





Abstract
The pellicles of alveolates (ciliates, apicomplexans, and dinoflagellates) share a common organization, yet perform very divergent functions, including motility, host cell invasion, and armor. The alveolate pellicle consists of a system of flattened membrane sacs (alveoli, which are the defining feature of the group) below the plasma membrane that is supported by a membrane skeleton as well as a network of microtubules and other filamentous elements. We recently showed that a family of proteins, alveolins, are common and unique to this pellicular structure in alveolates. To identify additional proteins that contribute to this structure, a pellicle proteome study was conducted for the ciliate Tetrahymena thermophila. We found 1,173 proteins associated with this structure, 45% (529 proteins) of which represented novel proteins without matches to other functionally characterized proteins. Expression of four newly identified T. thermophila pellicular proteins as green fluorescent protein-fusion constructs confirmed pellicular location, and one new protein located in the oral apparatus. Bioinformatic analysis revealed that 21% of the putative pellicular proteins, predominantly the novel proteins, contained highly repetitive regions with strong amino acid biases for particular residues (K, E, Q, L, I, and V). When the T. thermophila novel proteins were compared with apicomplexan genomic data, 278 proteins with high sequence similarity were identified, suggesting that many of these putative pellicular components are shared between the alveolates. Of these shared proteins, 126 contained the distinctive repeat regions. Localization of two such proteins in Toxoplasma gondii confirmed their role in the pellicle and in doing so identified two new proteins of the apicomplexan invasive structure—the apical complex. Screening broadly for these repetitive domains in genomic data revealed large and actively evolving families of such proteins in alveolates, suggesting that these proteins might underpin the diversity and utility of their unique pellicular structure.
Introduction
The infrakingdom Alveolata unites three major but very different eukaryotic phyla: Ciliata, Apicomplexa, and Dinoflagellata (Adl et al. 2005). This group encompasses predators that are among the largest and most complex single-celled organisms (e.g., the ciliate Spirostomum), tiny obligate intracellular parasites (e.g., the causative agent of malaria, the apicomplexan Plasmodium), and remarkably ornate photosynthetic organisms (e.g., the dinoflagellate Ceratium). Despite such diversity of form, lifestyle, and environmental niche, molecular phylogenies firmly group these phyla in a monophyletic lineage (Wolters 1991; Baldauf et al. 2000; Burki et al. 2007; Hampl et al. 2009). Furthermore, all alveolates share a common ultrastructural organization of their pellicle, namely a layer of flattened membrane sacs called alveoli that subtend the plasma membrane (Wolters 1991). A membrane skeleton that can appear either amorphous or fibrous is closely associated with the alveoli, and a microtubular cytoskeleton is in turn associated with these membranes in addition to other filamentous elements (Dodge 1987; Hausmann and Bradbury 1996; Morrissette et al. 1997). This pellicular organization is common to all Alveolata, and a progenitor version was presumably present in the common ancestor of this group and has been maintained throughout alveolate diversification. This is supported by the presence of a unique family of proteins named “alveolins”; proteins associated with alveoli and common to alveolates (Gould et al. 2008). This family of proteins is characterized by a conserved repeat, in Toxoplasma gondii alone encoding a dynamic set of 14 proteins localizing to the subpellicular network (Anderson-White et al. 2010).
The alveolate pellicle performs a structural role of maintaining cell shape, evident by its resilience after cell lysis or death. For example, detergent extraction of ciliates recovers the pellicle as a cell “ghost” that maintains the cell size and shape (Williams 2004). Similar extracts of apicomplexa recover a subpellicular network of interwoven filaments, subpellicular microtubules, and associated pellicular structures (Mann and Beckers 2001). Dead dinoflagellates also leave ghosts of the cell pellicle (called amphiesma in this group) that retain the original cell shape and ornamentation. These observations reveal the mechanical strength of these complex structures. Further to this basic structural role, the pellicle also serves to anchor and coordinate several dynamic structures that have been developed in each of the phyla. In ciliates, the many cilia are anchored into the pellicle and are coordinated by a complex network of microtubules and other fibrilar arrays that are integral to this structure (Allen 1967; Satir and Wissig 1982). Ciliate oral apparatuses are also specialized structures that are coordinated within the pellicular architecture and typically consist of compound ciliary structures organized around a cytosome and anchored by dedicated cytoskeletal elements (fig. 1). Apicomplexans have developed at least two specialist pellicular invasion structures: 1) the glideosome, which comprises motile complexes that span the plasma membrane and alveoli to enable gliding motility of cells as they seek out new host cells and 2) the conoid of the apical complex, which is essential for host cell penetration and invasion (Morrissette and Sibley 2002; Santos et al. 2009). In dinoflagellates, vast arrays of ejectile organelles (trichocysts, also seen in ciliates) occur within the pellicle. These are coordinated among the alveoli, that in many dinoflagellates form an armor of protective plates reinforced with cellulose in the alveoli lumen (Dodge 1987). Hence, it is apparent that each phylum has customized its alveolar pellicle to suit particular requirements. Little is known, however, about the basic common construction of this unifying cell feature.
