruits CTF1 and the octamer factor to their binding sites within the chromatin modified by the receptor (44). After transient GR activation, these binding sites are reincorporated into the positioned nucleosome, and transcription is repressed (45). A CTF2-dependent hormone activation of this promoter has also been shown in yeast (42). A porcine CTF2 that lacks 69 aa at its C terminus, which is produced through differential splicing of the NFI gene transcript (38), was used and shown to bind its site within a nucleosome. The strongly synergistic interaction between GR and CTF2 was dependent on low-level expression of GR, low ligand concentration, and high-level expression of CTF2 (42).
The variety of results from in vitro and in vivo analyses of the chromatin structure of the MMTV promoter (42–49) may reflect the effects of flanking sequences on nucleosome positioning (34). Consistently, in our analyses we observe that the position of the most promoter-proximal nucleosome is influenced by the minimal yeast promoter fused to the vitellogenin B1 sequences (compare Fig. 6, A and B). We were obliged, however, to use yeast sequences for the minimal promoter elements (either CYC1 or DED1) because the proximal region of the vitellogenin B1 promoter (−41 to +1) confers a very high basal activity in yeast (data not shown). The exchange of yeast for Xenopus sequences in this region may also account for the shift of the nucleosome from its position mapped in vitro (−300 to −140) to the position from high-resolution mapping in yeast (−250 to −80).
Surprisingly, despite the strong activating potential of hER during estrogen stimulation, no qualitative differences were observed by end-labeling analyses on the positioned nucleosome (−270 to −100± 20) in the presence or absence of hER and estradiol. Our analysis suggested the presence of a second nucleosome from −430 to −270 (±20), a region of the vitellogenin B1 promoter that contains the ERU, suggesting that hER can bind to the ERU even when wrapped around a nucleosome. Alternatively, the hER-inducible expression might derive from only a small subpopulation of plasmids.
From our previous in vitro studies, we postulated that the folding of DNA around a positioned nucleosome on the vitellogenin B1 promoter creates a static loop, which facilitates the interaction of the ER with transcription factors bound to the proximal promoter elements (6). We show here that the deletion of most of the nucleosome positioning sequences enhances transcription about 5-fold, as compared with the full-length promoter, indicating that the nucleosome-positioning sequences are not essential for activation in yeast. However, the deletion of these nucleosome-positioning sequences does not significantly increase estrogen-dependent transcription of the same promoter in transient transfection studies using primary hepatocytes (50). The nucleosome-induced loop should not only favor the interaction of ER with CTF1, but also with proximal promoter-bound factors such as the liver-enriched factors CCAAT/enhancer-binding protein (C/EBP) and hepatic nuclear factor 3 (HNF3) (see Ref. 15), that bind between the −100 boundary of the positioned nucleosome and the TATA box. Although these factors are lacking in yeast, it should now be possible to test whether a nucleosome loop aids activation by hER when HNF3 and/or C/EBP are coexpressed with hER in yeast carrying the vitellogenin gene B1 promoter. It must, of course, be kept in mind that several different aspects of chromatin structure influence the access of transcription factors.
In conclusion, we have investigated the importance of a positioned nucleosome on the hormonal activation of the vertebrate vitellogenin gene B1 promoter in yeast, in particular with respect to the synergism between the hER and CTF1. The hER appears to bind and activate transcription without disrupting a positioned nucleosome downstream of the EREs. This same nucleosome appears to mask the CTF/NFI site and prevent functional interaction between the hER and CTF1 in yeast. In view of these results, it is now of interest to define the position of nucleosomes in hepatocytes with the vitellogenin B1 promoter embedded in its natural context, to determine whether the mechanism of vitellogenin gene B1 induction relies on the interaction of the ER with members of the CTF/NFI family, or with those of the C/EBP and HNF3 families. This should contribute to our further understanding of the tissue-specific expression of this gene.
