





Abstract
The evolution of a placenta is predicted to be accompanied by rapid evolution of genes involved in processes that regulate mother–offspring interactions during pregnancy, such as placenta formation, embryonic development, and nutrient transfer to offspring. However, these predictions have only been tested in mammalian species, where only a single instance of placenta evolution has occurred. In this light, the genus Poeciliopsis is a particularly interesting model for placenta evolution, because in this genus a placenta has evolved independently from the mammalian placenta. Here, we present and compare genome assemblies of two species of the livebearing fish genus Poeciliopsis (family Poeciliidae) that differ in their reproductive strategy: Poeciliopsis retropinna which has a well-developed complex placenta and P. turrubarensis which lacks a placenta. We applied different assembly strategies for each species: PacBio sequencing for P. retropinna (622-Mb assembly, scaffold N50 of 21.6 Mb) and 10× Genomics Chromium technology for P. turrubarensis (597-Mb assembly, scaffold N50 of 4.2 Mb). Using the high contiguity of these genome assemblies and near-completeness of gene annotations to our advantage, we searched for gene duplications and performed a genome-wide scan for genes evolving under positive selection. We find rapid evolution in major parts of several molecular pathways involved in parent–offspring interaction in P. retropinna, both in the form of gene duplications as well as positive selection. We conclude that the evolution of the placenta in the genus Poeciliopsis is accompanied by rapid evolution of genes involved in similar genomic pathways as found in mammals.
Introduction
The origin of biological innovations is one of the most tantalizing questions in evolution—How does something seemingly complex emerge? All biological innovations ultimately find their origin in the genome. The evolution of genes, and the selective pressures on them to modify traits, or even generate novelties, can be studied by comparative analysis. With an increase in the number of sequenced genomes, and improvements in cost and quality, the power of comparative genomic methods is similarly increasing, bringing this approach within reach to study well-established, classic, evolutionary models of biological innovation.
The fish family Poeciliidae (sensu Parenti 1981) constitutes such an evolutionary model. The Poeciliidae form a large family of livebearing fish, consisting of ∼275 species (Van Der Laan et al. 2014). They are widely distributed across the American continents, living in South-, Middle- and North America, as well as in the Caribbean (Reis et al. 2003). Species within this family are models for a variety of research areas, including cancer research (Meierjohann and Schartl 2006), invasion biology (Hoffberg et al. 2018), life history evolution (Reznick et al. 1996), phenotypic plasticity (Trexler et al. 1990), sexual selection (Basolo 1990), and placenta evolution (Pollux et al. 2009). Because of their use in various research areas, the genomes of several poeciliid species have been sequenced and assembled, including members of four major genera Poecilia, Xiphophorus, Gambusia, and Poeciliopsis (Schartl et al. 2013; Künstner et al. 2016; Shen et al. 2016; Hoffberg et al. 2018; Warren et al. 2018; Mateos et al. 2019).
Within the Poeciliidae, the genus Poeciliopsis plays a particularly important role as a model for the evolution of the placenta. Within this genus, the placenta evolved independently three times (Reznick et al. 2002). As a consequence, this genus contains closely related species that either have or lack a placenta (referred to as placentotrophic and lecithotrophic species, respectively). Additionally, the species that have placentas show a more or less continuous variation in the complexity of the placenta (Turner 1940; Kwan et al. 2015). This remarkable variation within a single genus allows for a comparison between closely related species that differ in the degree of placentation.
The implications of creating an interface between mother and offspring—that is, a placenta—have been the subject of a number of theories. For instance, it has been predicted that the evolution of the placenta should result in parent–offspring conflict: Although it is in the mother’s interest to balance the reproductive investments among her offspring, it is in each of the individual offspring’s interest to claim more for itself than would be in the interest of the mother to give (Trivers 1974; Zeh and Zeh 2000; Crespi and Semeniuk 2004). The placenta is the site where this conflict is most apparent, because of the intimate contact between mother and offspring (Zeh and Zeh 2000). On a genomic level, parent–offspring conflict has been predicted to manifest in rapid antagonistic coevolution of genes involved in placenta formation, embryonic development, and nutrient transfer to offspring (Haig 1993; Zeh and Zeh 2000). In mammals, several studies support this hypothesis. For example, placental Cadherin (CDH) genes evolve under positive selection in humans (Summers and Crespi 2005). It is hypothesized that this rapid change is driven by antagonistic coevolution between CDH genes and genes that modify their binding or expression, due to the influence of CDH genes on nutrient transfer from mother to offspring. In addition to positive selection, it has been shown that this rapid evolution of placental genes can also manifest in the form of gene duplications (Knox and Baker 2008). Examples of these duplications are for instance the duplication of BMP8 in mice (Zhao and Hogan 1996) and the expansion of the pregnancy-associated glycoprotein gene family in artiodactyl species (Hughes et al. 2000). Within the Poeciliidae, evidence for this rapid genomic change as a consequence of placenta evolution is scarce (but see O’Neill et al. 2007).
