





Abstract
Enzymes catalyze the chemical reactions of life. For nearly half of known enzymes, catalysis requires the binding of small molecules known as cofactors. Polypeptide-cofactor complexes likely formed at a primordial stage and became starting points for the evolution of many efficient enzymes. Yet, evolution has no foresight so the driver for the primordial complex formation is unknown. Here, we use a resurrected ancestral TIM-barrel protein to identify one potential driver. Heme binding at a flexible region of the ancestral structure yields a peroxidation catalyst with enhanced efficiency when compared to free heme. This enhancement, however, does not arise from protein-mediated promotion of catalysis. Rather, it reflects the protection of bound heme from common degradation processes and a resulting longer lifetime and higher effective concentration for the catalyst. Protection of catalytic cofactors by polypeptides emerges as a general mechanism to enhance catalysis and may have plausibly benefited primordial polypeptide-cofactor associations.
Introduction
Life involves a vast network of chemical reactions, most of which would not occur at a sufficient rate in the absence of effective enzyme catalysis. With the obvious exception of ribozymes, enzymes are based on protein scaffolds. Most, if not all, of modern enzyme activities have evolved from prior enzyme activities (Ohno 1970; Khersonsky and Tawfik 2010). This evolutionary process is reasonably well-understood and protein engineers can mimic it in the lab using directed evolution (Campbell et al. 2016; Zeymer and Hilvert 2018). On the other hand, it is a logical necessity that the first enzymes did not arise from previously existing “older” enzymes. That is, the first primordial enzymes must have necessarily arisen de novo in previously noncatalytic protein scaffolds. Reconstructions of the gene content of the last universal common ancestor (LUCA) support that a diversity of enzymes already catalyzed many different reactions at a very early evolutionary stage (Weiss et al. 2016). It seems reasonable, therefore, that there are efficient mechanisms for the de novo emergence of completely new enzymes. Such mechanisms, however, remain elusive to protein engineers, who have found the generation of efficient de novo enzymes in the lab to be extremely challenging (Blomberg et al. 2013; Risso et al. 2017; Donnelly et al. 2018; Lovelock et al. 2022; Gutierrez-Rus et al. 2022; Yeh et al. 2023). Our limited grasp of the evolutionary mechanisms of de novo enzyme generation implies a serious gap in our understanding of the origin of life.
A substantial fraction (around 50%) of enzymes relies on cofactors for catalysis (Fischer et al. 2010). This statement is true for many modern enzymes, but it also very likely holds for the most ancient enzymes, as indicated by reconstructions of the gene content of LUCA (Weiss et al. 2016). Many cofactors, in particular metals and metal-containing organic cofactors, display by themselves significant levels of catalysis with a diversity of chemical reactions (Andreini et al. 2008; Fischer et al. 2010). Both polypeptides and cofactors were likely available already at a prebiotic stage (Frenkel-Pinter et al. 2020; Goldman and Kacar 2021). Therefore, the recruitment of catalytic cofactors by polypeptides would appear to provide a simple and straightforward mechanism for the de novo emergence of many of the primordial enzymes (although certainly not of all of them). On closer examination, however, this mechanism has a serious shortcoming: the driving force for recruitment is not apparent at all. Certainly, once a polypeptide-cofactor complex has been formed, new possibilities arise in a Darwinian evolution scenario, including the enhancement of catalysis through mutations in the protein moiety. Yet, we know evolution has no foresight (Jacob 1977), so any driver of primordial polypeptide-cofactor association must have provided an immediate selective advantage, such as an instant enhancement in catalysis. However, the opposite would seem reasonable on general grounds. Association of a cofactor with a “naïve” polypeptide, that is a polypeptide that has not evolved to enhance catalysis by the cofactor, would simply decrease cofactor exposure to the environment and therefore its accessibility to substrates, thus likely impairing catalysis.
Here, we report experimental studies on the emergence of cofactor-based catalysis in an ancestral TIM-barrel protein. Our experiments reveal a general mechanism of immediate catalysis enhancement that may have plausibly provided a driving force for the polypeptide-cofactor association during a primordial period.
