





Abstract
In Drosophila melanogaster, a key germline stem cell (GSC) differentiation factor, bag of marbles (bam) shows rapid bursts of amino acid fixations between sibling species D. melanogaster and Drosophila simulans, but not in the outgroup species Drosophila ananassae. Here, we test the null hypothesis that bam’s differentiation function is conserved between D. melanogaster and four additional Drosophila species in the melanogaster species group spanning approximately 30 million years of divergence. Surprisingly, we demonstrate that bam is not necessary for oogenesis or spermatogenesis in Drosophila teissieri nor is bam necessary for spermatogenesis in D. ananassae. Remarkably bam function may change on a relatively short time scale. We further report tests of neutral sequence evolution at bam in additional species of Drosophila and find a positive, but not perfect, correlation between evidence for positive selection at bam and its essential role in GSC regulation and fertility for both males and females. Further characterization of bam function in more divergent lineages will be necessary to distinguish between bam’s critical gametogenesis role being newly derived in D. melanogaster, D. simulans, Drosophila yakuba, and D. ananassae females or it being basal to the genus and subsequently lost in numerous lineages.
Introduction
In most sexually reproducing animals, egg and sperm production begins with the differentiation of germline stem cells (GSCs). These GSCs divide both to self-renew to maintain the germline and to produce daughter cells that differentiate to produce gametes (Kahney et al. 2019). The cellular processes of daughter cell differentiation and stem cell self-renewal are the most fundamental functions of stem cells and are therefore highly regulated and presumed to be conserved. In the germline, the mis-regulation of either of these processes leads to sterility (Gleason et al. 2018). However, many of the genes that control GSC maintenance and GSC daughter differentiation in Drosophila melanogaster are rapidly evolving due to positive selection for amino acid diversification (Civetta et al. 2006; Bauer DuMont et al. 2007; Choi and Aquadro 2015; Flores, DuMont, et al. 2015).
One such gene is bag-of-marbles (bam) which exhibits a rapid burst of amino acid substitutions between the sister species D. melanogaster and Drosophila simulans, acts as the key switch for GSC daughter cell differentiation in D. melanogaster females, and is necessary for terminal differentiation of spermatogonia in males (McKearin and Spradling 1990; Civetta et al. 2006; Bauer DuMont et al. 2007; Insco et al. 2009). Despite 60 fixed amino acid differences between D. melanogaster and D. simulans, a D. simulans bam transgene is sufficient for GSC daughter cell differentiation in D. melanogaster females and males. The intriguing observations that bam is both rapidly evolving due to positive selection and functions as a switch for GSC daughter differentiation in D. melanogaster has led us to investigate what could be driving its positive selection between species (Flores, Bubnell, et al. 2015).
In females, bam is repressed in GSCs, and upon its expression promotes differentiation by binding to the protein product of benign gonial cell neoplasm (bgcn) as well as a series of other protein partners to repress the translation of self-renewal factors nanos and eIF4a (Li et al. 2009; Ting 2013). The GSC daughter cell differentiates into a cystoblast, which then goes through four rounds of mitotic divisions while bam localizes to the fusome, a cellular structure that interconnects the resulting dividing cells that make up the cyst. In these cells, Bam functions with Bgcn to regulate mitotic synchrony (Chen and McKearin 2003). Before meiosis begins, bam expression is repressed. In males, bam is expressed in GSCs and its expression increases as differentiation progresses (Insco et al. 2009). In early dividing spermatogonia, Bam protein must reach a threshold to bind with partners Bgcn and tumorous testis (tut) to repress mei-P26 and switch the cellular program from proliferation to terminal differentiation to begin meiosis (Chen et al. 2014). After this switch, bam expression is rapidly repressed. Loss of bam function in either sex results in the over-proliferation of either GSCs in females or spermatogonia in males causing tumors and sterility (McKearin and Spradling 1990).
One possible explanation for the observed signal of positive selection at bam is a change of function. As bam regulates gametogenesis, it is possible that life history or environmental factors drive positive selection at bam. It is even possible that the genetic interaction between the endosymbiotic bacteria Wolbachia pipientis and bam has led to such changes in bam’s function in different lineages, as Wolbachia infects the GSC niche and is propagated through the germline (Flores, Bubnell, et al. 2015). While functional divergence of bam seems less likely due to its essential role in GSC maintenance and differentiation in D. melanogaster, bam sequence is highly divergent among species. It is even difficult to confidently align bam sequence between D. melanogaster and Drosophila ananassae, both members of the melanogaster species group (subgenus Sophophora), let alone the many more divergent Drosophila species (Bauer DuMont et al. 2007). Additionally, bam appears to be a novel gene in the genus Drosophila as no orthologues are reported in any non-Drosophila genomes using orthoDB v10 (Kriventseva et al. 2019) and Ensembl Metazoa (Howe et al. 2020) (both accessed 11 April 2021).
Here, we investigate the possibility that the observed signals of positive selection in D. melanogaster and D. simulans at bam are due to selection for a novel function for bam in gametogenesis in these lineages. To do this, we ask if bam is necessary for gametogenesis in diverse species within the Drosophila genus. We generate bam loss-of-function alleles in five Drosophila species in the melanogaster species group (D. melanogaster, D. simulans, Drosophila yakuba, Drosophila teissieri, and D. ananassae). Surprisingly, we find that bam is not necessary for gametogenesis in one or both sexes in two species within the melanogaster species group. We also analyze additional population samples of species mainly within the melanogaster species group and a few more distant lineages for nucleotide sequence variation at bam. Within the melanogaster species subgroup (D. melanogaster, D. simulans, D. yakuba, Drosophila santomea, and D. teissieri), we find evidence of lineage-specific signals of adaptation for three species, but not for D. teissieri or D. santomea. Outside of the melanogaster species subgroup, we find similar patterns of lineage-specific signals of positive selection. Together with our bam loss-of-function results, these observations raise the possibility that the critical role for bam in GSC differentiation is newly derived in the melanogaster species group and the signatures of positive selection we observe in some of those species is associated with species-specific refinements of bam’s GSC differentiation function or species-specific adaptive pressures acting on bam’s GSC differentiation function.
Results
Generating bam Null Alleles for non-melanogaster Species
To ask if bam’s GSC function is conserved in species outside of D. melanogaster, we designed and generated a series of bam null alleles in five species in the melanogaster species group to assay for GSC daughter differentiation (fig. 1A). To do this, we used CRISPR/Cas9 to introduce a 3xP3-YFP or a 3xP3-DsRed gene cassette into the first exon of bam, thereby disrupting the bam coding sequence and introducing a premature termination codon (fig. 1B). This also generates an allele that is trackable by eye color, which is necessary for the non-melanogaster species of which we do not have balancer chromosomes to maintain alleles that cause sterility. To generate the homozygous bam null genotype, we cross the 3xP3-YFP line to the 3xP3-DsRed line and select flies with both DsRed and YFP positive eyes. This scheme also allows us to use the same cross to assay the heterozygous and wildtype bam siblings (table 1).

Species chosen to generate bam null alleles using CRISPR/Cas9 to disrupt bam with a trackable eye marker cassette. (A) The five species we chose to generate bam null alleles span approximately 30 million years of divergence. (B) Schematic for generating null alleles with trackable eye markers using CRISPR/Cas9. We generate two null alleles using different color eye markers and cross the heterozygotes together to generate an easily trackable null genotype.