MATERIALS AND METHODS
Chemicals
Linkers, restriction, and modification enzymes were purchased from Boehringer Mannheim (Indianapolis, IN); poly (dI·dC)·poly (dI·dC) was purchased from Pharmacia (Piscataway, NJ). ICI 164.384 (N-n-butyl-11-(3,17β-dihydroxyoestra-1,3,5(10)-trien-7α-γl)-N-methylundecanamide) was a gift from Dr. A. Wakeling, (ICI Pharmaceuticals, Alderley Park, Macclesfield, UK). 17β-Estradiol, tamoxifen, nafoxidine, and general reagents were purchased from Sigma Chemical Co. (St. Louis, MO). Yeast medium components were obtained from Difco Laboratories (Detroit, MI).
Yeast Strains
The protease-deficient strain GA24 (MATa GAL+ pep4–3 ura3 his3 bar1 suc2Δ9) was used for all experiments. All transformations into this strain were accomplished by using the lithium acetate transformation protocol (51).
Construction of Plasmids
The expression vector p2HG-hER was obtained as follows. The hER vector, ER (hERval400-mutant), and p2HG (52) were gifts of D. Picard. The vector ER was digested with BamHI, and the hER cDNA was subcloned into the BamHI-digested plasmid pGAL1, resulting in pGAL1-hER. The plasmid pGAL1 contains a 907-bp SalI-BamHI fragment of the yeast GAL1-GAL10 promoter (53) subcloned into the SalI and BamHI sites of YEp24 (50). pGAL1-hER was digested with SalI and ApaI, and the resulting 3.6-kb fragment containing the promoter GAL1-GAL10, hER, and part of YEp24 was subcloned into the SalI- and ApaI-digested p2HG, yielding plasmid p2HG-hER. The human CTF1 expression plasmid was constructed as follows. The human CTF1 cDNA was isolated as a 1.7-kb NcoI-XhoI fragment from pCTF1 (54) and subcloned into the NcoI and XhoI sites of p2HG-GAL1. To obtain p2HG-GAL1, a MluI linker was inserted into the filled BamHI site of pGAL1, and NcoI and XhoI sites were added before the 907-bp yeast GAL1-GAL10 promoter was inserted as a SalI-XhoI fragment into p2HG. p2HG-CTF1/hER resulted from subcloning the 3.6-kb SalI-ApaI fragment, containing the hER under the control of the yeast GAL1 promoter, from pGAL1-hER into the XhoI-ApaI sites of p2HG-CTF1. The hybrid protein (CTF1-GAL4) comprises the first 398 aa of human CTF1 fused to the C-terminal 129 aa of yeast GAL4. The expression vector for the fusion protein (CTF1-GAL4) was derived as follows. A 444-bp PvuII-EcoRI fragment containing the acidic activation domain of GAL4 was isolated from pSG4 (55) and ligated into the Klenow-filled BglII and EcoRI sites of pCTF1, replacing the proline-rich transcription activation domain of CTF1. The resulting plasmid, pBS-CTFGAL4, was digested with NcoI and XhoI, and the 1.7-kb fragment was subcloned into p2HG-CTF1 to create p2HG-CTFGAL4. p2HG-CTFGAL4/hER arose then from p2HG-CTFGAL4 as p2HG-CTF1/hER above. The fusion protein (VP16-CTF1) comprises the C-terminal 80 aa of the herpes simplex virus protein VP16 fused N-terminal to the human CTF1. The expression vector for the hybrid protein (VP16-CTF1) was constructed as follows. The CTF1 cDNA was isolated from pCTF1 as a Klenow-filled SpeI and XhoI fragment and inserted downstream from the acidic transcription activation domain of VP16 into the Klenow-filled EcoRI site of pSD10 (56), resulting in pSD-VP16CTF1. Then the KpnI and SacII sites in pSD-VP16CTF1 were flush-ended with T4 DNA polymerase, and the GAL1 promoter and VP16CTF1-containing fragment were isolated and ligated into the Klenow-filled and dephosphorylated SalI site of p2HG-hER, resulting in p2HG-VP16CTF1/hER. The fusion protein (VP16-CTFΔ) has the C-terminal 100 aa long proline-rich domain deleted by digesting pSD-VP16CTF1 with BglII and XhoI and religation of the Klenow blunt-ended sites resulting in pSD-VP16CTFΔ. p2HG-VP16CTFΔ/hER arose then from pSD-VP16CTFΔ as p2HG-VP16CTF1/hER above.