Another predicted consequence of placenta evolution is a shift from precopulatory sexual selection to postcopulatory or even postzygotic mechanisms of selection (Zeh and Zeh 2000). Indeed, it has been shown that the evolution of the placenta in the Poeciliidae correlates with phenotypic and behavioral male traits that are associated with a reduced reliance on precopulatory female mate choice (e.g., an absence of, or less intense, body coloration, courtship behavior, and ornamental display traits) (Pollux et al. 2014). The placenta is further associated with smaller male bodies and longer genitalia, traits that facilitate sneak mating, and aid in circumventing precopulatory female mate choice (Pollux et al. 2014). If placental species indeed rely more on postcopulatory or postzygotic means of selection, then this should be reflected in the selective pressures on genes involved in these processes. An example of a postcopulatory process is sperm competition, which in mammals is known to drive the rapid evolution of sperm proteins (Torgerson et al. 2002). However, it is currently unknown whether increased postcopulatory selection also drives rapid evolution of sperm proteins in placental species from the Poeciliidae.
Placental evolution is thus predicted to be accompanied by rapid evolution of genes involved in placenta formation, embryonic development, and nutrient transfer to offspring, as well as sperm proteins. However, whether placentation universally results in similar selection on the same pathways is unknown—The placenta in mammals evolved only once, making it very difficult to disentangle the genomic innovations and drivers behind this evolutionary novelty. To test whether similar drivers operate in the evolution of the placenta in fish, we sequenced and assembled the genomes of two species from the genus Poeciliopsis (family Poeciliidae): P. retropinna and P. turrubarensis. Although these two species are from the same genus, P. retropinna has a complex placenta, whereas P. turrubarensis completely lacks a placenta (Reznick et al. 2002). Comparing these genomes allows us to identify differences between these two closely related species that may stem from selection on genes associated with placentation. We employ two novel sequencing techniques to sequence these genomes: PacBio long read sequencing for P. retropinna (Rhoads and Au 2015), and 10× Genomics linked read sequencing, generating “pseudo long reads,” for P. turrubarensis (Weisenfeld et al. 2017). Both of these techniques allow for very high contiguity assemblies, either by generating very long reads (PacBio) or by using barcodes to mark sequencing reads originating from the same DNA molecule (10× Genomics).
As more high-quality genomes become available, it is becoming increasingly clear that some evolutionary novelties are based on structural variations, such as gene duplications. The high contiguity expected from our study should enable us to investigate such gene duplications and assess their potential role in generating evolutionary innovations such as the placenta. Additionally, genome annotations generated in this study allow us to perform a genome-wide scan for genes evolving under positive selection in these two genomes. Using these methods, we can test the hypothesis that the evolution of the placenta in the genus Poeciliopsis is accompanied by rapid evolution of genes involved in placenta formation, embryonic development, nutrient transfer to offspring, and sperm proteins.
Results
Genome Assembly
We sequenced and assembled the genomes of P. retropinna and P. turrubarensis, using two different sequencing techniques. The genome of P. retropinna was sequenced and assembled using ∼80× coverage PacBio sequencing, supplemented with ∼40× coverage Illumina 150-bp paired-end data for assembly polishing. The genome of P. turrubarensis was sequenced and assembled using 10× Genomics linked reads, sequenced on an Illumina platform. Both assemblies show good quality metrics. Total assembled bases are congruent with estimated genome size based on k-mer analysis, with the P. turrubarensis genome estimated to be slightly smaller than the P. retropinna genome (table 1). Both genomes show good contiguity, although the P. retropinna assembly is more contiguous than the P. turrubarensis assembly, having a scaffold N50 of 21.6 Mb, with half of the assembly contained within just 13 scaffolds. A Blast search for telomeric repeat sequences showed that for three scaffolds, telomeres could be found on both sides, indicating that these scaffolds were completely assembled chromosomes (fig. 2B and supplementary fig. 1, Supplementary Material online). In terms of expected genes, both genome assemblies are very complete: 97.5% and 95.5% of universal single-copy orthologs are present and full-length in the assembly of P. retropinna and P. turrubarensis, respectively (table 1). The genomes of P. retropinna and P. turrubarensis have similar repeat contents of 20.8% and 18.5%, respectively. This is comparable to other sequenced poeciliid species (Künstner et al. 2016; Hoffberg et al. 2018).
Assembly Statistics for the Poeciliopsis retropinna and Poeciliopsis turrubarensis Assemblies.