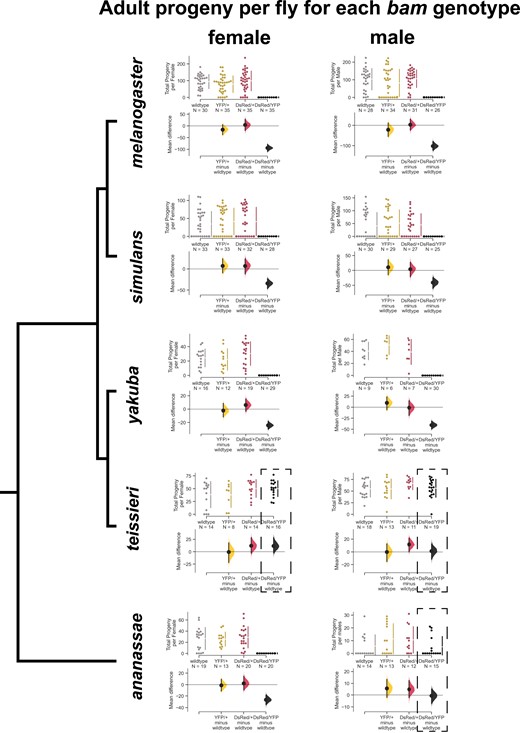
bam is necessary for female and male fertility in Drosophila melanogaster, Drosophila simulans, and Drosophila yakuba, but not for Drosophila ananassae males nor both Drosophila teissieri males and females. Swarm plots showing the total progeny per female or per male for each bam genotype and Cumming estimation plots showing the 95% confidence interval effect size. Dotted boxes show genotypes where the phenotype did not match that of D. melanogaster. For D. melanogaster, D. simulans, and D. yakuba, the bam null DsRed/YFP females and males were sterile (P < 0.0001, permutation test), and the heterozygous genotypes did not have a significant effect on fertility (P > 0.05, permutation test). For D. teissieri, the bam null DsRed/YFP females and males did not have significantly different fertility from wildtype, nor did the heterozygotes. For D. ananassae, the bam null DsRed/YFP females were sterile (P < 0.0001, permutation test), and the heterozygotes did not affect fertility. However, the bam null DsRed/YFP males were fertile and there was no significant difference between the null and heterozygotes and wildtype fertility (P > 0.05, permutation test).
Abbreviations for bam Genotypes for all Species and the Expected Phenotypes as Observed in Drosophila melanogaster.
| Drosophila melanogaster Phenotype Expectations | ||||
|---|---|---|---|---|
| Genotype | Abbreviation | Eye Color | Fertility | Cytological |
| bam+/bam+ | wildtype | white | Wildtype | Wildtype |
| bam3xP3-DsRed/bam+ | DsRed/+ | DsRed | Wildtype | Wildtype |
| bam3xP3-YFP/bam+ | YFP/+ | YFP | Wildtype | Wildtype |
| bam3xP3-DsRed/bam3xP3-YFP | DsRed/YFP | DsRed & YFP | Sterile | Tumorous ovaries and testes |
| Drosophila melanogaster Phenotype Expectations | ||||
|---|---|---|---|---|
| Genotype | Abbreviation | Eye Color | Fertility | Cytological |
| bam+/bam+ | wildtype | white | Wildtype | Wildtype |
| bam3xP3-DsRed/bam+ | DsRed/+ | DsRed | Wildtype | Wildtype |
| bam3xP3-YFP/bam+ | YFP/+ | YFP | Wildtype | Wildtype |
| bam3xP3-DsRed/bam3xP3-YFP | DsRed/YFP | DsRed & YFP | Sterile | Tumorous ovaries and testes |
Abbreviations for bam Genotypes for all Species and the Expected Phenotypes as Observed in Drosophila melanogaster.
| Drosophila melanogaster Phenotype Expectations | ||||
|---|---|---|---|---|
| Genotype | Abbreviation | Eye Color | Fertility | Cytological |
| bam+/bam+ | wildtype | white | Wildtype | Wildtype |
| bam3xP3-DsRed/bam+ | DsRed/+ | DsRed | Wildtype | Wildtype |
| bam3xP3-YFP/bam+ | YFP/+ | YFP | Wildtype | Wildtype |
| bam3xP3-DsRed/bam3xP3-YFP | DsRed/YFP | DsRed & YFP | Sterile | Tumorous ovaries and testes |
| Drosophila melanogaster Phenotype Expectations | ||||
|---|---|---|---|---|
| Genotype | Abbreviation | Eye Color | Fertility | Cytological |
| bam+/bam+ | wildtype | white | Wildtype | Wildtype |
| bam3xP3-DsRed/bam+ | DsRed/+ | DsRed | Wildtype | Wildtype |
| bam3xP3-YFP/bam+ | YFP/+ | YFP | Wildtype | Wildtype |
| bam3xP3-DsRed/bam3xP3-YFP | DsRed/YFP | DsRed & YFP | Sterile | Tumorous ovaries and testes |
We used D. melanogaster as a control to ensure our method would generate a loss of function bam phenotype, as the bam null phenotype has been well characterized in this species (table 1). We also generated bam nulls in four other species within the melanogaster species group D. simulans, D. yakuba, D. teissieri, and D. ananassae (fig. 1A).
bam is necessary for female and male fertility in D. melanogaster, D. simulans, and D. yakuba, but only in D. ananassae females, and in neither D. teissieri males nor females.
In D. melanogaster, both male and female bam nulls are sterile as early germline cells fail to differentiate and proceed through gametogenesis. In classic null alleles of D. melanogaster bam (bamΔ59 and bamΔ86), one copy of functional bam is sufficient to rescue the bam null sterility phenotype. We first asked if our novel bam null alleles behaved this way in D. melanogaster. Heterozygotes of each 3xP3-DsRed or 3xP3-YFP disruption line showed similar fertility to wildtype bam in both males and females (mean difference of adult progeny, not significant, permutation test) and transheterozygous males and females carrying both the 3xP3-DsRed and the 3xP3-YFP disruptions (which we will term the bam null genotype) were completely sterile (mean difference of adult progeny, P < 0.0001, permutation test) (fig. 2, supplementary file S2, Supplementary Material online). We observed this same pattern for D. simulans and D. yakuba (fig. 2, supplementary file S2, Supplementary Material online). We observed some sterile D. simulans females and males across all bam genotypes, which we attribute to the strain and unlikely to be related to bam (fig. 2, supplementary file S2, Supplementary Material online). In contrast, in D. teissieri we found that there was no difference in fertility between any of the bam genotypes tested for females or males (mean difference of adult progeny, not significant, permutation test) (fig. 2, supplementary file S2, Supplementary Material online). The bam null genotype did not result in sterility, or even reduced fertility. In D. ananassae, bam null females were sterile (mean difference of adult progeny, P < 0.0001, permutation test), and heterozygotes of each 3xP3-DsRed or 3xP3-YFP disruption did not significantly reduce fertility (mean difference of adult progeny, not significant, permutation test). However, in D. ananassae males, bam nulls were fertile, with no significant mean difference in progeny across the tested bam genotypes (fig. 2, supplementary file S2, Supplementary Material online). Therefore, bam is not necessary for fertility in all species in the melanogaster species group, or even all species within the melanogaster species subgroup.
Consistent with the fertility results, bam is also necessary for germ cell differentiation in D. simulans and D. yakuba females and males, but not in D. teissieri males or females, and only D. ananassae females.
The ovaries of D. melanogaster bam null females feature over-proliferating GSCs that fail to differentiate into cystoblasts (McKearin and Spradling 1990; Ting 2013). This results in the germarium and developing downstream cysts accumulating small, GSC-like cells resembling tumors (fig. 3A). In the testes of D. melanogaster bam null males, spermatogonia differentiation is blocked. The resulting testes feature over-proliferating spermatogonia that fail to develop into terminally differentiated spermatocytes (fig. 3A) (Insco et al. 2009, 2012).