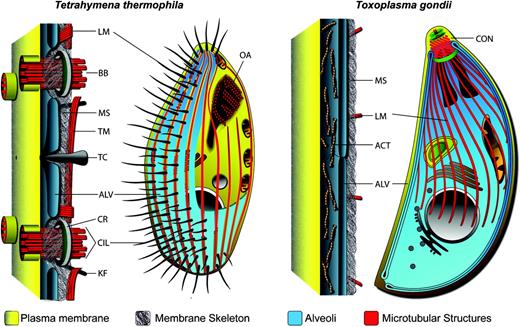
Schematic of the pellicles of ciliate Tetrahymena thermophila and the apicomplexan Toxoplasma gondii. Both share a common pellicle organization characterized by alveolar sacs (ALV) beneath the plasma membrane supported by a membrane skeleton (MS) and associated with microtubules (longitudinal, LM; transverse TM) and other filamentous elements. BB, basal body; TC, trichocyst; CR, circumciliary rings; CIL, cilia; KF, kinetodesmal fibers; CON, conoid; ACT, actin filaments.
Ciliates and apicomplexa have received more scientific attention with respect to their pellicular composition and architecture than dinoflagellates, largely because they are more tractable experimentally. Much more is known, however, about the specialist elements of the pellicle than its underlying basic construction. Ciliates such as Tetrahymena and Paramecium have been models for many cell processes, including motility systems owing to their arrays of cilia. For instance, protein profiling of isolated basal bodies has greatly expanded our knowledge of constituent proteins (Kilburn et al. 2007). Similarly, electron microscopy has detailed how these cilia are set between alveolar sacs, which in turn are supported by an amorphous membrane skeleton (also called epiplasm) on the cytoplasmic face of the alveoli (Allen 1967; Satir and Wissig 1982). In Tetrahymena, bundles of microtubules run lengthways (longitudinal) between the epiplasm and alveoli and crossways (transverse) under the epiplasm and together with other fibers and strands of unknown protein composition, these coordinate and anchor the arrays of cilia (fig. 1).
From Tetrahymena epiplasm extracts, three major high molecular weight protein species have been identified, and these are located continuously across the membrane skeleton (although with subtle differences) (Williams et al. 1995). Gene disruptions of the most abundant of these, EPC1, result in misshaped cells and disorganized cilia and oral apparatuses (Williams 2004). In Paramecium, similar epiplasm extracts recover 20–50 major proteins that, in vitro, show remarkable abilities to reassemble into filaments (Nahon et al. 1993; Coffe et al. 1996). A multigene family (51 genes) named the epiplasmins encodes these major proteins, and gene disruptions result in similar phenotypes seen with Tetrahymena EPC1 knockouts (Pomel et al. 2006; Damaj et al. 2009). Four homologs of the epiplasmins occur in Tetrahymena; however, these are distinct from the three major protein bands (including EPC1), and the Tetrahymena epiplasmins (EpiT1-3, 5) are yet to be localized (Damaj et al. 2009). Two further distinct classes of proteins are known from the Tetrahymena epiplasm: a pair of small calmodulins (Numata et al. 2000) and the so-called “K antigens,” that are located on amorphous shirts (circumcilliary rings and terminal plates) around the basal bodies that appear to bind them directly to the epiplasm layer (fig. 1) (Williams et al. 1990). In addition, proteins specific to the ciliate oral apparatus (tetrins) have been shown to form small filaments (3–4 nm) (Honts and Williams 1990). Despite these insights, however, it is clear that much remains to be discovered regarding the composition and organization of this remarkably complex cortical structure.
Among apicomplexa, the pellicles of the human parasites Plasmodium and Toxoplasma have been most closely examined. A fibrous membrane skeleton (subpellicular network) supports the alveoli (also known as the inner membrane complex) on the cytoplasmic side and is associated with a cage of microtubules that in Toxoplasma extends for two-thirds of the cell length (fig. 1) (Morrissette and Sibley 2002). Extraction of intact full-length Toxoplasma pellicles confirms the structural role of the membrane skeleton, and freeze fracture reveals linear arrays of intramembrane particles within the alveoli that imply integral association of both of these structures with the alveoli (Meszoely et al. 1982; Morrissette et al. 1997; Mann and Beckers 2001). Immunoidentification of two membrane skeleton proteins provided the first description of alveolin proteins (originally named IMC proteins) that are now recognized to be common to all alveolate phyla (Mann and Beckers 2001; Gould et al. 2008). Gene disruption of alveolins results in loss of cell pellicle strength, yet it is unresolved if alveolins themselves contribute to the filamentous character of the membrane skeleton (Mann et al. 2002; Khater et al. 2004; Tremp et al. 2008). Little more is known about the basic structure of the pellicle, with far more attention given to the invasion-related conoid structure and the glideosome that is essential for motility associated with host cell invasion and egress (Morrissette and Sibley 2002; Santos et al. 2009).