CEN-based expression vectors were obtained by replacing the 2μ containing 2.6-kb SpeI-NotI fragments in p2HG-hER, p2HG-CTFGAL4/hER, and p2HG-VP16CTF1/hER with a 2.5-kb SpeI-NotI fragment containing the ARS1 and CEN4.
Reporter plasmids were constructed as follows. The parent vector used for all reporter plasmids was pLG669Z (51). To obtain pLGΔSmaI-XhoI, pLG669Z was digested by SmaI and XhoI, the XhoI site was filled by Klenow DNA polymerase, and the SmaI and filled XhoI sites were religated, restoring the filled XhoI site. pLGERE was obtained from D. Picard and is the same ERE reporter plasmid as used in Ref. 52 . The plasmid pLGCTF/NF1 was obtained by introducing a synthetic oligonucleotide corresponding to the consensus CTF/NFI site into the Klenow-filled XhoI site of pLGΔSmaI-XhoI. pLGERE-CTF/NFI was constructed by cloning the consensus CTF/NF1 site oligomer into the HincII and SmaI sites of pBSKS+ (51), resulting in pBS-CTF/NF1. Then a 75-bp BamHI fragment, containing two consensus EREs, was ligated into the BamHI site of pBS-CTF/NFI. The resulting pBSERE-CTF/NFI was then digested with SpeI and the site was filled with Klenow polymerase and further digested with XhoI. The XhoI and filled SpeI fragment, containing the (ERE)2-CTF/NFI DNA elements, was then subcloned into the XhoI-SmaI-digested pLG669Z, yielding pLGERE-CTF/NFI. The VitB1-based reporter plasmids were constructed by adding a XhoI linker into the Klenow-filled BglII site of pB1(−596/+8)CAT8+ (57) and then subcloning the VitB1 promoter as a HincII-XhoI fragment into the SmaI-XhoI sites of pLG669Z, resulting in pVitB1-CYC1. pLGΔVitB1 was obtained by subcloning the 0.8-kb ApaI-SspI fragment from pVitB1-CYC1 into the ApaI and Klenow-filled XhoI sites of pLGΔSmaI-XhoI, and pVitB1-DED1 was obtained by subcloning the 1.1-kb ApaI-XhoI fragment fom pVitB1-CYC1 into the ApaI-XhoI sites of pLS168 (58). CEN-based reporter plasmids were obtained by replacing the 2μ containing the 1.9-kb SpeI fragment in pVitB1-CYC1 and pVitB1-DED1 with a 2.5-kb SpeI fragment containing the ARS1 and CEN4.
Growth of Yeast Cells and Preparation of Protein Extracts
Cells were grown and protein extracts prepared as follows. Transformed yeast cells were grown in synthetic drop-out medium lacking uracil and histidine (51) and supplemented with 2% (wt/vol) glucose, 3% (vol/vol) glycerol, and 2% (wt/vol) lactate. At late-log phase the cultures were diluted (1:100) into the same medium without glucose and the growth continued for 24 h, after which galactose was added to 2% (wt/vol) to induce the yeast GAL1 promoter and estrogen or antagonists as needed. After 8 h, cells were harvested, washed first with water and then with buffer A [50 mm Tris-HCl (pH 7.5), 1 mm EDTA, 1 mm EGTA, 10% (vol/vol) glycerol, 15 mm MgCl2, 5 μm ZnSO4, 50 mm NaCl, 20 mm Na-molybdate, 25 mm sodium fluoride, 1 mm sodium bisulfite, 1 mm dithiothreitol, 1 mm phenylmethylsulfonyl fluoride (PMSF), and 1 μg/ml each of the following protease inhibitors: antipain, benzamidine, chymostatin, leupeptin, and pepstatin A]. The washed cells from a 15-ml culture were resuspended in 0.5 ml buffer A in 15-ml round bottom plastic tubes and frozen rapidly in liquid nitrogen. Cells were thawed in ice and 0.5 g glass beads (250 μm) was added, after which the cells were vortexed at maximum speed in the cold, 6 × 1 min bursts with cooling in ice between. Glass beads and cell debris were removed by centrifugation (10 min at 15,000 × g, 4 C). The supernatants were transferred into 1.5-ml microcentrifuge tubes, and the centrifugation was continued for 30 min in a microcentrifuge, 13,000 × g, 4 C. The protein concentration in the extracts was determined by the Bradford method, using the Bio-Rad protein assay (Bio-Rad, Richmond, CA).