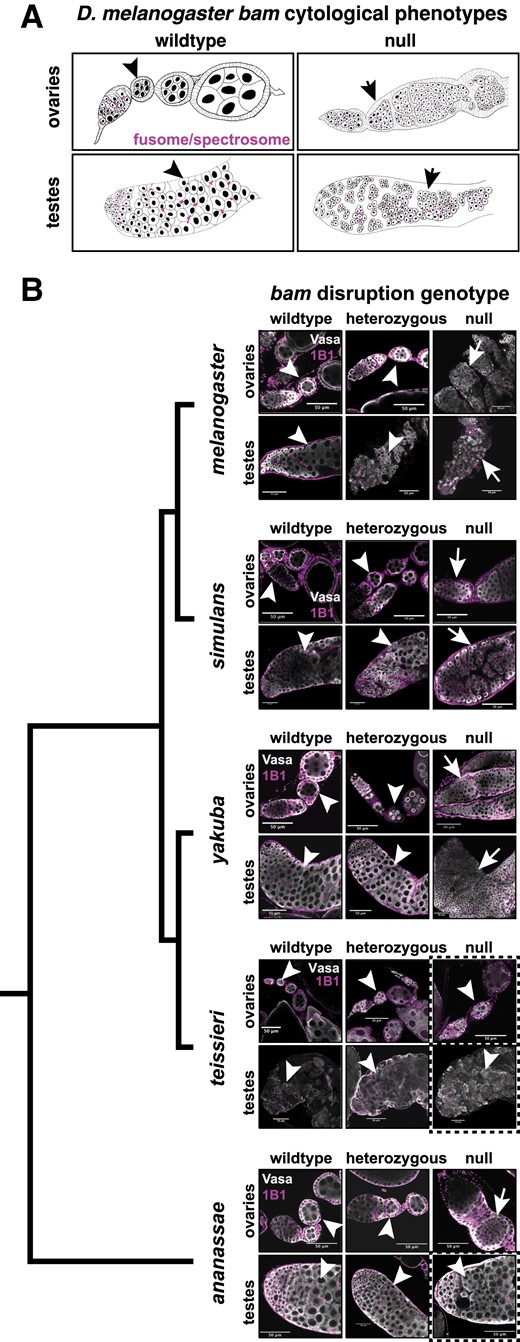
bam is necessary for female and male GSC differentiation in Drosophila melanogaster, Drosophila simulans, and Drosophila yakuba, but not for Drosophila ananassae males nor Drosophila teissieri males and females. (A) Schematics of ovaries and testes showing the bam wildtype and null cytological phenotypes in D. melanogaster. Wildtype bam ovaries exhibit developing egg chambers with differentiated nurse cells and testes exhibit large, differentiated spermatocytes. Null bam ovaries exhibit egg chambers filled with small GSC-like tumors that contain undeveloped fusomes and testes exhibit cysts filled with small over-proliferating spermatogonia. (B) Ovaries and testes from all five species of wildtype (bam+/bam+), heterozygous (YFP/+ or DsRed/+), and null (DsRed/YFP) bam genotypes. All tissues are labeled with antibodies to Vasa (germline) and Hts-1B1 (fusome). Dashed boxes indicate genotypes and tissue that are not consistent with the D. melanogaster phenotype. Ovaries and testes from all five species exhibit developing nurse cells or spermatocytes (respectively) for wildtype and heterozygous bam genotypes. We did not observe any tumorous cysts (arrowheads). Ovaries and testes from bam null D. melanogaster, D. simulans, and D. yakuba all exhibited cysts filled with GSC-like tumors (ovaries) or over-proliferating spermatogonia (testes) (arrows). The bam null ovaries exhibit undeveloped fusomes. D. ananassae bam null ovaries exhibited the tumorous egg chamber phenotype (arrow), however testes exhibited large developing spermatocytes (arrowhead) and we did not observe any tumors. The bam null D. teissieri ovaries and testes exhibited nurse cell positive egg chambers and developing spermatocytes (respectively) (arrowheads) and we did not observe any tumors.
We imaged ovaries from 2- to 5-day-old D. melanogaster females from each bam genotype. We assayed for the characteristic bam null tumor phenotype using antibodies to Vasa to visualize the morphology of the germline and Hts-1B1 to visualize the fusome, which fails to fully form in bam mutants (Lavoie et al. 1999; Hinnant et al. 2020). We found that the bam null genotype resulted in the characteristic bag of marbles phenotype consistent with classic bam null alleles, with tumorous ovaries filled with undifferentiated GSC-like cells (fig. 3B). One copy of wildtype bam was sufficient to rescue the bam null phenotype, as has been previously observed for other bam null alleles (fig. 3B) (Lavoie et al. 1999; Flores, Bubnell, et al. 2015). Therefore, our 3xP3-DsRed and 3xP3-YFP disruptions successfully recapitulate the bam null phenotype in females. We imaged testes from 3- to 5-day-old D. melanogaster males and also immunostained with antibodies to Vasa to label the germline and Hts-1B1 to label the fusome. We found that the bam null genotype resulted in tumorous testes filled with small over-proliferating cells that fail to differentiate into spermatocytes (fig. 3B). We found that one copy of wildtype bam was sufficient to rescue the bam null tumor phenotype, as has been previously reported for classic bam null alleles (Gönczy et al. 1997; Shivdasani and Ingham 2003) (fig. 3B). Therefore, our bam null alleles also recapitulate the previously described bam null phenotype in D. melanogaster males.
We repeated the same analysis for the other species in the study. We found for D. simulans and D. yakuba both female and male homozygous bam null genotypes exhibited the characteristic ovarian and testes tumors as described in D. melanogaster (fig. 3B). In these two species for both males and females, one copy of bam was also sufficient to rescue the bam null phenotype as observed in D. melanogaster (fig. 3B). However, in the D. teissieri bam null genotype, we observed no evidence of GSC over-proliferation in females or spermatogonia over-proliferation in males. We observe the same phenotype for the heterozygous alleles in both males and females, as well as wildtype (fig. 3B). This indicates that bam is not necessary for GSC daughter differentiation in D. teissieri females nor is bam necessary for spermatogonia differentiation in males. In D. ananassae bam null females, we observed the characteristic bam null phenotype observed in D. melanogaster with over-proliferating GSC-like cells in the germarium and cysts and undeveloped fusomes (fig. 3B). One copy of bam was sufficient to rescue the bam null tumorous phenotype (fig. 3B). Therefore, the D. ananassae female bam null phenotype is consistent with that of D. melanogaster, D. simulans, and D. yakuba. In contrast, in D. ananassae males of the bam null genotype we did not observe over-proliferating spermatogonia (fig. 3B). The homozygous bam null genotype resulted in the same phenotype as the heterozygous bam null genotypes and wildtype indicating that bam is not necessary for the switch from proliferating spermatogonia to differentiated spermatocytes in D. ananassae males. Therefore, these cytological data are fully consistent with our fertility assay observations.
Confirming the Loss-of-Function Status of bam in the bam Null Disruption Design
The only available Bam antibody is weakly cross-reactive in D. melanogaster and D. simulans thus we cannot directly demonstrate the absence of Bam protein in all the species tested (Flores 2013; Flores, Bubnell, et al. 2015). Although we used the same design to generate the bam null alleles across this study and therefore expect the 3xP3-DsRed and 3xP3-YFP disruptions to have the same effect on bam in all five species, we further confirmed the loss-of-function status of the bam disruption alleles in D. ananassae and D. teissieri given their functional divergence.
First, we asked if bam had undergone duplication events in D. ananassae and D. teissieri using publicly available sequence data and found no evidence of bam duplication in either species (details described in supplementary file S1, supplementary table S8, Supplementary Material online). Next, since the 3xP3-DsRed and 3xP3-YFP insertions introduce a premature termination codon, we expect the mRNA of the disrupted bam allele to be degraded through nonsense-mediated mRNA decay (NMD), but if the premature termination codon induced exon skipping, the disrupted bam exon could be alternatively spliced out resulting in expression of a truncated bam allele, which may be sufficient for function (Brogna and Wen 2009; Mou et al. 2017; Chen et al. 2018; Sui et al. 2018). We used RT-qPCR to ask whether patterns of expression in the bam null alleles in D. ananassae males and D. teissieri females and males were consistent with expectations for NMD. We found that for D. ananassae and D. teissieri, the expression of the bam null disruption alleles were reduced relative to the bam wildtype allele to levels consistent with NMD (∼1–50%, details in supplementary file S1, supplementary figs S1 and S2, Supplementary Material online) (Pereverzev et al. 2015). Therefore, the mRNA of the bam disruption alleles are likely targeted for degradation. Additionally, since both bam null D. teissieri males and females show wildtype phenotypes we looked for evidence of exon skipping. We asked if the exon disrupted by 3xP3-YFP or 3xP3-DsRed in the bam null genotypes was expressed in proportion to the non-disrupted exons relative to the wildtype genotype. We found that the ratio of exon 1 to exon 2 expression was not significantly different between D. teissieri nulls and D. teissieri wildtype genotypes for males and females (details in supplementary file S1, supplementary fig S3, Supplementary Material online).