Results
The TIM-barrel is the most widespread enzyme fold, providing a structural scaffold for a large diversity of modern enzyme functionalities (Wierenga 2001; Nagano et al. 2002). In fact, the TIM-barrel fold has been proposed to have played an essential role in early metabolism (Goldman et al. 2016). We recently reported an ancestral reconstruction exercise targeting family 1 glycosidases, which are of the TIM-barrel fold (Gamiz-Arco et al. 2021). We found a putative common ancestor of eukaryotic and bacterial family 1 glycosidases that displayed unusual properties, including tight and stoichiometric binding of heme at a conformationally flexible region distinct from the glycosidase active site (figs. 1A and 1B and 1C). Remarkably, heme binding is extremely rare among modern TIM-barrel proteins (Gamiz-Arco et al. 2021).
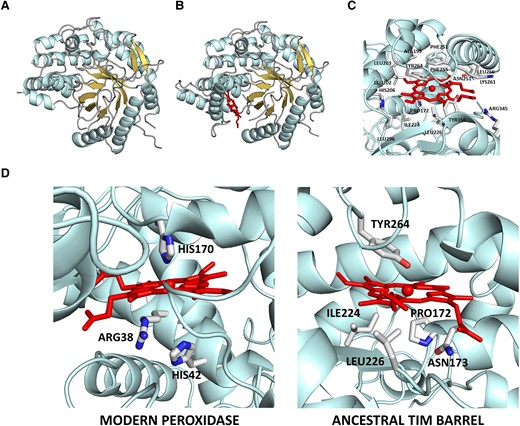
3D-structure of the ancestral TIM-barrel glycosidase studied in this work as determined by X-ray crystallography (Gamiz-Arco et al. 2021). (A) Structure of the protein without heme bound (PDB ID 6Z1H). Note that the region where heme binds displays substantial conformational flexibility and that, as a result, there are missing sections in the electronic density maps (for further details, see Gamiz-Arco et al. 2021). (B) Structure of the protein with heme bound (PDB ID 6Z1M). Heme binding rigidifies the protein and allows a larger part of the structure to be determined from the density maps, as it is apparent by comparing the structure shown in B with that shown in A (for further details, see Gamiz-Arco et al. 2021). (C) Zoom-in of the heme-binding region providing amino acid detail. (D) Comparison of the molecular environments of the heme in horseradish peroxidase (PDB ID 1HCH) and the ancestral TIM-barrel (PDB ID 6Z1M). Critical residues for peroxidase catalysis (the proximal histidine and distal histidine and arginine) are highlighted in the peroxidase structure. In the ancestral TIM-barrel structure, the proximal residue is not a histidine and the residues at the opposite side of the heme ring (labeled) do not include histidines or arginines.
Free heme is known to display a low but measurable peroxidase activity (Brown et al. 1970). Therefore, heme binding may be expected to confer some peroxidase activity to the ancestral TIM-barrel. This would be a de novo redox activity, distinct from the natural, non-redox activity of glycosidases (i.e., cleavage of glycosidic bonds). We have used the common peroxidase substrate o-dianisidine (fig. 2A) to compare the peroxidase activity of free heme with that of heme bound to the ancestral scaffold. Peroxidation of o-dianisidine leads to absorption in the visible region of the spectrum, which can be used to follow the chemical reaction (Jenkins et al. 2021). As is customary in enzyme kinetics studies, we started with determinations of the initial rates of the reaction, which can be easily calculated from the initial increases of the absorbance due to the peroxidation product (fig. 2B). We performed a large number of experiments in solutions at different pH values. In most of the studied pH ranges, the peroxidase activity of the free heme is substantially higher than that of the heme-bound ancestral scaffold (fig. 2B). This result is not surprising, given that burial of the heme in the ancestral protein structure (see figure 7C in Gamiz-Arco et al. 2021) should hinder the substrate access to the cofactor, thus impairing catalysis. Furthermore, the heme-binding region of the ancestral structure has not evolved to promote cofactor-based catalysis. Enhancement of cofactor-based catalysis in modern peroxidase enzymes is linked to a specific molecular machinery that includes the proximal histidine and distal histidine and arginine residues (Poulos and Kraut 1980; Ortiz de Montellano 2010). Such assistance molecular machinery is lacking from the ancestral TIM-barrel (fig. 1D).