Antibody Preparation
An Escherichia coli β-galactosidase (Met1-Asp374) and X. laevis ER (Asp276-His508) fusion protein was cloned into pEX2 (59) and expressed in E. coli. The overexpressed fusion protein precipitated inside the bacterial cell and could be easily purified as insoluble protein. The fusion protein was separated by SDS-PAGE (51), and the corresponding 67-kDa band was cut out and lyophilized. About 10 μg of fusion protein were rehydrated, mixed with complete Freund’s adjuvant, and injected ip into a rabbit. The animal was boosted at 6-week intervals with a mixture of fusion protein and incomplete Freund’s adjuvant and bled 1–2 weeks after each boost.
Immunoblot Analysis
Protein extracts were separated on 8% SDS-polyacrylamide gels and transferred to nitrocellulose as described (51). The (1:1000) diluted polyclonal antibody against the X. laevis ER was used for the indirect detection of the yeast expressed hER. Specific antigen-antibody complexes were detected with the enhanced chemiluminescence (ECL) reagent (Amersham, Arlington Heights, IL).
DNA Mobility Shift Assay
Protein extracts in buffer A were preincubated with poly (dI·dC)·poly (dI·dC), (0.1 mg/ml) at 0 C for 15 min, after which the 32P-end-labeled DNA probe was added to a final volume of 20 μl and the incubation was continued at 20 C for 15 min. The protein-DNA complexes were analyzed on nondenaturing 5% polyacrylamide gels (15, 51).
Assay of β-Galactosidase
The analysis of the β-galactosidase activity in the protein extracts was performed as described in Ref. 51 using the given formula to calculate the specific activity.
Preparation of Yeast Nuclei and Nuclease Digestions
Transformed yeast cells were grown mainly as described above, except the growth in galactose and estradiol was reduced to 5–6 h. Nuclei were isolated from 2 liters of culture, about 16 g wet cells, following the method described (35). Cells were harvest by centrifugation and spheroplasted as described in Ref. 60 at 10 ml/g wet cells in synthetic drop-out medium lacking uracil and histidine and supplemented with 1 μm estradiol, 2% (wt/vol) galactose, 3% (vol/vol) glycerol, 2% (wt/vol) lactate, 1.1 m sorbitol, 0.15 mg/ml Zymolyase (Seikagaku, Chuo-ku, Tokyo), and 800 U/ml lyticase (60). Spheroplast formation continued at 30 C for 30 min with gentle agitation. After centrifugation at 4 C, spheroplasts were washed twice by gentle but thorough resuspension in 20 mm K-PO4 buffer (pH 6.5) supplemented with 1.1 m sorbitol and 0.5 mm PMSF at 4 C. Washed spheroplasts were resuspended in 80 ml buffer F [20 mm PIPES (pH 6.5), 18% (wt/vol) Ficoll 400, 0.125 mm spermidine, 0.05 mm spermine, 0.5 mm EDTA, and 1 mm PMSF] and divided into four 40-ml polycarbonate tubes. Each tube was vortexed at maximum speed for two 5-min bursts at 4 C. The sheared spheroplasts (25 ml each) were carefully layered onto 18 ml buffer GF [20 mm PIPES (pH 6.5), 20% (vol/vol) glycerol, 7% (wt/vol) Ficoll 400, 0.125 mm spermidine, 0.05 mm spermine, 0.5 mm EDTA, and 1 mm PMSF] and centrifuged at 22,500 × g for 30 min at 4 C. Supernatants were discarded and each pellet resuspended in 20 ml buffer F (without EDTA). Vortexing was repeated as above, and unlysed cells and cell debris were removed by centrifugation at 3,400 × g for 15 min at 4 C. Nuclei