Although bam null D. ananassae males and D. teissieri males and females exhibited wildtype phenotypes, together the genomic and RT-qPCR analyses indicate that the bam null alleles are loss-of-function alleles. This evidence therefore supports our conclusion that bam is not required for spermatogenesis in D. ananassae and D. teissieri nor oogenesis in D. teissieri.
D. ananassae bam Null Males Show Evidence of Germline Defects
In D. melanogaster, Fas3 expression in the testes is limited to the hub, the testes microenvironment that houses the somatic and GSC populations at the apical tip of the testes. As we found that bam was necessary for oogenesis in D. ananassae but not for spermatogenesis, we assayed for more subtle evidence of germline defects in D. ananassae males. We immunostained testes with an antibody to Fas3 to look for evidence of ectopic expression of Fas3 as in Gonzalez et al. (2015). In D. melanogaster bam null males, Fas3 is ectopically expressed, as the spermatogonia over proliferate (fig. 4A and B). In D. ananassae bam null males, we also observed ectopic expression of Fas3 in cells outside of the apical tip of the testes, although not nearly as severe as in D. melanogaster bam null males (fig. 4D and E). We then asked if there was any evidence of ectopic Fas3 expression in D. teissieri bam null males, as we also found that bam was not necessary for spermatogenesis in D. teissieri. In contrast to D. ananassae and D. melanogaster, we did not observe ectopic expression of Fas3 in D. teissieri bam null males (fig. 4B and C). This indicates that while bam is not necessary for differentiation in D. ananassae males, it may be playing a role in regulating the early differentiating germ cell population.
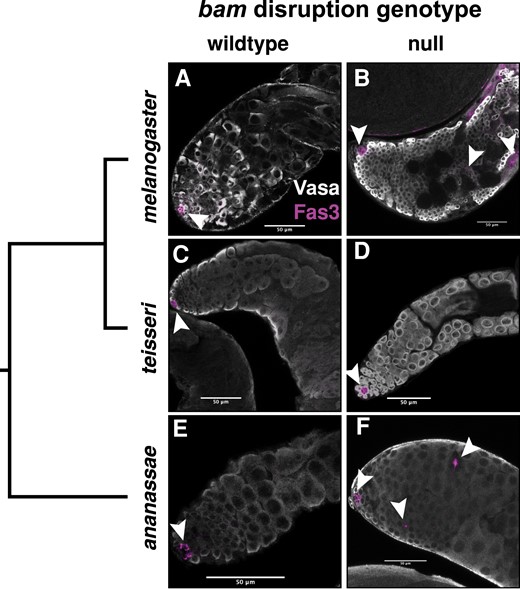
Loss of bam function in D. ananassae males shows ectopic expression of Fas3, a marker expressed in the hub, but not in Drosophila teissieri. All testes tissues are labeled with antibodies for Vasa (germline) and Fas3 (hub, arrowheads). Significance was assessed with a Fisher’s exact test comparing counts of testes with ectopic Fas3 expression between wildtype and bam null testes. (A) Drosophila melanogaster bam wildtype testes with Fas3 expression limited to the anterior tip of the testes hub (inset) and featuring large differentiating spermatogonia (Fas3 ectopic expression n = 0/13). (B) D. melanogaster bam null testes feature ectopic Fas3 expression and small over-proliferating spermatogonia (Fas3 ectopic expression n = 8/11, P < 0.000). (C) D. teissieri bam wildtype testes with Fas3 expression limited to the anterior tip of the testes (hub) and featuring large differentiating spermatogonia (Fas3 ectopic expression n = 0/20). (D) D. teissieri bam null testes with Fas3 expression limited to the anterior tip of the testes (hub) and large differentiating spermatogonia (Fas3 ectopic expression n = 0/16, P = 1). (E) Drosophila ananassae bam wildtype testes with Fas3 expression limited to the hub and featuring large differentiating spermatogonia (Fas3 ectopic expression n = 31). (F) D. ananassae bam null testes with ectopic Fas3 expression and large differentiating spermatogonia (Fas3 ectopic expression n = 19/27, P < 0.000).
bam Shows Lineage-specific Bursts of Positive Selection Throughout the Drosophila Genus
Previous studies have reported that bam is evolving under strong positive selection detected by the McDonald–Kreitman test (MKT) in D. melanogaster and D. simulans, but not in D. ananassae (Civetta et al. 2006; Bauer DuMont et al. 2007; Choi and Aquadro 2014). As these three species span about 30 million years of divergence, we wanted to know if there was evidence of positive selection in lineages more closely related to D. melanogaster and D. simulans, or if this signal was unique to the individual D. melanogaster and D. simulans lineages. We expanded the species sampled to include three closely related species from the yakuba complex: D. yakuba, D. santomea, and D. teissieri. We also took advantage of published population genomic data from samples of inbred lines to expand our assessment of bam variation in additional population samples for D. melanogaster (Lack et al. 2015) and D. simulans and to newly assess bam variation in population samples in the montium species group (Drosophila serrata, Drosophila bunnanda, Drosophila birchii, and Drosophila jambulina) and the ananassae species subgroup (Drosophila bipectinata, Drosophila pseudoananassae, and Drosophila pandora) (Li et al. 2021).
In addition to further sampling closely related species to D. melanogaster and D. simulans, we also wanted to ask if there was any evidence of adaptive evolution at bam in lineages outside of the melanogaster group. We resequenced bam from population samples of three more distantly related Drosophila species: Drosophila pseudoobscura, Drosophila affinis, and Drosophila mojavensis which span approximately 70 million years of divergence (Li et al. 2021). We also used published population genomic data from samples of inbred lines to assess bam variation in Drosophila immigrans and Drosophila rubida, both members of the immigrans species group (Li et al. 2021).
To ask if any population samples showed departures from selective neutrality consistent with positive selection at bam similar to what we previously observed for D. melanogaster and D. simulans, we used a lineage-specific MKT (Siddiq et al. 2017). The null hypothesis of the MKT is that the ratio of nonsynonymous to synonymous polymorphism within a species is equal to the ratio of nonsynonymous to synonymous fixed differences between species (or in our case, a predicted common ancestral sequence). A previous study reported signatures of positive selection at bam as an excess of nonsynonymous fixations in the D. melanogaster and D. simulans lineages (Bauer DuMont et al. 2007). As bam appears to be experiencing episodic signals of adaptive evolution restricted to individual lineages, we measured lineage-specific divergence. We used codeml to generate reconstructed ancestral sequences at the node of interest by maximum likelihood (Yang 1997, 2007). We then used this sequence to calculate nonsynonymous and synonymous divergences for each population sample. We excluded polymorphisms below 12% as low-frequency mutations are likely to be slightly deleterious and reduce the power of the MKT to identify positive selection (Fay et al. 2002; Charlesworth and Eyre-Walker 2008).
For D. melanogaster, we utilized the Zambia population sample from the Drosophila genome Nexus to measure bam variation (Lack et al. 2015). The Zambia sample contains the largest sample size of intact bam sequence (few missing calls), and represents variation found in the ancestral range of D. melanogaster. We used the predicted common ancestor of D. melanogaster and D. simulans and found similar patterns of polymorphism and divergence to the smaller samples of D. melanogaster from the USA and Zimbabwe previously reported by Bauer DuMont et al. (2007) (fig. 5). The D. melanogaster lineage shows a strong signal of positive selection (alpha = 0.943, P = 0.001) (fig. 5).