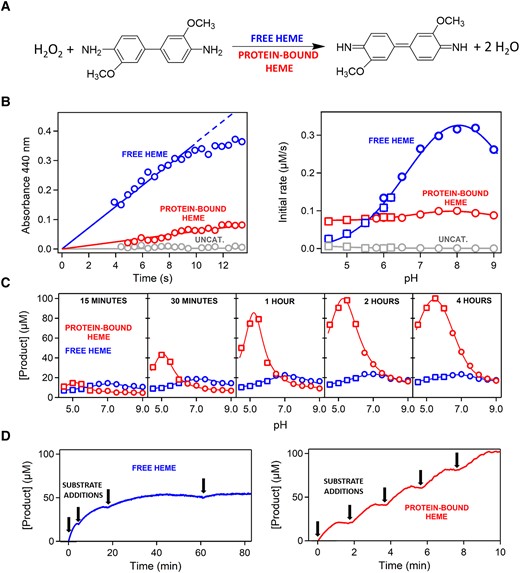
Peroxidase activity of free heme versus heme bound to the ancestral TIM-barrel. (A) Reaction used to test peroxidase activity. The kinetics of o-dianisidine peroxidation can be determined by following the increase of absorbance at 440 nm upon peroxidation. (B) Left: representative examples of the profiles of absorbance at 440 nm versus time used to calculate the initial rates of the reaction. The example shown corresponds to pH 7, heme concentration of 0.4 µM (either free- or protein-bound) and initial concentrations of hydrogen peroxide and o-dianisidine of 10 and 0.1 mM, respectively. The data show that the initial reaction rate is higher with free heme as a catalyst. Right: plot of initial rate versus pH for free heme, protein-bound heme, and a control experiment in the absence of heme (labeled “uncat”). In most of the studied pH range, initial rates are higher with free heme as a catalyst. Heme and initial substrate concentrations are the same as in B. Symbols refer to the buffer used: squares, 200 mM acetate, 150 mM NaCl; circles, 200 mM phosphate, 150 mM NaCl. (C) Profiles of the amount of peroxidation product formed versus pH over long reaction times (shown inside the plots). As the reaction time increases, bound heme progressively becomes a more efficient peroxidation catalyst than free heme. Buffers, heme concentration, and initial substrate concentrations are the same as in B. (D) Peroxidation kinetics over long reaction times with free heme (left) and protein-bound heme (right) as catalysts with repeated additions of 0.02 mM o-dianisidine substrate (labeled with arrows). Degradation of free heme catalysis is apparent by the progressive absence of peroxidation of freshly added o-dianisine. By contrast, no such degradation is observed with protein-bound heme. The data correspond to pH 7, heme concentration of 2 µM (either free- or protein-bound), and initial concentration of hydrogen peroxide of 10 mM.
Subsequently, we determined the total yield of peroxidation product for several pH values at times ranging from 15 min to 4 h. Strikingly (fig. 2C) we found a much higher conversion of substrate to product with the heme-bound ancestor when compared to free heme, in particular for reaction times on the order of hours. A reasonable explanation for these results is that free heme undergoes time-dependent processes that impair its peroxidase activity, while heme bound to the protein is protected and retains its peroxidase activity for a much longer time. To test this hypothesis, we performed experiments at longer durations and with repeated additions of a substrate (fig. 2D). We found that, unlike the heme bound to the ancestral protein, free heme gradually loses its capability to peroxidize the freshly added substrate.
To further explore the mechanisms of heme protection upon binding to the ancestral protein, we performed kinetic experiments in which heme, either free or protein-bound, was incubated for given times in the reaction buffer before substrate addition. Profiles of product concentration versus reaction time for these experiments are shown in figure 3A. Interestingly, we found the final substrate yield of the reaction catalyzed by free heme to decrease with incubation time. This decrease is most likely linked to heme self-association during incubation time and the consequent reduction in the concentration of the more active heme monomer. In support of this interpretation, the alteration in the Soret band known to be linked to heme self-association (Inada and Shibata 1962) occurs in the same time scale as does the decrease in peroxidase activity observed upon incubation (figs. 3B and 3C). Heme bound to the ancestral protein cannot associate with other heme molecules and it is therefore protected from the loss of peroxidase activity caused by self-association.