The conoid is an open cone structural unit of the apical complex that is intimately associated with the apical terminus of the alveoli and subpellicular network (fig. 1). It plays a mechanical role during host cell invasion but is also apparently involved in cell replication as one of the seminal features of developing daughter cells and biogenesis of the pellicle (Morrissette and Sibley 2002). A proteomic analysis of the conoid characterized several proteins associated with this tubulin-based structure and indicated over 50 further unknown proteins that putatively associate with this structure (Hu et al. 2006). The glideosome describes the motor complex responsible for the characteristic gliding motility of these parasites. Glideosomes coordinate interaction between transplasma membrane adhesive proteins, actin–myosin motors, the plasma membrane, alveoli, and anchoring proteins that themselves are believed to span the two membranes of the alveoli. Several glideosome protein groups and their interactions have been described (Mann et al. 2002; Gaskins et al. 2004; Baum et al. 2008; Bullen et al. 2009).
Although these targeted functional studies have led to great progress in understanding some of the important functions of the alveolate pellicle, it is clear that there is still some way to go in understanding the assembly and molecular interactions that underpin pellicular organization and function. Here, we undertake a proteomic analysis of the pellicle of the ciliate Tetrahymena. We compare this data set with predicted proteins of other alveolates to identify new pellicle candidates in parasites, and we identify and validate novel pellicle proteins in both ciliates and apicomplexans. We also identify a conspicuous new class of proteins—abundant in alveolates but present in most eukaryotes—that are characterized by distinct repetitive elements constituted of particular amino acids. This provides a very rich new data set to pursue the essential functions of alveolate pellicles.
Materials and Methods
Cell Strains and Culture
Tetrahymena thermophila strains CU522 and CU428 were cultured in SPP medium (2% proteose-peptone, 0.1% yeast extract, 0.2% Glucose, 0.003% Fe-EDTA) routinely at 16 °C and transferred to 30 °C for overnight growth before harvesting the cells for all downstream experiments. RNA, DNA, and total protein were isolated using Trizol (Invitrogen, Australia), following the manufactures protocol. RH stain T. gondii parasites lacking the Ku80 gene ([Huynh and Carruthers 2009]; a kind gift from Mae Huynh and Vern Carruthers, University of Michigan) were grown in human foreskin fibroblasts as previously described (Striepen and Soldati 2007).
Pellicle Preparation
The pellicle of T. thermophila was isolated on ice from 2 × 107 pelleted cells based on a protocol by Williams et al. (1987) with minor modification. The cell pellet was resuspended in 5 ml of cold 0.25 M sucrose and subsequently 15 ml of cold SEMT solution (l M sucrose, l mM EDTA, 0.1% 2-mercaptoethanol, 10 mM Tris–HC1 pH 9) and 2.5 ml of 10% Triton X-100 in phosphate buffer added in sequence with continuous agitation. This released the pellicles/epiplasms and separated them from membranous structures. They were harvested at 4,000 × g for 20 min, and the pellet resuspended in phosphate buffer at pH 7, containing Complete Midi tablets (Roche, Australia) against protease degradation. The pellicles were then concentrated at 20,000 × g for 10 min and washed three times in phosphate buffer at pH 7. For Na2CO3 extraction, we then additionally incubated the sample for 30 min on ice with 0.1 M Na2CO3 buffer. All preparations were stained with DAPI to check for DNA contamination and with anti-alveolin (αALV1.2, Gould et al. 2008) to analyze the integrity of the pellicle structures.
Proteomics
Proteomics Profiling Experiment
The pellicle proteins were digested with trypsin, and strong cation exchange (SCX) chromatography was performed as previously described in Natera et al. (2008). Fractions obtained from SCX-HPLC were reduced under vacuum and resuspended in 0.1% formic acid (60 μl), filtered through a minisart 0.2 μm membrane (Sartorius Stadim Biotech, Aubagne, France), and one quarter of each fraction was loaded onto a 300 μm × 5 mm Zorbax 300SB-C18 (Agilent Technologies, Palo Alto, CA) reversed-phase precolumn attached to a Shimadzu Prominence nano LC system (Shimadzu Corporation, Kyoto, Japan). The precolumn was washed with 0.1% formic acid in 5% acetonitrile for 15 min before placing in-line with a 75 μm × 150 mm Zorbax 300SB-C18 (Agilent Technologies) reversed-phase column. Peptides were eluted using a gradient of 5–65% (v/v) acetonitrile in 0.1% formic acid over 60 min, at a flow rate of 0.25 μl/min. Peptides were analyzed via electrospray ionization on a QSTAR Elite hybrid quadrupole time-of-flight mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA). Each fraction was analyzed twice.
The mass spectrometer was operated in the positive ion mode, ion source voltage of 2,200 V, using 10 μm uncoated SilicaTips (New Objective, Woburn, MA). Analyst QS 2.0 software (Applied Biosystems/MDS Sciex) was used to collect data in a data-dependent acquisition mode for the three most intense ions fulfilling the following criteria: m/z between 450 and 2,000; ion intensity 40 counts; and charge state between +2 and +5. After MS/MS analysis, these ions were dynamically excluded for 18 s, using a mass tolerance of 50 mDa. Mass spectrometry (MS) scans were accumulated for 0.5 s, and MS/MS scans were collected in automatic accumulation mode for a maximum of 2 s. Mass and charge state-dependent rolling collision energy were used, and the MS instrument was calibrated daily with [Glu]-fibrinopeptide B (Sigma-Aldrich, St Louis, MO).