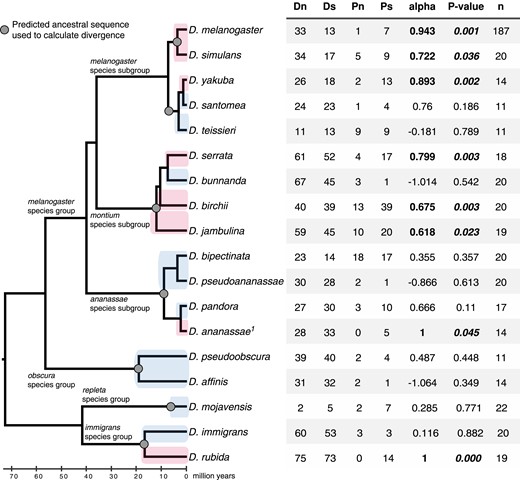
bam shows lineage-specific signals of positive selection scattered across the Drosophila genus. Cladogram of Drosophila species we sampled in this study. Lineages where we detect evidence of positive selection at bam with the MKT are D. melanogaster, D. simulans, D. yakuba, D. serrata, D. birchii, D. jambulina, D. ananassae, and D. rubida and are highlighted in pink. Lineages of which we could not reject neutrality with the MKT are D. teissieri, D. bunnanda, D. bipectinata, D. pseudoananassae, D. pandora, D. pseudoobscura, D. affinis, D. mojavensis, and D. immigrans and are highlighted in blue. The 2×2 table from the MKT is shown on the right (N = nonsynonymous, S = synonymous). Alpha is the proportion of nonsynonymous fixations predicted to be due to positive selection. P-value is from the χ2 test, n is the sample size. “1” indicates polymorphism data from Choi and Aquadro (2014).
For D. simulans bam, we utilized the Burnley, Victoria population sample from Li et al. (2021). We used the same common ancestral sequence as we did for the D. melanogaster analysis to calculate divergence. The D. simulans lineage shows a signal of positive selection for bam (alpha = 0.722, P = 0.036) consistent with results previously reported by Bauer DuMont et al. (2007) (fig. 5). We also assessed bam variation in the D. simulans CA, USA population sample from Signor et al. (2018), but due to elevated levels of genome-wide intermediate frequency polymorphisms caused by a recent population bottleneck, this sample violates key assumptions for the MKT (Fay 2011; Messer and Petrov 2013). For completeness, we report our findings for the CA sample in supplementary file S1, Supplementary Material online.
We used the predicted common ancestor of the yakuba complex (D. yakuba, D. santomea, D. teissieri) to estimate divergence for bam in each of the population samples. For D. yakuba we also observed a significant signal of positive selection for bam (alpha = 0.893, P = 0.002) (fig. 5). In contrast, we could not reject neutrality for D. teissieri bam or for D. santomea bam. The estimate of alpha is very low for D. teissieri (alpha = −0.011, P = 0.983), but for D. santomea the alpha value was high, although not significant (alpha = 0.76, P = 0.186), which suggests it may be informative to sample bam from additional D. santomea populations with larger sample sizes (n = 11 here) (fig. 5).
In the montium species subgroup, we used the predicted common ancestor of D. serrata, D. bunnanda, D. birchii, and D. jambulina to calculate divergence for bam. We found a significant signal of positive selection for bam in D. serrata (alpha = 0.799, P = 0.003), D. birchii (alpha = 0.675, P = 0.003), and D. jambulina (alpha = 0.618, P = 0.023) (fig. 5). However, we could not reject neutrality for D. bunnanda (alpha = −1.014, P = 0.542) (fig. 5).
In the ananassae species group, we used the predicted common ancestor of D. bipectinata, D. pseudoananassae, D. pandora, and D. ananassae to calculate divergence for bam. We reassessed lineage-specific divergence at bam for the D. ananassae population sample previously reported by Choi and Aquadro (2014) as they reported divergence between D. ananassae and Drosophila atripex. We did not reject neutrality at bam for D. bipectinata (alpha = 0.355, P = 0.357), D. pseudoananassae (alpha = −0.866, P = 0.613), or D. pandora (alpha = 0.666, P = 0.11) (fig. 5). However, we found a significant signal of positive selection for bam in D. ananassae (alpha = 1, P = 0.045) (fig. 5). Therefore, the signals of positive selection at bam previously reported for D. melanogaster and D. simulans are not unique to the melanogaster species group.
In the obscura species group, we calculated divergence for bam from population samples of D. pseudoobscura and D. affinis from their predicted common ancestor and observed similar patterns of polymorphism and divergence (D. pseudoobscura: alpha = 0.487, P = 0.448), (D. affinis: alpha = −1.064, P = 0.349), neither of which were close to rejecting neutral expectations (fig. 5). We used the predicted common ancestor of D. mojavensis and D. arizonae bam to calculate divergence in the D. mojavensis lineage and again did not observe a trend towards an excess of nonsynonymous divergence (alpha = 0.285, P = 0.771) (fig. 5). For the immigrans species group, we calculated divergence for bam from the predicted common ancestor of D. immigrans and D. rubida. We did not reject neutrality for bam in D. immigrans (alpha = 0.116, P = 0.882), but we did find a strong signal of positive selection for bam in D. rubida (alpha = 1, P < 0.000) (fig. 5). Therefore, the signals of positive selection at bam are heterogeneously distributed throughout the Drosophila genus.
Discussion
bam's Role in GSC Maintenance and Differentiation Shows Remarkable Variation in the D. melanogaster Species Group
Our comparative assessment of bam requirement in gametogenesis across diverse Drosophila species indicates that the functional evolution of bam is unexpectedly complex. We discovered that D. teissieri and D. ananassae do not require bam for gametogenesis in one or both sexes. A single orthologous copy of bam is found in all Drosophila species examined to date, but is absent in outgroups for example, house fly (Musca) and the sheep blow fly (Lucinia). These results raise the question as to whether bam’s critical role in GSC differentiation originally characterized in D. melanogaster is in fact a phylogenetically recent gain of function, or whether bam’s critical role is basal to the genus and that bam’s role has been recently (and possibly repeatedly) lost in specific lineages. For example, assuming bam’s role in GSC differentiation is basal, then the most parsimonious explanation of our functional data is that bam was required for oogenesis and spermatogenesis in the common ancestor of the species tested (fig. 1A), then bam function was lost in D. ananassae males, and then lost again independently in D. teissieri males and females. These alternative scenarios can be distinguished by future follow-up analyses of bam function in more divergent Drosophila species.
Evidence of Positive Selection for Protein Diversification at Bam is Strikingly Heterogeneous Across Divergent Drosophila Lineages
We analyzed additional population samples of species mainly within the melanogaster species group and a few more distant lineages for nucleotide sequence variation at bam. We found new evidence of lineage-specific signals of positive selection for accelerated protein sequence evolution at bam in three of the melanogaster subgroup species (D. melanogaster, D. simulans, and D. yakuba), three of the montium subgroup species (D. serrata, D. birchii, and D. jambulina), and one of the ananassae subgroup species (D. ananassae). Additionally, we found evidence of positive selection in the D. rubida lineage, indicating that the protein diversification at bam is repeatedly occurring in species spanning over 70 million years of divergence.
While we could not reject neutrality for bam for the remaining lineages, it is important to note as well that the MKT has limited power statistically when applied to single genes (Akashi 1999; Zhai et al. 2009) and thus a failure to reject selective neutrality does not prove that positive selection is not acting or has not acted on this gene.
Are Stem Cell Genes Hotspots for Adaptive Evolution?