| P. retropinna | P. turrubarensis | |
|---|---|---|
| Assembly size (Mb) | 621.8 | 597.0 |
| Predicted genome size (k-mer analysis) (Mb) | 621 | 586 |
| Scaffold N50 (Mb) | 21.6 | 4.2 |
| Scaffold L50 | 13 | 35 |
| Scaffold N90 (Mb) | 6.0 | 0.34 |
| Scaffold L90 | 31 | 195 |
| Largest scaffold (Mb) | 30.7 | 17.0 |
| Total number of scaffolds | 78 | 5,398 |
| GC content (%) | 38.8 | 39.5 |
| Repeat content (%) | 20.8 | 18.5 |
| Heterozygosity (%) | 0.22 | 0.34 |
| BUSCO score (%) | 97.5 | 95.5 |
| Number of predicted genes | 25,375 | 24,077 |
| P. retropinna | P. turrubarensis | |
|---|---|---|
| Assembly size (Mb) | 621.8 | 597.0 |
| Predicted genome size (k-mer analysis) (Mb) | 621 | 586 |
| Scaffold N50 (Mb) | 21.6 | 4.2 |
| Scaffold L50 | 13 | 35 |
| Scaffold N90 (Mb) | 6.0 | 0.34 |
| Scaffold L90 | 31 | 195 |
| Largest scaffold (Mb) | 30.7 | 17.0 |
| Total number of scaffolds | 78 | 5,398 |
| GC content (%) | 38.8 | 39.5 |
| Repeat content (%) | 20.8 | 18.5 |
| Heterozygosity (%) | 0.22 | 0.34 |
| BUSCO score (%) | 97.5 | 95.5 |
| Number of predicted genes | 25,375 | 24,077 |
Note.—BUSCO score represents the percentage of complete and full-length gene models from a collection of conserved vertebrate genes.
Assembly Statistics for the Poeciliopsis retropinna and Poeciliopsis turrubarensis Assemblies.
| P. retropinna | P. turrubarensis | |
|---|---|---|
| Assembly size (Mb) | 621.8 | 597.0 |
| Predicted genome size (k-mer analysis) (Mb) | 621 | 586 |
| Scaffold N50 (Mb) | 21.6 | 4.2 |
| Scaffold L50 | 13 | 35 |
| Scaffold N90 (Mb) | 6.0 | 0.34 |
| Scaffold L90 | 31 | 195 |
| Largest scaffold (Mb) | 30.7 | 17.0 |
| Total number of scaffolds | 78 | 5,398 |
| GC content (%) | 38.8 | 39.5 |
| Repeat content (%) | 20.8 | 18.5 |
| Heterozygosity (%) | 0.22 | 0.34 |
| BUSCO score (%) | 97.5 | 95.5 |
| Number of predicted genes | 25,375 | 24,077 |
| P. retropinna | P. turrubarensis | |
|---|---|---|
| Assembly size (Mb) | 621.8 | 597.0 |
| Predicted genome size (k-mer analysis) (Mb) | 621 | 586 |
| Scaffold N50 (Mb) | 21.6 | 4.2 |
| Scaffold L50 | 13 | 35 |
| Scaffold N90 (Mb) | 6.0 | 0.34 |
| Scaffold L90 | 31 | 195 |
| Largest scaffold (Mb) | 30.7 | 17.0 |
| Total number of scaffolds | 78 | 5,398 |
| GC content (%) | 38.8 | 39.5 |
| Repeat content (%) | 20.8 | 18.5 |
| Heterozygosity (%) | 0.22 | 0.34 |
| BUSCO score (%) | 97.5 | 95.5 |
| Number of predicted genes | 25,375 | 24,077 |
Note.—BUSCO score represents the percentage of complete and full-length gene models from a collection of conserved vertebrate genes.
Synteny between the two assemblies is highly conserved, and large collinear blocks can be observed when aligning the assemblies (supplementary table 1, Supplementary Material online). When aligning the five longest scaffolds from P. turrubarensis to the P. retropinna assembly, all but one of these scaffolds fall within a single P. retropinna scaffold, without signs of chromosomal rearrangements (fig. 1A). The five biggest P. retropinna scaffolds align to a number of P. turrubarensis scaffolds each, which is a logical consequence of the difference in contiguity between the two assemblies. Again, almost no large-scale rearrangements are evident (fig. 1B). By contrast, when aligning the P. retropinna assembly to the published Poecilia reticulata genome assembly, many within-chromosome inversions and rearrangements can be observed. However, on the interchromosomal level, synteny is still highly conserved (fig. 1C).

(A) Dotplot of the five biggest scaffolds of the Poeciliopsis turrubarensis assembly, compared with the P. retropinna assembly. (B) Dotplot of the five biggest scaffolds of the P. retropinna assembly, compared with the P. turrubarensis assembly. (C) Dotplot of the five biggest scaffolds of the P. retropinna assembly, compared with the published Poecilia reticulata assembly. Only alignments longer than 1 kb from scaffolds longer than 1 Mb were plotted. Color codes: blue, forward alignment; red, reverse alignment.
Genome Annotation
The genome assemblies of P. retropinna and P. turrubarensis were annotated using the BRAKER2 pipeline (Hoff et al. 2019), with subsequent filtering steps to filter out predicted genes that were likely false positives (see Materials and Methods). In total, 25,375 protein-coding genes were predicted for P. retropinna, and 24,077 for P. turrubarensis. These numbers are slightly higher compared with genome assemblies from the related Poecilia reticulata and Xiphophorus maculatus (Schartl et al. 2013; Künstner et al. 2016), but similar to Poecilia formosa and other members from the subgenus Mollinesia (Warren et al. 2018). The annotations of the P. retropinna and P. turrubarensis genomes contained 94.5% and 95.4% of all universal single-copy orthologs, respectively, as determined by running BUSCO on the predicted transcriptome.