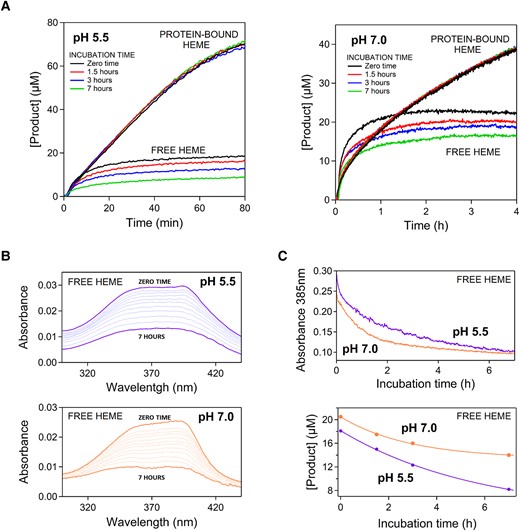
Degradation of catalysis by free heme during and before the peroxidation reaction. (A) Peroxidation kinetics over long reaction times with free heme and protein-bound heme as catalysts for pH 5.5 and pH 7. In all experiments, the heme concentration (either free- or protein-bound) was 0.4 µM and the initial concentrations of o-dianisidine and hydrogen peroxide were 0.1 and 10 mM, respectively. The experiments differ in the incubation duration, that is the time free heme or protein-bound heme were kept in the solution before the addition of the substrates. The curvature of the kinetic profiles for free heme reveals severe catalysis degradation during the peroxidation reaction. Yet, substantial degradation also occurs during the incubation time as shown by decreasing final product yields with increasing incubation time. (B) Self-association of heme as revealed by flattening of the Soret band of free heme in solution. Spectra were collected approximately every 30 min from the dilution of the heme stock in the buffer (zero time) to seven hours. See legend to figure 2 and Methods for buffer composition. (C) Plots of absorbance at 385 nm versus time (from the data given in B) and plots of final product yield versus incubation time (from the data given in A for free heme). Self-association occurs in the same time scale as (and it is the likely cause of) the degradation of catalytic potential of free heme during the incubation time.
It is also clear from the strong curvature and the leveling-off of the kinetic profiles at long reaction times (figs. 3A and 2D) that the degradation of the peroxidase activity of free heme is substantially accelerated after substrate addition. This points to an additional mechanism of inactivation acting during the catalytic cycle, likely involving chemical alterations of the heme caused by reactive oxygen species, as it has been described for heme in peroxidases (Valderrama et al. 2002) and for free heme in solution (Brown et al. 1968; Brown and Jones 1968). Remarkably, the loss of peroxidase activity during the catalytic cycle appears to be considerably limited in the restricted and defined molecular environment provided by the ancestral TIM-barrel for the bound heme (figs. 3A and 2D).
Discussion
Heme binding at a conformationally flexible region of an ancestral TIM-barrel leads to an enhancement in the efficiency of heme as a peroxidation catalyst, as shown by the increase in peroxidation reaction yield over a time scale of hours. Determination of initial reaction rates, however, indicates that binding does not improve the intrinsic peroxidase activity of heme. In fact, the opposite is true within most of the studied pH ranges: initial reaction rates are somewhat lower with the protein-heme complex as compared with free heme, likely reflecting limited substrate access to the substantially buried bound heme. Therefore, the enhancement in the efficiency of peroxidation catalysis upon heme binding to the protein scaffold cannot be attributed to the promotion of cofactor catalysis by the protein moiety, as is the case with modern peroxidases (fig. 1D). In fact, our experimental data (figs. 2 and 3) clearly support that the enhancement is due to the retardation of processes that impair the peroxidase activity of heme, resulting in a much longer lifetime and a higher effective concentration for the catalyst when it is bound to the protein.
The ancestral TIM-barrel we have used as a protein scaffold in our experiments is a putative ancestor of bacterial and eukaryotic family-1 glycosidases (Gamiz-Arco et al. 2021) and may perhaps probe an early stage in the evolution of this enzyme family. The phylogeny we used is presented in Gamiz-Arco et al. (2021), which also provides in supplementary table S1 and supplementary figure S1, Supplementary Material online the relevant details of the Bayesian analysis and the set of modern glycosydases used for ancestral sequence reconstruction. Family 1 glycosidases span the three domains of life (see the page for family 1 glycosidases in the CAZy database: http://www.cazy.org/GH1.html). Therefore, they likely existed already in the LUCA. Furthermore, they are of the TIM-barrel fold, which is a very ancient fold, proposed to have played a key role in the early evolution of metabolism (Goldman et al. 2016). Unlike the natural activity of these enzymes (glycosidic bond cleavage), the de novo activity generated in our ancestral TIM-barrel by heme binding is obviously of the redox type. However, it does not involve molecular oxygen, but it is a peroxidation activity that requires hydrogen peroxide (H2O2) as a reactant. A number of geochemical sources of H2O2 in the early Earth have been suggested (see He et al. 2021; Stone et al. 2022; He et al. 2023, and references therein), including both processes in the atmosphere and processes on mineral surfaces. Furthermore, it has been hypothesized that early life used H2O2 as an electron donor before the emergence of oxygenic photosynthesis (see Olson 2001; He et al. 2021, and references therein). In general, H2O2 availability at primordial times may have driven the emergence of enzymes that used H2O2 as a reactant and also the emergence of detoxification enzymes that simply decomposed H2O2. In view of all this, it cannot be ruled out that molecular events akin to that characterized in this work, that is heme binding to a TIM-barrel with concomitant protection and enhancement of heme-based catalysis, did actually occur at some primordial stage. However, our results have general implications beyond this specific possibility. That is, our experiments reveal a general mechanism of immediate enhancement of cofactor catalysis that points to and mimics a plausible scenario for the primordial emergence of enzymes. We briefly elaborate on this scenario below.