Peak lists from the MS/MS spectra were made using ProteinPilot software version 2.0.1 (Applied Biosystems/MDS Sciex) and searched against T. thermophila proteins (September 2009 release) using MASCOT (Perkins et al. 1999), X!Tandem (Craig and Beavis 2004), and the Paragon algorithm (Shilov et al. 2007). The MASCOT and X!Tandem search parameters were: enzyme: trypsin; fixed modifications: carbamidomethyl; MS peptide tolerance: 0.25 Da; MS/MS tolerance: 0.15 Da; number of missed cleavages: up to 1. The Paragon algorithm parameters were: sample type: identification; Cys alkylation: iodoacetamide; Digestion: trypsin; Search effort: Thorough ID. The outputs from all search algorithms were combined, and only proteins with two or more peptides with a P < 0.05 in all three search algorithms were reported. The false positive rate determined using a randomized version of the T. thermophila protein database was 0.2%.
Reporter Protein-Fusion Protein Expression
For GFP-fusion expression in T. thermophila, we designed a new plasmid, pTtag (accession FJ789658), which is based on a previous GFP-tagging plasmid described by Shang et al. (2002). The main modifications of pTtag are: 1) a codon-optimized GFP, 2) a 1 kb shorter MTT-promoter, 3) optimized restriction sites, and 4) a multiple cloning site. For C-terminal GFP fusion, the target genes were cloned into pTtag via the HindIII and XhoI restriction sites. Tetrahymena thermophila was transformed using a PDS-1000 gene gun (Bio-Rad, Australia) with 900 psi rupture discs and 20 μg of linearized plasmid DNA according to the published method (Gaertig and Kapler 2000). Transformed cells were selected for 10 days in SPP supplemented with 20 μg/ml paclitaxel (LC Labs, MA). For GFP-fusion protein expression, cells were induced for at least 2 h using 2 μg/ml CdCl2.
For generation of genetically modified T. gondii strains, we tagged the 3′ end of the endogenous genes of the viral A-like repeat/charged repetitive motif proteins (CRMP) TgME49_044470 and TgME49_052880 with 3xHA epitope tags. We polymerase chain reaction (PCR) amplified approximately 1.1 kb of the 3′ end of TgME49_044470 using the primers 5′-TACTTCCAATCCAATTTAGCGGAGACGCCATCAAACAAATCCGA and 5′-TCCTCCACTTCCAATTTTAGCGTTTGTTGATGCGTCCGAGACAAC, and RHDHXGPT genomic DNA as a template. We PCR amplified approximately 1.1 kb of the 3′ end of TgME49_052880 with the primers 5′-TACTTCCAATCCAATTTAGCTGAAGAAAGTCCTTGAGGGTC and 5′-TCCTCCACTTCCAATTTTAGCGAAGCCCCAGAAGTATCCAGCAATGGAC, using RHDHXGPT genomic DNA as a template. The TgME49_044470 product was cloned into the vector pHA3.LIC.DHFR (a kind gift from Michael White, University of South Florida) by ligation independent cloning as previously described (Huynh and Carruthers 2009). This construct was linearized by digestion with NsiI and transfected into pyrimethamine-sensitive RH strain KU80 knockout parasites as previously described (Striepen and Soldati 2007). We selected for pyrimethamine-resistant parasites and cloned parasites by limiting dilution. We modified the pHA3.LIC.DHFR vector by replacing the DHFR selectable marker with a HXGPRT marker to generate the vector pHA3.LIC.HX (Giel van Dooren, unpublished data). We cloned the TgME49_052880 PCR product into this vector by ligation independent cloning as previously described (Huynh and Carruthers 2009). This construct was linearized by digestion with ApaI and transfected into mycophenolic acid-sensitive RHDHXGPT strain KU80 knockout parasites as previously described (Striepen and Soldati 2007; Huynh and Carruthers 2009). We selected for mycophenolic acid-resistant parasites and cloned parasites by limiting dilution. Successful modification of the genetic locus for each gene was confirmed by PCR analysis of isolated clones (not shown).
Immunofluorescence assays were performed as previously described (van Dooren et al. 2009). We used rat anti-HA antibodies (Roche) at a dilution of 1:100, mouse anti-IMC antibodies (mAB 45.36; kind gift from Gary Ward, University of Vermont) at 1:500, and mouse antitubulin antibodies (mAb 12G10, Developmental Studies Hybridoma Bank, University of Iowa) at 1:10. We use anti-rat AlexaFluor 488 and anti-mouse AlexaFluor 546 (Invitrogen) at 1:200 as secondary antibodies. To extract the parasite pellicle, we performed deoxycholate treatments as previously described (Mann and Beckers 2001). Briefly, we filtered parasites through a 3-μm filter and resuspended them in phosphate-buffered saline. We attached parasites to coverslips with 0.1% polyethyleneimine and extracted in 10 mM deoxycholate and 0.5 mM MgCl2 for 10 min at room temperature. We fixed parasites in 4% paraformaldehyde for 10 min and then proceeded to label extracted pellicles with antibodies as described above.