We characterized repeat content and composition using the RepeatModeler and RepeatMasker programs (Smit and Hubley 2008; Smit et al. 2013). Furthermore, satellite DNA was identified using Tandem Repeat Finder (Benson 1999). Repeat content is similar for both genome assemblies (20.8% for P. retropinna and 18.5% for P. turrubarensis). DNA elements are the main repeat class for both genomes (supplementary table 2, Supplementary Material online). When looking at the repeat content throughout the largest scaffolds, some repetitive “hotspots” can be observed near the edges of these scaffolds (fig. 2C and supplementary fig. 1, Supplementary Material online). Differences in occurrence are even more pronounced when looking only at satellite DNA (fig. 2D and supplementary fig. 1, Supplementary Material online). Regions with high density of satellite DNA likely correspond to centromeric regions, as centromeric DNA is usually satellite rich (Lee et al. 1997). Additionally, the location of these satellite “hotspots” corresponds well to the most likely location of the centromeres, as chromosomes of Poeciliidae species are acrocentric (Cimino 1973; Haaf and Schmid 1984).

Overview of scaffold 2 of the P. retropinna assembly, a presumed completely assembled chromosome. Statistics are displayed in bins (bars) and moving average (line). (A) Gene density as percent of bins covered by genic region, 200-kb bins. (B) Blast hits for telomeric repeats, 200-kb bins. (C) Repeat content as identified by RepeatMasker, 100-kb bins. (D) Satellite content as identified by Tandem Repeat Finder, 100-kb bins. (E) Pairwise nucleotide divergence between P. retropinna and aligned P. turrubarensis assembly, 100-kb bins. (F) Dotplot between P. retropinna scaffold 2 and aligned scaffolds of the P. turrubarensis assembly.
Whole-Genome Alignment and Gene Duplications
We aligned the genome assemblies of P. retropinna and P. turrubarensis to each other and to the published genome of the guppy, Poecilia reticulata, (Künstner et al. 2016) using the reference-free whole-genome aligner ProgressiveCactus (Paten et al. 2011). The resulting alignments were postprocessed into pairwise synteny blocks (see Materials and Methods for details). Pairwise average nucleotide divergence between the P. retropinna and P. turrubarensis assemblies was computed, and likewise between these two assemblies and the Poecilia reticulata assembly (fig. 2 and supplementary fig. 2, Supplementary Material online). Nucleotide divergence between pairs of species was higher in regions with high repeat density. On average, nucleotide divergence between Poecilia reticulata and P. turrubarensis was higher than nucleotide divergence between Poecilia reticulata and P. retropinna (supplementary fig. 2, Supplementary Material online), although both species pairs have the same most recent common ancestor (Pollux et al. 2014).
Gene duplications are an important driving force in adaptive evolution. Duplicated segments from the whole-genome alignment were extracted and checked for duplicated genes. Using this method, we identified 151 duplicated segments in the P. retropinna genome, and 102 duplicated segments on the P. turrubarensis genome. On these segments, 29 duplicated genes were identified in P. retropinna, and 4 duplicated genes in P. turrubarensis (supplementary table 3, Supplementary Material online).
Within the duplicated genes in P. retropinna, we found a number of genes that code for sperm-associated proteins. Three duplicated proteins (spag6, cfap58, and cep162) are a part of sperm flagella or cilia in mammals (Sapiro et al. 2002; Wang et al. 2013; Nixon et al. 2018), and their expression (in humans) is either restricted to testis (spag6 and cfap58) or biased toward testis (cep162) (Uhlén et al. 2015). One more gene that is duplicated in P. retropinna (gcnt1) is involved in sialyl Lewis X antigen presentation, which is the antigen responsible for sperm-to-egg cell recognition (Pang et al. 2011).
Another interesting result is the expansion of the Vitellogenin (vtg) gene family in P. retropinna. Vitellogenin is an egg yolk precursor, but in placental teleosts may be involved in postfertilization nutrient provisioning (Vega-López et al. 2007). Other poeciliid fish have three vtg genes: two similarly sized vtg genes that are placed in tandem (vtg1 and vtg2) and a smaller vtg on a different location on the same chromosome (vtg3). However, in P. retropinna, vtg1 is duplicated, leading to a cluster of three vtg genes placed in tandem, along with the additional vtg3 gene (fig. 3).

(A) Phylogeny of the vtg gene family in five poeciliid fish species. (B) Conserved synteny of the vtg gene cluster in five poeciliid species.
Positive Selection
We performed likelihood tests for positive selection in phylogenetic branches leading to P. retropinna and P. turrubarensis using the codeml program from the PAML package (Yang 2007). Briefly, the hypothesis was tested that a coding sequence shows an elevated ratio of nonsynonymous to synonymous mutations at certain codon sites, in one of the tested branches, implemented in PAML as the branch-site model. For this, we used 1:1 orthologous genes found in four poeciliid genomes, of which the coding sequence was aligned using a codon-aware aligner (see Materials and Methods). We found 180 genes evolving under positive selection in P. retropinna, and 165 genes evolving under positive selection in P. turrubarensis, using a 5% false discovery rate (supplementary table 4, Supplementary Material online). Gene Ontology (GO) term enrichment analysis showed that the genes evolving under positive selection in P. retropinna were significantly enriched for genes involved in “neural system development” and “protein targeting to peroxisome” (supplementary table 5, Supplementary Material online). We found that five out of seven genes with GO term “protein targeting to peroxisome” in this gene set are involved in peroxisomal lipid metabolism, whereas the other two genes are involved in regulating the transport of enzymes to the peroxisomes.