Reasonable chemical mechanisms for the prebiotic formation of polypeptides have been proposed (Frenkel-Pinter et al. 2020). Indeed, it has been hypothesized that polypeptides already existed in the primordial RNA world, where they served as enhancers of ribozyme activity (Wolf and Koonin 2007; Romero et al. 2016). Regarding protein cofactors, their extreme evolutionary conservation strongly suggests that they are very ancient (Chu and Zhang 2020). The likely origin of inorganic cofactors in the geochemical environment in which life began and that of organic cofactors (coenzymes) as parts of primordial ribozymes have been noted (Goldman and Kacar 2021). Overall, there can be little doubt that polypeptides and cofactors co-existed at some primordial stage. Then, their association may have immediately promoted catalysis by increasing the lifetime and the effective concentration of catalytic cofactors. It follows that, as soon as some means of transmission of genetic information, even if rudimentary and error-prone, was available, natural selection for polypeptides with increasing capability to protect catalytic cofactors became possible. This selection could have also favored cofactors that interact efficiently with polypeptides and become readily protected. Finally, the stable and catalytically competent polypeptide-cofactors complexes thus formed provided starting points for the Darwinian evolution of a protein molecular machinery that assisted and further enhanced cofactor-based catalysis.
It is interesting to view the evolutionary narrative proposed above in light of recent protein engineering efforts aimed at generating cofactor-based enzymes capable of catalyzing new reactions. These studies demonstrate that laboratory-directed evolution can readily find mutations that stabilize the transition state of a targeted reaction, thus converting low activities to levels comparable to those of modern natural enzymes. For instance, Studer et al. (2018) prepared a zinc-binding peptide with a very low esterase activity and achieved a 10,000-fold enhancement in activity over nine rounds of laboratory evolution. More recently, Bhattacharya et al. (2022) used NMR to guide low-throughput variant library screening and reported a 62,000-fold enhancement in catalytic-efficiency for a Kemp-elimination activity linked to myoglobin-bound heme. Overall, it emerges that the bottleneck in the evolution of efficient de novo enzymes is the emergence of a low-level of catalysis on which selection can act. The protection mechanism may have contributed to bypassing the bottleneck at primordial times, because it brings about an immediate and, therefore, selectable enhancement in cofactor catalysis. Of course, once cofactor-polypeptide complexes with significant activity are formed, natural selection can be invoked to further enhance catalysis through mutations in the protein moiety and eventually lead to highly efficient enzymes in many cases.
The protection mechanism for catalysis enhancement has been demonstrated in this work based on the peroxidase activity of heme, but it should apply to many other cofactors. The emergence of enzymes based on catalytic iron-sulfur clusters may be a particularly relevant example. Iron and sulfur were abundant in the geochemical environment that likely hosted primordial life (Beinert 2000) and enzymes based on iron-sulfur clusters abound in reconstructions of the gene content of LUCA (Weiss et al. 2016). Free iron-sulfur clusters have been found to mimic the basic features of protein-bound clusters, but in nonaqueous media and in the absence of oxygen (Beinert 2000). Recent phylogenetic analyses (Jablonska and Tawfik 2021) of oxygen-utilizing and oxygen-producing enzymes support that oxygen was available well before the great oxidation event ∼2.4 billion years ago. Indeed, according to recent proposals (Taverne et al. 2020; He et al. 2023) life may have emerged in weakly oxic micro-environments. Yet, even if life evolved in a completely anaerobic environment (Weiss et al. 2016) and destruction by reaction with oxygen was not an issue at an early evolutionary stage, iron-sulfur clusters still appear as obvious candidates for primordial catalysis enhancement through protection by polypeptides, as supported by recent engineering studies (Kim et al. 2018).