Cells were analyzed on a Leica TCS SP2 confocal laser-scanning microscope, and only the brightness/contrast ratio of the images was modified using Adobe Photoshop CS4.
CRMP Prediction
XSTREAM v1.7 was used to identify repeats in protein sequence data using the following parameters: seed lengths 2, 5, and 7; DP Match Score 7; DP Miss/Gap Penalty −1, −5, respectively; Consecutive Gap Max (g*) 4; minimum period 10; maximum gaps 4; minimum period 10; minimum copy number 3; minimum word and consensus match to 0.5. No sequence splitting was performed. Results were processed using the Konstanz Information Miner (knime.org) and proteins sorted according to amino acid composition satisfying: E ≥ 6%, K ≥ 6%, Q ≥ 3%, and the sum of I, L, and V ≥ 10%. Repeats with any single amino acid component >30% were removed as low complexity sequence. Analysis of whole-genome data was preceded by removal of duplicate sequences and predicted sequences trailing stop codons.
Results and Discussion
Pellicle Protein Isolation and Identification
To identify novel proteins that associate with the pellicle of alveolates, we performed a proteomic investigation of the pellicle of the model alveolate T. thermophila. Tetrahymena has the advantage that it can be grown axenically, has a sequenced genome, and has established procedures for isolation of the pellicle (Williams et al. 1987; Eisen et al. 2006). Pellicles were prepared by detergent extraction and differential centrifugation (Williams et al. 1987) in which membrane proteins and lipids are solubilized by TX-100 but the semirigid membrane skeleton that lies immediately beneath the alveolar membranes is retained and purified by low-speed centrifugation. A second extraction used Na2CO3 that is known to form membrane sheets and dislodge peripheral membrane proteins and therefore enriching for strongly associated and integral membrane proteins. This extraction aimed to identify contamination with nonpellicle proteins during cell lysis and specimen preparation. Two samples were prepared using each method and analyzed using 2D liquid chromatography-tandem mass spectrometry following trypsin digestion. Proteins were identified as positive only if two separate peptides were detected by each of the three prediction algorithms that matched mass spectra to the T. thermophila protein database.
From the total detergent-resistant T. thermophila pellicle preparation, we identified 5,720 peptides that corresponded to 1,119 predicted proteins. Each protein was represented by at least two peptides and on average five peptides per protein were identified, but up to 80 peptides were identified from a single protein (TTHERM_00578520). The equivalent analysis of the Na2CO3-extracted pellicular material identified 2,085 peptides representing 516 proteins that were retained after removal of approximately half of the total detergent-resistant proteins. Of these, 90% (462 proteins) were common to both preparation types, with only 54 new proteins identified in the Na2CO3-extracted material proving that this second extraction was indeed a more stringent measure. Combining these two data sets gave 1,173 putative pellicle-associated proteins (for complete protein list, see supplementary table S1, Supplementary Material online).
The pellicle proteome was annotated according to Blast matches to known protein homologs in public protein databases (minimum E value e−5) and relevant proteomics studies, namely those of the basal bodies and cilia (Pazour et al. 2005; Kilburn et al. 2007). Accordingly, the putative pellicular proteins and possible false positives were classified into six broad functional categories: 1) structural proteins, including proteins associated with cilia, the membrane skeleton, and other known cytoskeletal proteins; 2) cytosolic metabolic functions (including translation); 3) mitochondrial function, 4) nuclear function; 5) endomembrane-associated proteins; and 6) novel proteins (any protein without Blast match <e−5 to proteins of known function) (supplementary fig. S1, Supplementary Material online). The novel proteins represented the largest fraction (45%, 529 proteins). Of these, 62% (329 proteins) shared similarity (E values > e−5) with proteins in other nonciliate eukaryotes, and 38% (200 proteins) are apparently ciliate specific.
The recovery of proteins previously implicated in pellicle structure and function confirms the success of the pellicle isolation procedures. These proteins include both alveolins (TtAlv1 and TtAlv2), the major epiplasm protein Epc1p, cortical calmodulins (TCBP-25, -23), tetrins of the oral apparatus, proteins of the cilia and basal bodies, and numerous cytoskeletal components, including tubulins and motor proteins (supplementary table S1, Supplementary Material online). We also recovered the Paramecium epiplasmin homologs (EpiT1-3 and 5) that were implicated in pellicular function based on homology but whose location had not been determined (Damaj et al. 2009).
The recovery of proteins associated with cytosolic metabolism, mitochondria, the nucleus, and the endomembrane system, totaling 38.4% of our proteome, likely represent contamination as many of these proteins are abundant and Na2CO3 extraction did not preferentially reduce any of the functional categories (supplementary fig. S1, Supplementary Material online). Such contaminants were also present in the basal body proteome of Kilburn et al. (2007) where 48% of proteins represented these other functional groups. Electron microscopy indicates that the mitochondrion and a layer of rough endoplasmic reticulum are closely appressed to the membrane skeleton of the pellicle (Satir and Wissig 1982), and enzymes associated with cytosolic energy metabolism have been previously found to associate with cilia in T. thermophila and other organisms (Pazour et al. 2005; Kilburn et al. 2007). Thus, these metabolic functions and compartments might have been recovered in this pellicle preparation in part due to their functional association with the pellicle. In any case, the “novel” fraction of the pellicle proteome presumably contains a similar portion of proteins from these metabolic functions.