Shown in figure 4 is the probability of positive selection for each amino acid of one of these peroxisomal enzymes, peroxisomal multifunctional enzyme type 2 (hsd17b4). This enzyme is involved in peroxisomal beta oxidation of fatty acids. Positive selection is most apparent in three subsequent residues inside the N-terminal MaoC-like dehydratase domain. This domain contains no catalytic sites but instead determines substrate specificity (Koski et al. 2004). Therefore, positive selection within this domain may change the substrates on which the enzyme acts, without impairing catalytic function.
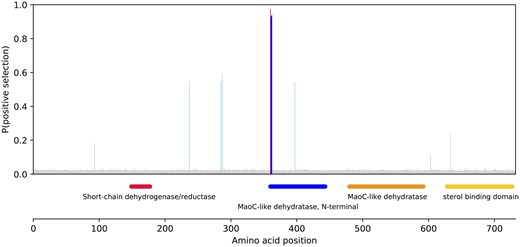
Likelihood of positive selection for each codon in P. retropinna peroxisomal multifunctional enzyme 2. Conserved domains are plotted on the bottom panel. Color codes for probability of positive selection: red > 95% > blue > 90% > light blue > 50% > gray.
Additionally, we found several genes coding for parts of the Wnt signaling pathway to be positively selected in P. retropinna (wnt6, wnt7b, cxxc4, rspo2, and ctnnd1), suggesting changes in this pathway as well. The Wnt signaling pathway is an essential signaling cascade in embryonic development throughout the vertebrate lineage (Clevers 2006) and a regulator of placenta formation in mammals (Sonderegger et al. 2010).
For P. turrubarensis, significant enrichment among positively selected genes was found for the GO term “phosphate-containing compound metabolic process” (supplementary table 6, Supplementary Material online). Genes belonging to this GO term seemed to be involved in a variety of metabolic processes and pathways, in which we did not observe a clear trend.
It is predicted that mate choice prior to copulation plays a more important role in sexual selection in nonplacental (lecithotrophic) species compared with their placental relatives. In our analysis, we find four genes involved in vision that are positively selected in P. turrubarensis (eya4, rlbp1, opso, and rp9). Positive selection on genes involved in vision could facilitate optimizing signaling of visual cues from potential mating candidates. Additionally, we found evidence of positive selection in P. turrubarensis for the melanin concentrating hormone receptor 1 (mchr1) gene. Melanin concentrating hormone is involved in pigment pattern formation in teleosts (Nagai et al. 1986).
Discussion
Studying the origin of biological innovations by comparative approach requires high-quality genomes and fairly complete gene sets. To meet this requirement, we sequenced and assembled the genomes of two livebearing fish from the same genus: P. retropinna and P. turrubarensis. The resulting genomes are of high contiguity and quality and will be valuable for future genomic analyses that involve these species.
Although both assemblies showed good quality metrics, the P. retropinna assembly, based on PacBio sequencing, outperformed the P. turrubarensis assembly both in terms of contiguity and gene completeness, especially in regions that are rich in repeats (fig. 2 and supplementary fig. 1, Supplementary Material online). An additional advantage of the PacBio assembly is that we could validate tandem duplications directly by mapping reads to the assembly (see supplementary fig. 3, Supplementary Material online, for example). However, these advantages come at a cost, as PacBio is considerably more expensive in raw sequence generation and computational resources for assembly, even with an efficient assembler such as wtdbg2. A possible reason for the difference in contiguity is that 10× Genomics based assemblies, even more so than PacBio based assemblies, rely heavily on the extraction of very high molecular weight DNA. DNA extraction based on red blood cells allows for the highest molecular weight DNA, since no mechanical degradation of tissue is necessary for DNA extraction. Sampling blood, however, is not feasible for these small fishes as single fish simply do not contain sufficient quantities. Extracting DNA from tissues, as was done in this study, results in additional mechanical stress on DNA, resulting in a shorter average molecule size (∼40 kb). Finally, the 10× Genomics method is primarily optimized for mammalian genomes; nonmammalian genomes are known to result in reduced contiguity. Despite the observed difference in contiguity, the quality of both genomes allows for a comprehensive and systematic assessment of gene duplications, as well as a genome-wide scan for genes evolving under positive selection.
The evolution of the placenta in mammals was accompanied by an increased rate of evolution of genes involved in mother–offspring interaction: genes involved in nutrient transfer to offspring, embryonic development, and placenta formation (Clark et al. 2003; Crespi and Semeniuk 2004; Nielsen et al. 2005). Furthermore, sperm selection is predicted to intensify in placental species (Zeh and Zeh 2000) and is hypothesized to drive rapid evolution of sperm proteins in placental mammals (Torgerson et al. 2002). Our results indicate that similar processes are ongoing in the placental P. retropinna.