The results of past studies of GSC evolution (Bauer DuMont et al. 2007; Choi and Aquadro 2014, 2015; Flores, DuMont, et al. 2015) and our population genetic and functional results in this study lead to the question of why GSC developmental functions are frequent targets of adaptation. It has been documented that selfish transposable elements are upregulated in the absence of piRNAs, implying that piRNAs act to protect the germline genome thereby leading to an evolutionary conflict that may drive positive selection at piRNA genes (Aravin et al. 2007; Simkin et al. 2013). There is natural variation in the gene Bruno that provides tolerance to deleterious P-element expression in the genome (Kelleher et al. 2018). In a scRNA-seq study of male D. melanogaster testes, Witt et al. (2019) report many fixed de novo genes expressed in GSCs and early spermatogonia (where bam is expressed) at similar levels to testis-specific genes. In contrast, segregating de novo genes show lower expression in GSCs and early spermatogonia implying that cell type expression may impact the likelihood for a de novo gene to become fixed. As de novo gene evolution is increasingly recognized as a critical mode of adaptation, especially in reproduction, GSCs/early spermatogonia may be generating necessary variation for adaptation (Witt et al. 2019). A recent finding that the insect gene oskar, which is critical for germ cell formation and oogenesis, arose de novo through a horizontal gene transfer event from a prokaryote, resulting in a fusion between prokaryotic and eukaryotic sequence illustrates yet another mechanism by which novel genes can take on key developmental roles in reproduction (Jenny et al. 2006; Blondel et al. 2020).
The bam sequence itself has many properties of novel genes: it does not contain any known DNA or RNA binding motifs, it binds with multiple protein partners, contains structurally disordered regions, and is expressed in a tight spatiotemporal manner (Chen et al. 2010; Jiang and Assis 2017; Wilson et al. 2017; Klasberg et al. 2018; Vakirlis and McLysaght 2019). Novel genes that become fixed often gain critical roles in conserved gene networks. Their new role may lead to selection to fine tune their critical role thereby providing additional functional flexibility for these conserved processes (Chen et al. 2010). Such a process could represent what has been called developmental systems drift, a model to explain how developmental systems remain phenotypically conserved while the genetic pathways that determine them diverge (True and Haag 2001).
It is possible that bam was a gene novel to the common ancestor of Drosophila (and possibly some other dipterans) and has functionally diversified across the Drosophila genus, only gaining its role in GSC differentiation in the lineage leading to the melanogaster species group. Additionally, it is possible that bam quickly evolved a role in GSC daughter differentiation in Drosophila, but that function has been shaped by germline conflicts in specific lineages. Conflict with germline agents (such as W. pipientis) may even drive the fixation of novel genes in new roles that successfully evade their control. As W. pipientis must recognize host cues during oogenesis to successfully infect the developing oocyte, it is possible that novel host genes that regulate differentiation might then be under selection for their new function if it evades unfavorable manipulation by W. pipientis.
It certainly seems plausible that genetic conflict is one way for new genes to evolve required functions in conserved processes such as GSC daughter differentiation. The D. melanogaster testes and the accessory gland show enriched expression of novel genes, many of which show signals of adaptive evolution, and some of which are predicted to be driven by sexual conflict (Levine et al. 2006; Zhou et al. 2008; Findlay et al. 2009; Kaessmann 2010; Gubala et al. 2017; Witt et al. 2019). While the developmental processes of gametogenesis and reproduction must be conserved, our results demonstrate that the individual genes that function in that process may vary among even closely related species.
Materials and Methods
Fly Stocks and Rearing
We raised fly stocks on standard cornmeal-molasses food at room temperature. We used yeast-glucose food for fertility assays. The D. melanogaster w1118 isogenic line and w1118 balancer lines were gifts from Luis Teixeira, and the D. simulans w501 line (stock 14021-0251.011), the D. yakuba white eyed line (14021-226.03) and the D. ananassae reference genome (14024-0371.14) lines are from the Drosophila species stock center (http://blogs.cornell.edu/drosophila/). The W. pipientis-free D. ananassae line Nou83 was a gift from Artyom Kopp. The D. teissieri line (teissieri syn) was a gift from Daniel Matute (Turissini et al. 2015).
We tested all lines for W. pipientis infection status by qPCR (NEB Luna Universal qPCR kit) using primer sets for wsp and arm (supplementary table S1, Supplementary Material online). Lines that tested positive (D. teissieri, D. ananassae, D. simulans) were treated with tetracycline (200 μg/mL) for three generations to remove W. pipientis (Glover et al. 1990). We allowed these lines to recover for at least three generations before using them in any analyses. We were unable to fully clear the D. ananassae reference line of W. pipientis with tetracycline, so to obtain a W. pipientis-free stock, we crossed males of our bam null lines to females from the Nou83 line, which we confirmed to be W. pipientis-free by qPCR.
Cloning for Bam Null Constructs
We used Flybase to obtain the nucleotide sequence information for designing constructs in D. melanogaster (Dmel 5.57), D. simulans (dsim r2.01), D. yakuba (dyak r1.04), and D. ananassae (dana r1.04) (Thurmond et al. 2019). We obtained sequence information for the D. teissieri sequence from Turissini and Matute 2017 (accession SAMN07407364-SAMN07407376) (Turissini and Matute 2017). We used Geneious for all cloning design.
We used the NEB Q5 High Fidelity 2× master mix to generate all PCR products. We gel extracted and purified PCR products using the NEB Monarch DNA gel extraction kit or Qiagen MinElute gel extraction kit. IDT primers were used for PCR, sequencing, and cloning. We generated the donor plasmids for the 3xP3-DsRed and 3xP3-YFP bam disruption lines using the NEB HiFi Assembly Cloning kit into the pHD-attP-DsRed vector from flyCRISPR (Gratz et al. 2014) as follows: we amplified 1.5 kb homology arms from genomic DNA of the appropriate species stock flanking the insertion site for 3xP3-DsRed or 3xP3-YFP. 3xP3-DsRed was amplified from the pHD-attP-DsRed plasmid and 3xP3-YFP was amplified from the D. simulans nos-Cas9 line, a gift from David Stern. We then gel extracted the two homology arms and the appropriate 3xP3 marker, purified them, and assembled them into the pHD vector backbone using the manufacturer’s protocol (supplementary table S3, Supplementary Material online).
We used the U6:3 plasmid from CRISPRflydesign (Port et al. 2014) to express gRNAs for generating the D. yakuba lines. The gRNA target sequence was generated by annealing primers with the gRNA sequence and BbsI site overhangs, and ligating into the BbsI ligated U6:3 vector (T4 ligase NEB, BbsI NEB) (supplementary table S3, Supplementary Material online). We used NEBalpha competent cells for transformations.
We prepped and purified all plasmids for embryo injections with the Qiagen plasmid plus midi-prep kit. We additionally purified plasmids for injections with phenol-chloroform extraction to remove residual RNases. We confirmed plasmid sequences with Sanger sequencing (Cornell BRC Genomics Facility).
CRISPR/Cas9
gRNA Selection
We chose gRNAs using the flyCRISPR target finder (Gratz et al. 2014) for D. melanogaster, D. simulans, D. yakuba, and D. ananassae. For D. teissieri, we used one completely conserved gRNA from D. yakuba. We also chose an additional gRNA from D. yakuba that had two divergent sites between D. yakuba and D. teissieri, thus we modified them to the D. teissieri sites. We selected gRNAs that had no predicted off-targets in the reference genome (supplementary tables S4 and S5, Supplementary Material online). For D. yakuba, we used the U6:3 gRNA plasmid from CRISPRfly design as described above. For the remaining species (D. melanogaster, D. simulans, D. teissieri, and D. ananassae), we used synthetic gRNAs (sgRNAs) from Synthego. We used 1–3 gRNAs per injection to increase the chances of a successful CRISPR event. We used multiple gRNAs if there were suitable options for that site to increase efficiency (multiple gRNAs within 50 bp with 0 predicted off-targets).