containing supernatants were transferred into fresh tubes and were divided appropriately so that some could be resuspended in digestion buffer and some in control buffer. Nuclei were recovered by centrifugation: 22,500 × g for 30 min at 4 C. Supernatants were discarded, and the crude nuclei-containing pellets were resuspended as follows. For undigested controls the nuclei were resuspended in 0.5 ml/g wet cells control buffer [10 mm HEPES (pH 7.5), 0.125 mm spermidine, 0.05 mm spermine, 10 mm EDTA, and 1 mm PMSF], divided into 220-μl aliquots, and DNA was purified as described below. Nuclei to be digested were resuspended in 0.5 ml/g wet cells digestion buffer[ 10 mm HEPES (pH 7.5), 0.5 mm MgCl2, 3 mm CaCl2, and 1 mm PMSF], divided into 200-μl aliquots, and preincubated at room temperature or at 37 C for 5 min. Nuclease digestions were started by adding 10 μl MNase or DNase I diluted in digestion buffer. The digestions were stopped after 10 min by the addition of 10 μl 0.2 m EDTA. DNA from nondigested and digested nuclei was purified by incubation for 30 min at 37 C with 10 μl (10 mg/ml) ribonuclease A, after which 30 μl 22% (vol/vol) sarkosyl, 30 μl 1.7 m NaCl, and 30 μl (10 mg/ml) proteinase K were added in this order with mixing between. The proteinase K digestion was performed at 37 C for 3–6 h, after which 30 μl 5 m NaClO4 were added, followed by extensive phenol and phenol/chloroform extractions. DNA was precipitated with 990 μl ethanol at room temperature, washed with 75% (vol/vol) ethanol, and resuspended in 100 μl 10 mm HEPES (pH 7.5).
Indirect End-Label Analysis
The cleavage sites of MNase-digested yeast nuclei were located by the indirect end-label method (61, 62). Purified DNA from digested nuclei and digested DNA controls were cleaved by XbaI, and the DNA fragments were separated by agarose gel electrophoresis and blotted onto nylon filter according to Southern (51). Vitellogenin B1-specific DNA sequences were probed with a 32P-end-labeled 150-bp XbaI-AccI fragment (−596 to −446 of the vitellogenin B1 promoter).
Primer Extension Assay
MNase and DNase I cleavage sites were located by primer extension assay mainly as described (35). The oligonucleotide used as primer includes the X. laevis vitellogenin B1 promoter up to bp −53 and overlaps the CYC1 gene (SG102: 5′-CCTGGCGGATCTGCTCGAGGGATCTGGCGCATGTGC-3′). The DNA/primer mix was denatured at 93 C for 90 sec, annealed at 62 C for 4 min, and then extended at 72 C for 3 min. This cycle was repeated 35 times with 10 min at 72 C for final extension. Reactions were terminated by chloroform extraction and ethanol precipitation. Dideoxy sequencing reactions using Taq polymerase were performed on plasmid DNA as described (35), using the same primer extension reactions as above. The DNA products were analyzed on 8% polyacrylamide/7 m urea sequencing gels with wedge spacers (51).
We thank D. Picard and N. Mermod for gifts of plasmids and A. Wakeling for providing ICI 164,384. We are also grateful to N. Mermod and D. Robyr for comments on the manuscript.
This work was supported by grants from the Swiss National Science Foundation to S.M.G. and to W.W. and by the Etat de Vaud. M.T.-P. was supported by the Swiss Institute for Experimental Cancer Research.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}