The high rate of evolution of genes involved in nutrient transfer in P. retropinna may provide insights into the metabolic mechanisms that play a role in the evolution of the poecilid placenta. Specifically, five enzymes involved in peroxisomal lipid metabolism evolve under positive selection in P. retropinna, as well as two genes involved in protein transport to the peroxisomes, highlighting a very specific metabolic pathway. Lipids metabolized in the peroxisome include very long chain fatty acids and certain steroids (Mannaerts and van Veldhoven 1996). The expression of these enzymes is regulated by the peroxisome proliferator receptor α (PPARα) nuclear receptor (Reddy and Chu 1996), and it has been reported that PPARα regulates placental lipid metabolism through the proliferation of peroxisomes in rats (Martinez et al. 2008). Coupregulation of peroxisomal lipid metabolizing enzymes and Vitellogenin was observed after treatment with estrogenic compounds in zebrafish (Ortiz-Zarragoitia and Cajaraville 2005). These results may suggest that changes in peroxisomal lipid metabolism in P. retropinna result from a shift from pre- to post-fertilization provisioning that is inherent to the evolution of a placenta. Directly related to this may be the expansion of the Vitellogenin (vtg) gene family in P. retropinna. Vitellogenin proteins are egg yolk precursors that, in nonplacental poeciliids, are synthesized in the liver before being transferred to the oocyte to build up nutrient supply, a process called vitellogenesis (Rocha et al. 2008). Placental poeciliids such as P. retropinna supply only a very small amount of yolk before fertilization, which makes an expansion of the vtg gene family seems somewhat counterintuitive at first. However, it has been shown that Vitellogenin is also used as a means of postfertilization maternal nutrient provisioning in placental fish from the family Goodeidae (Vega-López et al. 2007). This duplication suggests that Vitellogenin may fulfill a similar function in P. retropinna. At the same time, the egg, although small, does contain some yolk. One of the vtg gene copies may be involved new role in embryo provisioning, whereas the other remains active in yolking the egg. Although we have no further evidence to support this, it can be hypothesized that the two copies therefore have different timing in expression.
Innovations in tissue development, as clearly the case during placental evolution, require novel recruitment of signaling and other pathways involved in cell differentiation and cell migration. In this study, we report the rapid evolution of genes involved in embryonic development in P. retropinna. The most striking finding is five genes involved in the Wnt signaling pathway that were found to evolve under positive selection. This pathway is crucial in early embryonic development and tissue formation, and its function is conserved across vertebrates as well as some invertebrates (Clevers 2006). By contrast, no members of the Wnt pathway were found to evolve under positive selection in P. turrubarensis, implying that these changes are not characteristic for the genus Poeciliopsis but rather a feature of the lineage leading to the placental P. retropinna.
Another compelling finding is that four genes encoding sperm-associated proteins are duplicated in P. retropinna. Three out of four of these proteins are located in sperm flagella of cilia. This may be an indication for increased postcopulatory selection in P. retropinna, for instance on sperm quantity or quality. However, we also find some evidence for rapid evolution of sperm proteins in the nonplacental P. turrubarensis in the form of positive selection on three sperm-associated genes (spata4, spata17, and spag16). These proteins are involved in sperm production (Liu et al. 2004; Nie et al. 2013) and motility (Zhang et al. 2007). Although it is predicted that sperm selection intensifies in placental species, sperm selection plays a role in nonplacental poeciliids as well (Brown et al. 2018). These results indicate a continuing selection on sperm-related traits, with possible different traits related to the different life histories in the two species under study.
The rapid evolution of genes involved in mother–offspring interactions in mammals shows a high degree of overlap, in the pathways involved, with accelerated evolution in genes in P. retropinna. On the gene level, we do not find convergence between our results and studies on the genomic consequences of mammalian placenta evolution (Clark et al. 2003; Crespi and Semeniuk 2004; Nielsen et al. 2005; Hou et al. 2009). This may be explained by the fact that although the mammalian and poeciliid placentas perform a similar function (e.g., postfertilization nutrient transfer to offspring), the morphological and physiological characteristics of these placentas are in fact quite different (Griffith and Wagner 2017). For example, the vtg1 gene that is duplicated in P. retropinna, and which may be involved in lipid transfer to offspring in that species, is not functional in mammals. In mammals, lipid transfer to offspring is mediated by other proteins such as apolipoprotein B-100 (Madsen et al. 2004). Similarly, many aspects of placentation may be functionally convergent but not molecularly homologous between the poeciliid and mammalian placenta.
Genomes of nonmodel species are increasingly accessible for accurate characterization and annotation, creating opportunities for comparative analyses by filling in crucial phylogenetic gaps providing powerful evolutionary contrasts. This study shows compelling evidence for selection on genes and pathways involved in a key life history trait, providing a valuable hypothesis on placental evolution in livebearing fish that can be readily tested in future studies.