Ferredoxins are iron-sulfur proteins that perform electron transfer in a diversity of biochemical transformations. About 60 years ago, Margaret Dayhoff noted the simplicity of ferredoxins, which consist of an inorganic active site and a very short polypeptide which she proposed to have emerged by duplication of smaller peptides (Eck and Dayhoff 1966). Modern ferredoxins may, therefore, be relics of primordial protection events. An intriguing possibility is that heme binding to TIM-barrel glycosidases is also a relic of primordial protection. In this case, however, while our ancestral TIM-barrel glycosidase binds heme tightly and stoichiometrically, heme binding is only observed in a vestigial form in some modern glycosidases (Gamiz-Arco et al. 2021). We could speculate that, at some stage, the emergence of more efficient peroxidases deprived heme binding to the ancestral TIM-barrel of its selective advantage and, consequently, the ancestral functional feature underwent evolutionary degradation, as it is generally the case with features that become vestigial during evolution (Darwin 1871; Gamiz-Arco et al. 2019).
Methods
The ancestral TIM-barrel glycosidase was prepared as previously described (Gamiz-Arco et al. 2021). Briefly, the gene for the His-tagged protein in a pET24 vector was cloned into E. coli BL21 (DE3) cells and the protein was purified using Ni-NTA chromatography. Protein prepared in this way typically has a very small amount of bound heme. Ancestral protein saturated with heme was prepared by incubation with a 5-fold excess of heme followed by size-exclusion chromatography to eliminate nonbound heme, as we have previously described in detail (Gamiz-Arco et al. 2021). The heme-to-protein ratio was found to be close to unity based on absorbance determinations for the protein band at 280 nm and the heme Soret band.
As previously described (Gamiz-Arco et al. 2021), heme solutions were prepared by high-dilution (typically 1:1,000) in the desired buffer of a stock solution in concentrated sodium hydroxide and used immediately. Stock solutions of heme were prepared daily. Heme concentrations in stock solution were determined from the absorbance of the heme Soret band at 385 nm using a known value of the extinction coefficient (Deniau et al. 2003). Stock solutions of o-dianisidine were prepared by weight. Stock solutions of hydrogen peroxide were prepared by dilution of commercially available stock solution and their concentrations were determined from the absorbance at 240 nm using a known extinction coefficient (Jiang et al. 1990). The peroxidation reaction was initiated by adding microliter volumes of the reactants to a 2-mL solution containing free heme or protein-bound heme. The reaction was followed by measuring the absorbance of the peroxidation product at 440 nm. A known value of the extinction coefficient (Jenkins et al. 2021) was used to calculate substrate concentration from absorbance values. Peroxidation experiments were performed in a wide pH range using the following buffers: 200 mM acetate, 150 mM NaCl for the pH range 4.5–6.2, and 200 mM phosphate, and 150 mM NaCl for the pH range 5.8–9.0. Since the peroxidation reaction does not involve molecular oxygen, we did not use degassed solutions for activity determinations. Yet, we confirmed at a number of representative conditions that degassing had no significant effect on peroxidation rates.
Acknowledgments
This work was supported by Human Frontier Science Program grant RGP0041/2017 (J.M.S.R. and E.A.G.), National Science Foundation grant 2032315 (E.A.G.), Department of Defense grant MURI W911NF-16-1-0372 (E.A.G.), National Institutes of Health grant R01AR069137 (E.A.G.), Spanish Ministry of Science and Innovation/FEDER Funds grant PID2021-124534OB-100 (J.M.S.R.), and grant PID2020-116261GB-I00 (J.A.G.).
Author Contributions
L.I.G.R. carried out protein preparation, and designed and performed the experimental determination of peroxidase activity under the supervision of V.A.R.; G.G.A. provided essential input regarding the catalytic properties of the ancestral glycosidase; J.A.G. provided essential input regarding the structural interpretation of the protection mechanisms; E.A.G provided essential input regarding the interpretation of the data in an evolutionary context; J.M.S.R. designed the research and wrote the first draft of the manuscript; all authors discussed the manuscript, suggested modifications and improvements and contributed to the final version.
Data Availability
Raw experimental data are available from the authors upon reasonable request.

{kind=link}
{kind=link}
{kind=link}