Injections
All CRISPR/Cas9 injections were carried out by Genetivision. Lines were injected with the plasmid donor, gRNAs (synthetic or plasmid), Cas9 protein (Synthego), and an siRNA for Lig4 (IDT DNA). The same siRNA was used for D. melanogaster, D. simulans, D. yakuba, and D. teissieri (pers communication with Daniel Barbash and David Stern supplementary table S6, Supplementary Material online). We created a D. ananassae-specific siRNA due to sequence divergence (supplementary table S6, Supplementary Material online).
For the bam disruption lines, we screened for eye color in F1s in-house using a Nightsea fluorescence system with YFP (cyan) and DsRed (green) filters. We backcrossed all CRISPR/Cas9 mutants to the stock line we used for injections for three generations. All mutants are maintained as heterozygous stocks. We confirmed all CRISPR insertions by Sanger sequencing (Cornell BRC Genomics Core Facility).
Genotypes for Generating bam Disruption Homozygotes
We generated two bam disruption lines in each species in order to create the genotypes for the comparative bam null analyses. We generated one by disrupting bam with 3xP3-DsRed and the other with 3xP3-YFP. We selected DsRed or YFP positive flies and crossed them to generate homozygous bam disruption flies (bam3xp3-DsRed/bam3xP3-YFP). This cross also results in DsRed or YFP-positive heterozygous bam null progeny as well as bam wildtype progeny without any fluorescent eye marker.
We targeted the first exon of bam in all species in order to disrupt the bam coding sequence and introduce an early stop codon. While we were successful in generating first exon disruptions in each species, in D. simulans we were only able to obtain a 3xP3-YFP first exon disruption line. However, we did find that a D. simulans line with a 3xP3-DsRed insertion in its second intron resulted in homozygous sterile females and males with tumorous ovaries and testes, while heterozygous females and males were fertile with wildtype ovary and testes phenotypes. As this edit was specific to the bam locus, we presumed it was an mRNA null allele and we used this line for our analyses. Thus, if the phenotype of the 3xP3-DsRed second intron allele over the 3xP3-YFP first exon allele is the same as the homozygous 3xP3-DsRed second intron allele, this is consistent with classification as a genetic null.
Fertility Assays
Female Fertility
All female fertility assays were performed following the approach of Flores et al. (2015a). Specifically, we collected virgin females and aged them based on their time to sexual maturity 2–3 days (D. melanogaster, D. simulans, D. yakuba, and D. teissieri) or 5–7 days (D. ananassae) (Markow and O’Grady 2006). We collected all genotypes from each bottle to control for bottle effects. We collected virgin males of the wildtype genotype for each species and aged them for the same timeframe as the females of that species. We evenly distributed males collected from different bottles across the female genotypes to control for any bottle effects. We crossed single females collected within 48 h of each other to two virgin males. We allowed the trio to mate for 9 days, and then flipped them to new vials. Except for D. ananassae and D. yakuba, as fertility was very low, we cleared the trio after day 9 and did not continue further. For all other species crosses, after an additional 9 days, we cleared the trio. Then, we counted the progeny for each trio every 2–3 days to get the total adult progeny per female.
Male Fertility
All male fertility assays were performed as described for the female fertility assays except that one tester male was crossed to two virgin females.
Food and Conditions for Fertility Assays
For D. melanogaster, D. simulans, D. ananassae, and D. teissieri, we performed all fertility assays on yeast-glucose food. For D. yakuba, we performed fertility assays on cornmeal-molasses food, as this line did not thrive on the yeast-glucose food. We kept all crosses and fertility experiments in an incubator at 25°C with a 12-h light–dark cycle.
Statistics
For the fertility assays we used estimation statistics to assess the mean difference (effect size) of adult progeny between the wildtype bam genotype and each individual additional bam genotype. For generating the estimation statistics and plots (Ho et al. 2019), we used the dabest package in Python (v. 0.3.1) with 5,000 bootstrap resamples. Estimation statistics provide a non-parametric alternative to other statistical tests of the difference of the mean (for example, ANOVA), and allow us to understand the magnitude of the effect of bam genotype on the fertility phenotype. In text, we report significance as a mean difference (effect size) outside the 95% bootstrap confidence interval. While one benefit of these statistics is that they are less biased than reporting traditional P-values, we also report P-values for approximate permutation tests with 5,000 permutations.
Immunostaining
We used the following primary antibodies, anti-vasa antibody from Santa Cruz biologicals (anti-rabbit 1:200), anti-Hts-1B1 from Developmental Studies Hybridoma bank (anti-mouse 1:4 for serum, 1:40 for concentrate item), and anti-Fas3 from DSHB (anti-mouse 1:50). We used the following secondary antibodies: Alexaflour 488, 568 (Invitrogen) at 1:500.
We performed immunostaining as described in Aruna et al. (2009) and Flores, Bubnell et al. (2015). Briefly, we dissected ovaries and testes in ice-cold 1× PBS and pipetted ovaries up and down to improve antibody permeability. We fixed tissues in 4% paraformaldehyde, washed with PBST (1X PBS, 0.1% Triton-X 100), blocked in PBTA (1X PBS, 0.1% Triton-X 100, 3% BSA) (Alfa Aesar), and then incubated in the appropriate primary antibody in PBTA overnight. We then washed (PBST), blocked (PBTA), and incubated the tissue in the appropriate secondary antibody for 2 h, then washed (PBST) and mounted in mounting media with DAPI (Vecta shield or Prolong Glass with NucBlue) for imaging.
Confocal Microscopy
We imaged ovaries and testes on a Zeiss i880 confocal microscope with 405, 488, and 568 nm laser lines at 40X (Plan-Apochromat 1.4 NA, oil) (Cornell BRC Imaging Core Facility). We analyzed and edited images using Fiji (ImageJ).
RNA Extraction
For RT-qPCR experiments we dissected ovaries from 10 one-day old females per biological replicate or testes from 15 one-day old males in ice-cold 1× PBS. The tissue was immediately placed in NEB DNA/RNA stabilization buffer, homogenized with a bead homogenizer, and then stored at −80°C until RNA extraction. We used the NEB Total RNA mini-prep kit for all RNA extractions according to the manufacturer’s protocol and included the on column DNAse treatment. We assessed RNA concentration and quality with a Qubit 4 fluorometer and a NanoDrop.
RT-qPCR
For all RT-qPCR analyses, we used the Luna One Step RT-qPCR kit from NEB according to the manufacturer’s instructions at 10 μl total volume in 384 well plates. We used a Viia7 qPCR machine with the appropriate cycle settings as described in the Luna One Step RT-qPCR kit.
For the analysis of bam expression in D. ananassae, we used primers to D. ananassae bam with D. ananassae Rp49 as a control (supplementary table S1, Supplementary Material online). To assess the possibility that the bam disruption allele induced exon skipping of the disrupted exon 1 in D. teissieri, we designed a primer pair that spanned exon 1–exon 2 and another pair that spanned exon 2–exon 3 (supplementary table S1, Supplementary Material online). We used the exon 2–exon 3 target as the control. For both the D. teissieri and D. ananassae assays described above, we included a standard curve for each target on every plate at a 1:2 dilution and then used the standard curve method to calculate the relative quantity of bam (to either Rp49 for D. ananassae or bam exon 2–exon 3 for D. teissieri) with the QuantStudio software using the wildtype bam genotype as a control for each species.
For the allele specific analysis in D. teissieri, we designed one primer pair specific to the bam 3xP3-DsRed and 3xP3-YFP insertion alleles and another primer pair specific to the wildtype allele. We then used these two targets to measure the relative expression of the insertion allele to the wildtype allele in a single heterozygous sample. The primer pair for the bam insertion alleles target the sequence between the SV40-polyA tail of the 3xP3-DsRed and 3xP3-YFP insertion and the bam sequence downstream of the insertion (supplementary table S1, Supplementary Material online). The primer pair for the bam wildtype allele target bam upstream and downstream of the 3xP3-DsRed and 3xP3-YFP insertion, and therefore will not amplify the large insertion allele under RT-qPCR conditions (supplementary table S1, Supplementary Material online). We included a standard curve for each target on every plate at a 1:2 dilution and then used the standard curve method in the QuantStudio software to calculate the Ct and Ct standard error for each target and each sample. We report the relative quantity of the 3xP3-DsRed or 3xP3-YFP allele using the wildtype allele in the same sample as a control by calculating 2-(nullCt-wildtypeCt).