Localization of Novel T. thermophila Proteins to Pellicular Structures
To validate our proteomic approach for identifying novel T. thermophila pellicular proteins and to determine specific pellicular locations, four proteins were selected from the novel proteins (TTHERM_00388620, TTHERM_00188980, TTHERM_00128280, and TTHERM_00474830) and expressed as GFP-fusion proteins in T. thermophila cells. These four proteins selected were all well represented by peptides in the proteomic data (8–17 peptides) and were present in the Na2CO3 extraction material indicating a strong association with the pellicle. The four also contained conspicuous charged repetitive regions and similarity to viral A-type inclusion proteins (discussed below). Expression of these proteins under the control of a cadmium-inducible promoter indicated that all are exclusively located to the cell periphery consistent with being associated with the pellicle, and all four showed distinct pellicular localization patterns (fig. 2).
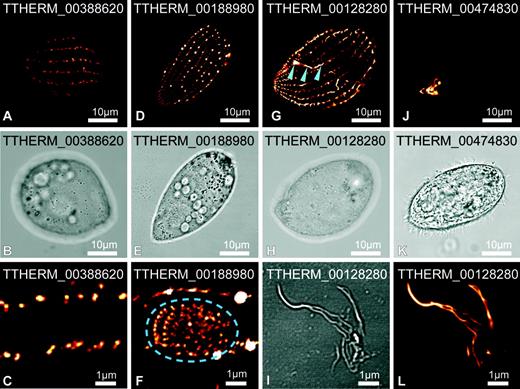
Location of four novel pellicle proteins in Tetrahymena thermophila. Proteins were GFP tagged and expressed in live cells (depicted orange to white according to increasing intensity). TTHERM_00388620 (A–C), TTHERM_00188980 (D–F), and TTHERM_00128280 (G–I, L) localize primarily along lines parallel to the rows of cilia and subpellicular/longitudinal microtubules. TTHERM_00188980 also localizes to the premature oral apparatus (F, indicated within dash ellipse) but not the mature apparatus. TTHERM_00128280 develops short filaments that emanate from the ciliary rows and also associates with the deep fiber bundle of the oral apparatus (G, arrowheads). Upon cell rupture TTHERM_00128280-labeled fibrous material remains intact (I, L). TTHERM_00474830 (J, K) localizes exclusively to the oral apparatus. Higher magnification detail of cell surface (C and F) and cell extracts (I and L) are shown.
Three of the T. thermophila proteins were generally organized into discontinuous longitudinal arrays beneath the plasma membrane that correspond in periodicity to the longitudinal rows of cilia and number of cilia within these rows (see fig. 1). The first, TTHERM_00388620, primarily occur as a row of dots organized along these linear arrays (fig. 2A–C), whereas TTHERM_00188980 and TTHERM_00128280 occur as a row of dashes forming near-continuous longitudinal lines (fig. 2D–H). This second protein, TTHERM_00188980, also associates with premature oral apparatuses (fig. 2F) but does not associate with the mature apparatus. The oral apparatus is a complex feeding structure consisting of four major compound ciliary structures organized around a cytostome with a “deep fiber” bundle of microtubules that radiates inward from this aperture (see fig. 1). Before cell division, a new oral apparatus forms de novo to be inherited by one of the daughter cells. TTHERM_00188980 apparently associates temporarily with the basal bodies of the nascent structure but dissociates as it matures (fig. 2F). The third protein, TTHERM_00128280, also associates with the oral apparatus, but in this case exclusively with the deep fiber bundle at the base of the mature oral apparatus (fig. 2G). This labeled structure remains intact after mechanical rupturing of the cell (fig. 2I and L). Furthermore, after prolonged induction of fusion protein expression, TTHERM_00128280 is located in fibrous structures emanating from the pellicle that extend into the cytoplasm (not shown). It is not known if these later observations represent overexpression-induced effects but, in any case, they indicate that this protein either generates or associates with filamentous elements. The fourth protein, TTHERM_00474830, associates exclusively with the mature oral apparatus and represents a novel protein of this ciliate structure (fig. 2J). Together, these data indicate that this proteomic strategy has been successful in identifying new proteins that function in the complex organization of the ciliate pellicular structures.
Known Structural Proteins and Many of the Novel Proteins Share Enrichment for Charged Repeat Motifs
The most conspicuous observation for the novel category of T. thermophila pellicle proteins was that 31% (162 proteins) recovered Blast matches to “viral A-type inclusion proteins” from either Trichomonas or Entamoeba as their best matches (E values as low as e−126) (see supplementary table S1, Supplementary Material online). Viral A-like proteins represent a collection of poorly characterized eukaryotic proteins mostly identified from similarity to pox virus proteins that generate inclusion bodies in the host cytoplasm into which viral A-like particles are sequestered. Almost nothing is known about their function in eukaryotes, but one viral A-like protein of the protist Trichomonas (p477, TVAG_012450) has been implicated in cytoskeletal function (Bricheux et al. 2007). The viral A-like proteins contain regions that are highly repetitive and enriched in charged residues, notably K and E, as well as hydrophobic residues, particularly L and I. The strong similarity of the T. thermophila proteins to viral A-like proteins was concentrated in these repetitive regions. The repeats observed in the viral A-like proteins are reminiscent of the repeat structure of the alveolin proteins, previously found to be common to all alveolates (Gould et al. 2008). Moreover, by scanning the structural proteins of the proteome data set with repeat finding algorithms (RADAR and XSTREAM, [Heger and Holm 2000; Newman and Cooper 2007]), we observed that many of these structural proteins also contained repeats with similar amino acid biases.