Materials and Methods
DNA Preparation and Sequencing
A male individual of P. retropinna and a male individual of P. turrubarensis were caught at Rio Cañas, Costa Rica. DNA was extracted using the Qiagen Genomic-tip 100/G DNA extraction kit, following the instructions on the manufacturer’s protocol. For the P. turrubarensis sample, extracted DNA was size selected for fragments >30 kb using a BluePippin system. The P. retropinna DNA was sequenced on a PacBio Sequel System (Rhoads and Au 2015), generating ∼6.75 million reads with an N50 read length of ∼17 kb. Additionally, a fraction of the same DNA was sequenced on an Illumina HiSeq 4000 sequencer, generating ∼240 million 150-bp paired-end reads. The P. turrubarensis DNA was processed by a Chromium controller chip, together with 10× Chromium reagents and gel beads following the manufacturer’s protocol. Subsequently, the DNA was sequenced in a single lane of an Illumina HiSeq X Ten sequencer, producing ∼344 million 150-bp paired-end reads.
For generating RNA-seq data, pregnant females were stored in RNAlater. Dissection of individual maternal follicles was performed in the laboratory and stored in TriZol. RNA was extracted from late stage maternal follicles in six P. retropinna and one P. turrubarensis females using a Direct-zol RNA miniprep kit. Total RNA was then depleted of rRNA as described by (Adiconis et al. 2013). This depletion was only partial, because human DNA oligos were used, as Poeciliopsis-specific DNA oligos tiling rRNAs do not exist. Libraries were subsequently prepared using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina for use with rRNA depleted RNA. In brief, 100 ng of each RNA sample was subjected to a 7-min fragmentation and primed using random primers, followed immediately by cDNA synthesis. Samples were end prepped and standard NEBNext adapters were ligated to the resulting cDNAs. Each sample was enriched for adapter ligated DNA using a seven-cycle polymerase chain reaction with each sample using a unique index primer, and then purified using Ampure XP beads. Library quality was assessed using both Qubit and Bioanalyzer technology. Libraries were multiplexed, and then sequenced on an Illumina NextSeq using one 150-cycle High Output v2 kit.
Poeciliopsis retropinna Assembly
Before assembly of the P. retropinna genome, genome size and heterozygosity were estimated using a k-mer analysis. K-mers from Illumina short reads were counted using KMC 3 (Kokot et al. 2017). The assembly was generated using a hybrid pipeline where reads were corrected using Canu v1.7.1 (Koren et al. 2017) using settings genomeSize = 625m, MhapSensitivity = normal, correctedErrorRate = 0.085, corMhapSensitivity = normal, and corOutCoverage = 50, and assembly was performed by wtdbg v2.1 (Ruan and Li 2020) using default settings. After assembly, redundant contigs corresponding to heterozygous regions in the genome were removed using Redundans v0.14a (Pryszcz and Gabaldón 2016), only using the reduction step of the pipeline. Then, the assembly was rescaffolded using SSPACE-longread v1-1 (Boetzer and Pirovano 2014). The reasons for rescaffolding an assembly already based on long reads are 2-fold: First, contigs corresponding to heterozygous regions of the genome were removed by running Redundans. These sections of the genome can decrease contiguity of an assembly by breaking contigs at heterozygous parts. After these contigs were removed this was no longer an issue, allowing further scaffolding of the assembly. Second, assembly was performed after read correction by Canu, which uses all reads to correct the longest reads corresponding to 50× coverage (as determined by the corOutCoverage parameter). Other reads that are not used for the assembly may still bridge multiple contigs. After rescaffolding, the draft assembly was polished by aligning Illumina reads to the genome using bwa v0.7.5, using the “mem” option (Li and Durbin 2009), after which the assembly was polished in two rounds using Pilon v1.22 (Walker et al. 2014). Gene completeness was assessed using BUSCO 3.0.2 (Simão et al. 2015). The assembly was checked for possible misassemblies by alignment to the published guppy (Poecilia reticulata) genome assembly (v1.0) (Künstner et al. 2016) using MUMmer 3.23 (Kurtz et al. 2004). One putative misassembly was manually corrected.
Poeciliopsis turrubarensis Assembly
For the P. turrubarensis assembly, Illumina basecall files were demultiplexed using bcl2fastq v2.20 (https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html, last accessed January 31, 2020), after which they were assembled using the Supernova assembler v2.1.1 (Weisenfeld et al. 2017), using setting –maxreads = 300,000,000, producing an assembly with a size of 618 Mb, and a scaffold N50 of 4.2 Mb. Although the Supernova assembler should separate haplotypes for the majority of the genome, we still observed a number of redundant contigs belonging to heterozygous regions in the genome that were assembled separately. We removed these using Redundans v0.14a (Pryszcz and Gabaldón 2016), using only the reduction step of the pipeline. Gene completeness was assessed using BUSCO 3.0.2 (Simão et al. 2015).