Genomic Analysis of bam Copy Number in D. teissieri
While bam is reported as a 1:1 syntenic ortholog on FlyBase for D. melanogaster, D. simulans, D. yakuba, and D. ananassae, data for D. teissieri is not available. To ask if bam has been recently duplicated in D. teissieri, we generated a blast database (blastn v 2.9.0) from a new de novo assembled D. teissieri reference genome (accession GCA_016746235.1) and blasted D. teissieri bam from the resequencing described above.
Additionally, to assess read coverage for bam compared to the surrounding loci and the entire chromosome 3R, we used publicly available short read WGS data for D. teissieri (SRR13202235) and used bwa-mem (v 0.7.17) to map reads to a new de novo assembled D. yakuba reference genome (accession GCA_016746365.1). We visualized the alignment and assessed read coverage using Geneious.
Population Genetic Analyses
The D. yakuba, D. santomea, and D. teissieri population sample lines were gifted by Brandon Cooper and Daniel Matute (Cooper et al. 2017). The D. yakuba and D. teissieri lines were collected in Bioko in 2013, the D. santomea sample was collected in Sao Tome in 2015. The D. affinis population sample lines were gifted by Robert Unckless (supplementary table S2, Supplementary Material online). The D. mojavensis population sample is from Organ Pipe National Monument, Arizona and DNA from this sample was gifted by Erin Kelleher and Luciano Matzkin. The D. pseudoobscura population sample is from Apple Hill, California, as described in (Schaeffer and Miller 1991). We used previously prepared DNA from the D. pseudoobscura population sample as described in (Schaeffer and Miller 1991).
We prepared genomic DNA from D. affinis, D. yakuba, D. santomea, and D. teissieri samples using the Qiagen PureGene kit. We used Primer3 to generate primers to amplify bam from D. affinis, D. mojavensis, D. pseudoobscura, D. yakuba, D. santomea, and D. teissieri population samples (supplementary table S7, Supplementary Material online). We used existing polymorphism data (Daniel Matute, personal communication) to generate primers for D. yakuba, D. santomea, and D. teissieri. We resequenced bam using Sanger sequencing from 5′UTR–3′UTR for D. yakuba, D. santomea, and D. teissieri and bam exons and introns for D. affinis (Cornell BRC Genomics Facility). We used Geneious for aligning the sequences and calling polymorphisms. We generated fasta files for each individual. We used DnaSP to phase individuals with heterozygous sites, and then randomly picked one allele for further analyses (Bauer DuMont et al. 2007). We will deposit all resequencing data described here in Genbank.
We used polymorphism data for D. melanogaster from the Zambia population sample from the Drosophila genome Nexus (Lack et al. 2015), as this is the largest population sample with intact bam sequence and represents a population in the D. melanogaster ancestral range. For the D. melanogaster Zambia population sample, we removed a single individual that contained missing nucleotide calls in the bam coding sequence (ZI117). As we filtered polymorphisms at <12% frequency, omitting this single individual would not affect our downstream analyses.
Polymorphism for D. simulans, D. serrata, D. bunnanda, D. birchii, D. jambulina, D. bipectinata, D. pseudoananassae, D. pandora, D. immigrans, and D. rubida as well as the single bam sequence for Z. bogoriensis that we used as an outgroup are from Li et al 2021 (accession PRJNA736147) (Li et al. 2021).
D. ananassae bam polymorphism, and single D. atripex bam sequences are from Choi et al. (Choi and Aquadro 2014). We obtained single bam sequences for species used for outgroups from FlyBase (D. erecta), and Genbank (D. eugracilis, D. arizonae, D. navojoa, D. miranda, D. subobscura).
We performed all multiple sequence alignments for the bam coding sequences using PRANK (v.170427) with the -codon and -F parameters and using the PRANK generated guide tree. As bam is highly divergent across the Drosophila genus, we generated multiple sequence alignments only among closely related clusters of species to ensure alignment quality. We generated a multiple sequence alignment of the melanogaster subgroup species (D. melanogaster, D. simulans, D. yakuba, D. santomea, D. teissieri, D. erecta, D. eugracilis) and montium subgroup species (D. serrata, D. bunnanda, D. birchii, D. jambulina), a multiple sequence alignment of D. ananassae, D. bipectinata, D. pseudoananassae, D. pandora, and D. atripex, a multiple sequence alignment of D. mojavensis, D. arizonae, and D. navajo, a multiple sequence alignment of D. affinis, D. pseudoobscura, D. miranda, and D. subobscura, and a multiple sequence alignment of D.immigrans, D. rubida, and Z. bogoriensis.
We used only unique sequences for input to codeml. We used the codeml package from PAML (v. 4.9) (Yang 1997, 2007) to generate the predicted common ancestor sequences to calculate lineage-specific divergence for bam with the MKT. We used the PRANK alignments and their corresponding gene trees as input to codeml with the following control file parameters (noisy = 9, verbose = 2, runmode = 0, seqtype = 1, CodonFreq = 2, clock = 0, aaDist = 0, model = 0, NSsites = 0, icode = 0, getSE = 0, RateAncestor = 1, Small_Diff = .5e-6, cleandata = 0, method = 1).
We used the http://mkt.uab.cat/mkt/mkt.asp webtool to perform the standard MKT comparing nonsynonymous vs. synonymous changes (Egea et al. 2008). We excluded polymorphic sites at <12% frequency, as these are likely slightly deleterious alleles that have not yet been purged by purifying selection (Charlesworth and Eyre-Walker 2008). For each species where we had polymorphism data, we performed the MKT using the specified predicted common ancestral sequence to calculate lineage-specific divergence. We report the values of the contingency table, the P-value of the χ2 test, as well as alpha, the proportion of fixations predicted to be due to positive selection (Eyre-Walker 2006).
Summary of Accession Numbers for Genomes Used in this Analysis
For read coverage analysis of bam: D. teissieri: SRR13202235, GCA_016746235.1. D. yakuba: GCA_016746365.1. For cloning D. teissieri bam null constructs: SAMN07407364-SAMN07407376. For bam polymorphism from D. simulans, D. serrata, D. bunnanda, D. birchii, D. jambulina, D. bipectinata, D. pseudoananassae, D. pandora, D. immigrans, and D. rubida: PRJNA736147.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Acknowledgments
We thank Santiago Herrera Alvarez for resequencing bam from Drosophila affinis and Jae Young Choi for resequencing bam from Drosophila pseudoobscura and Drosophila mojavensis. We thank Daniel Matute, Brandon Cooper, Robert Unckless, Erin Kelleher, and Luciano Matzkin for the population samples they provided for this study. We thank Fang Li for working to make the variant call files from Li et al. (2021) quickly available for this study. Additionally, we thank David L. Stern for his advice and guidance in designing the bam null alleles and for an extremely thoughtful and critical review of an earlier draft of the manuscript. We are grateful to Mariana F. Wolfner, Andrew G. Clark, Miwa Wenzel and Catherine Kagemann for valuable input and discussion of these experiments and results and helpful comments on this manuscript. Finally, we thank the three anonymous reviewers for their thoughtful and insightful feedback for improving this manuscript.
Data Availability
Sequence data generated in this study will be deposited in GenBank prior to publication. The remaining data underlying this article are available in the article and in its online supplementary material. Fly lines, CRISPR/Cas9 reagents, and plasmids used in this study are available upon request.
Funding
This work was supported by the National Institutes of Health grant R01-GM095793 to C.F.A. and by the National Science Foundation Graduate Research Fellowship Program DGE-114415 to J.E.B.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}