To test if a defining repeat could be observed more generally in pellicle structural proteins, we selected 20 characterized structural proteins that contained repetitive regions (as determined by RADAR and XSTREAM) and analyzed the amino acid composition of these repeats. This “test set” was selected for both the presence of conspicuously repetitive regions and to represent a range of functions within the pellicle (e.g., structural and motor proteins within the cilia and basal bodies, putative membrane skeleton proteins epiplasmins and alveolins, and oral apparatus proteins tetrins) (for full test set, see supplementary table S2, Supplementary Material online). Amino acid composition analysis of the test set repeat domains identified by XSTREAM21 showed a strong compositional bias (supplementary table S2, Supplementary Material online). Charged residues K (lysine, average 14.6%, minimum 6.1%) and E (glutamic acid, average 15.8%, minimum 6.6%) plus polar residue Q (glutamine, average 10.7%, minimum 3.3%) were dispersed among hydrophobic residues L, I, or V (leucine, isoleucine, valine, average sum of these residues 21.1%, minimum sum 10.1%) (supplementary table S2, Supplementary Material online). Thus, on average, at least 62.2% of repeats are composed of these residues (K, E, Q, L, I, V), which in turn is highly similar to the viral A-like proteins.
To measure how prevalent these repeats are among structural proteins as well as other proteins categories recovered in the proteome, we designed a bioinformatic screen for equivalent repeats. XSTREAM was used to identify all proteins that contained at least three repeats that are at least 10 residues long. These repeat sequences were then filtered for minimum average amino acid contents K = 6%, E = 6%, Q = 3%, and the sum of L, I, V = 10%. Using these search criteria, 34.4% of the known structural proteins (67 proteins, 5.7% of total proteome) and 28.0% of the novel putative pellicular proteins (148 proteins, 12.6% of total proteome) contain such repeats (fig. 3A). Conversely, only minor portions of the other protein categories contain such repeats—cytosolic metabolism (16 proteins), mitochondria (3 proteins), endomembrane (3 proteins), and nuclear function (13 proteins) (fig. 3A). Thus, the presence of these charged repeat motifs is apparently enriched in structural proteins of the pellicle and also in the novel proteins identified here further suggesting that they are a novel and important component of the ciliate pellicle. For the remainder of this report, we will refer to these proteins as CRMPs.
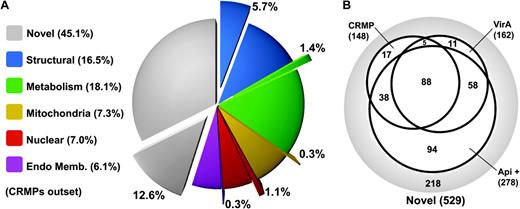
Distribution of CRMPs in Tetrahymena. (A) Distribution of CRMPs (outset wedges) in each of the functional categories from the Tetrahymena pellicle proteome (total percentage of each functional category is shown in the key). (B) Venn diagram of Tetrahymena novel proteins that are identified as CRMPs share Blast matches with viral A-like proteins and share Blast matches with apicomplexan proteins (Api +).
Intriguingly, there is substantial overlap between CRMPs and pellicular proteins with high sequence similarity to viral A-like proteins; 93 of the CRMP proteins (62.8%) are viral A-like proteins, and thus, 57.4% of the viral A-like proteins are CRMPs (fig. 3B). Therefore, there is broad overlap between the charged repeat motifs, identified from the known structural proteins and those proteins bearing similarity to the so-called viral A-like proteins. The higher portion of CRMPs (as well as the viral A-like proteins) in the “novel proteins,” compared with the other proteome categories, is consistent with numerous further structural proteins of the pellicle occurring among this group. The four GFP-tagged novel proteins that located to the pellicle (fig. 2) were all also identified as CRMPs and/or viral A-like proteins.
Novel CRMPs Are Broadly Conserved in Alveolates
How many putative ciliate pellicular proteins occur in other alveolates? We used the T. thermophila novel pellicular protein set as queries against apicomplexan genomes from Plasmodium falciparum and T. gondii (no genomic data is available for dinoflagellates). 54.4% (278 proteins) of the T. thermophila novel proteins recovered matches to uncharacterized apicomplexan proteins (E value < e−5), which represents 84.4% of the nonciliate-specific proteins. Of the proteins with apicomplexan matches, 66.2% were either identified as CRMPs or viral A-like, and the apicomplexan matches account for all but 23 novel CRMPs (15.5%) and 16 novel viral A-like proteins (9.9%) (fig. 3B). Thus, the overlapping classes of charged and repeat motif proteins are strongly represented in the common proteins of the alveolate phyla ciliates and apicomplexans.