Genome Annotation
Placental RNA-seq libraries of P. retropinna and RNA-seq libraries of the corresponding follicular tissue in P. turrubarensis were aligned to their respective genomes using HISAT2 2.1.0 (Sirén et al. 2014). Additionally, published RNA-seq libraries of Poeciliopsis prolifica (Jue et al. 2018) tissues were downloaded from GenBank and aligned in the same way. For both genomes, a de novo repeat library was constructed using RepeatModeler 1.0.11 (Smit and Hubley 2008). Subsequently, the genome was soft masked using RepeatMasker 4.0.7 (Smit et al. 2013), using these newly created species-specific repeat libraries. The soft-masked genomes were subsequently annotated using the BRAKER2 pipeline (Hoff et al. 2019). Besides the aligned RNA-seq, protein sets from Poecilia reticulata and X. maculatus were included as additional evidence for the annotation pipeline. After annotation, gene predictions were filtered by scanning all predicted proteins for functional domains against the protein family (pfam) database (Bateman et al. 2004), using HMMer 3.2.1 (Mistry et al. 2013). Predicted genes coding for a protein without any known protein domain were excluded from the final gene set. Additionally, predicted genes having a total length shorter than 500 base pairs were also excluded from the gene set. Functional annotations were added by performing protein Blast (Altschul et al. 1990) searches to the Swissprot database (Boeckmann et al. 2003) and running InterProScan (Jones et al. 2014) to add domain information for every predicted protein.
Whole-Genome Alignment
Soft-masked assemblies of P. retropinna and P. turrubarensis were aligned to each other and to the published genome assembly of Poecilia reticulata (Künstner et al. 2016) using ProgressiveCactus (Paten et al. 2011). Pairwise alignments were obtained by generating a multialignment format file for pairs of species using the hal2maf utility that is part of the hal toolbox (Hickey et al. 2013). Then, synteny blocks were formed by chaining the resulting pairwise alignments using UCSCs axtChain program (Kent et al. 2003). Because the ProgressiveCactus alignment is reference free, we could obtain these synteny blocks relative to each of the three genomes that we used, as well as from within-genome alignments (paralogous blocks). Chains were converted to GFF3 format for further analysis and visualization in Jbrowse (Skinner et al. 2009).
Gene Duplications
Gene duplications between the genomes of P. retropinna and P. turrubarensis were assessed using synteny blocks, applying the following criteria. First, regions of the reference genome where multiple synteny blocks of the query genome align to the same region of the reference genome were extracted. Then, these regions were filtered on the following criteria: 1) the overlapping synteny blocks should also overlap in the positions to which their “raw” alignments align, 2) the duplicated region must be at least 3-kb long, 3) no paralogous alignments should be present within the duplicated region, 4) the duplication must lie on a scaffold that is bigger than 100 kb, to prevent separate assembly of heterozygous regions of the genome to mimic a duplication, and 5) the average coverage of mapped short reads within the duplicated region must not differ more than 25% from both the surrounding region (10-kb upstream and downstream) and the genome-wide average. Genes that were found to reside within these duplicated regions were extracted from the genome annotation. To infer the most likely ancestral copy number of these genes, the Poecilia reticulata genome was included in the alignment as an outgroup. Using the same methodology as previously stated, the gene copy number was determined for Poecilia reticulata. Duplications were kept only if the Poecilia reticulata copy number matched the nonduplicated state. For an overview of the effect of distinct filtering steps on amount of duplicated segments passing the filtering, see supplementary table S7, Supplementary Material online.
Positive Selection
We detected orthologous genes shared between the genomes of P. retropinna, P. turrubarensis, Poecilia reticulata (Künstner et al. 2016), and X. maculatus (Schartl et al. 2013) using ProteinOrtho v5.16b (Lechner et al. 2011). To avoid comparing paralogs, we only selected genes displaying 1:1 orthology for all species. In total, we found 11,323 1:1 orthologs. A codon-aware alignment was made for the sequences of these orthologs using PRANK v.170427 (Löytynoja 2014). These alignments were tested for signs positive selection using the codeml program that is part of PAML 4.9 (Yang 2007). Alignment columns that have gaps in one of the species were excluded from analysis. All genes were tested for positive selection in the phylogenetic branch leading to P. retropinna, as well as in the branch leading to P. turrubarensis by applying the branch-site model. Additionally, we tested the genes for positive selection across the whole phylogeny using the site model. This way, a distinction was made between positive selection that is unique for a certain branch and positive selection that is shared between all poeciliid species. P values were obtained by performing likelihood ratio tests using a chi-square distribution, following recommendations from the PAML manual. Correction for multiple testing was performed with the Benjamini–Hochberg procedure (Benjamini and Hochberg 1995), using a 5% false discovery rate.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Sequencing reads used for both assemblies are available in the under accession number PRJEB3354. Genome assemblies are available in GenBank under accession numbers PRJNA555005 for Poeciliopsis retropinna and PRJNA555006 for P. turrubarensis. The P. retropinna and P. turrubarensis individuals used for sequencing were collected under permit number SINAC-SE-CUS-PI-R-005-2017 and SINAC-SE-005-2017.
Acknowledgments
This work was supported by a WIAS research grant from Wageningen Institute of Animal Sciences (to H.-J.M. and B.J.A.P.), a VIDI grant from the Netherlands Organisation for Scientific Research (864.14.008 to B.J.A.P.), a National Science Foundation Graduate Research Fellowship (2014185511 to M.W.G.), and a Diversifying Academia, Recruiting Excellence (DARE) Fellowship (to M.W.G.).
References
Author notes
Bart J.A. Pollux and Hendrik-Jan Megens contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}