To test if these candidate apicomplexan pellicular proteins identified by sequence similarity to the new T. thermophila pellicular proteins are indeed part of the parasite pellicle, we tagged two T. gondii proteins with C-terminal hemagglutinin (HA) epitopes. These two proteins (TgME49_044470 and TgME49_052880) both correspond to T. thermophila proteins identified as both CRMP and viral A-like proteins. TgME49_044470 shares strongest sequence similarity with T. thermophila protein TTHERM_00474830 shown above to locate to the oral apparatus (fig. 2J). It also shares strong similarity with a further ten of the T. thermophila novel proteins. TgME49_052880 corresponds to two T. thermophila proteins and itself contains several repetitive motifs similar to those of CRMPs. In T. gondii cells, both of these proteins are located at the cell periphery, specifically at the apical end of the parasite in close proximity to the conoid structure of the apical complex (fig. 4). TgME49_44470 is located in a ring-like structure at the apical end of the inner membrane complex (fig. 4A–I). We performed deoxycholate extractions to isolate the T. gondii pellicle (Mann and Beckers 2001) and colabeled TgME49_44470 with an antibody against tubulin to visualize the conoid and subpellicular microtubules. This revealed that TgME49_44470 appears to be located at one and possibly two rings at the apical complex (fig. 4G–I). These polar rings may represent two of three such rings identified in electron micrographs (see fig. 1). These structures are also apparent at the apical complex of newly formed daughter cells (fig. 4B and E). TgME49_44470 appears at a very early stage of daughter cell formation before any further development of the daughter cell pellicle is evident (fig. 4B and C). Thus, this protein is associated with a very early event of cell pellicle assembly.
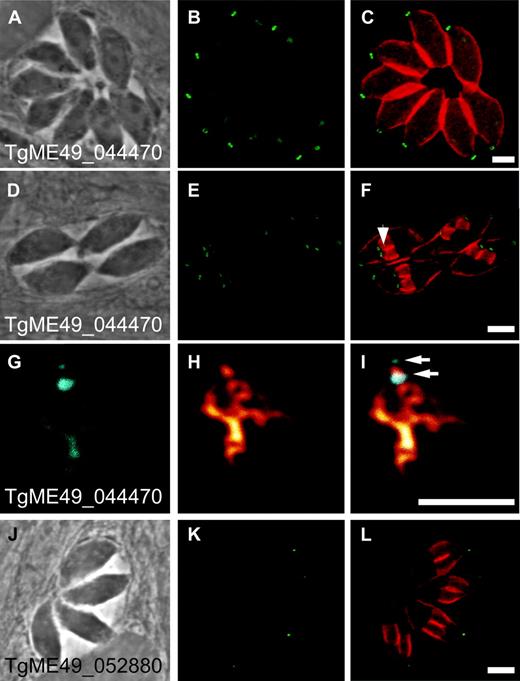
Location of novel pellicular proteins in Toxoplasma gondii. HA-tagged endogenous genes for TgME49_044470 (A–I) and TgME49_052880 (J–L) are immunostained (green/cyan). Cells are counterstained for the pellicle protein alveolin (orange-red, C, F, L). During cell division daughter cells assemble within the parent cell (endodyogeny), and the developing cell pellicles of daughter cells are evident in F (arrowhead) and L. Extracted pellicles (G–I) are counterstained for tubulin (orange, H, I) showing conoid and subpellicular microtubules, and the position of the two conoid-associated rings (arrows) labeled with TgME49_044470.
The second T. gondii protein, TgME49_052880, is also located at the apex of the cell in close vicinity of the conoid, but in this case forms a smaller focus than the rings formed by TgME49_044470 (fig. 4J and K). The expression pattern of TgME49_052880 is also very different to that of TgME49_044470, in that TgME49_052880 only appears in mature cells and is not present in the daughter cells formation (fig. 4K). TgME49_052880 therefore plays a different role to TgME49_044470. While TgME49_044470 was also identified in a proteomic study of the conoid (although its location was not independently verified), TgME49_052880 was not identified in that conoid study (Hu et al. 2006). Our identification of two new proteins that contribute exclusively to elements of the complex apicomplexan pellicle validates our approach of using ciliate proteomics to identify alveolate pellicle proteins.
Rapid but Constrained Evolution of CRMPs
Blast comparison of CRMPs among the various alveolate genome databases revealed two further conspicuous observations: 1) that this category of proteins can rapidly diversify by gene multiplication into small families of paralogs and 2) that evolution of the repetitive regions occurs in a peculiarly constrained manner. When the T. thermophila novel pellicle proteins were searched against either Plasmodium or Toxoplasma protein databases, multiple T. thermophila proteins often corresponded to a single Toxoplasma or Plasmodium protein. For example, the tagged Toxoplasma proteins TgME49_044470 and TgME49_052880 correspond to 11 and two T. thermophila proteins, respectively, and in one case, 33 T. thermophila proteins corresponded to a single T. gondii protein. This implies that, in such cases, CRMP genes have multiplied in T. thermophila subsequent to divergence of the two phyla (alternatively multiple paralogs would have had to have been lost in apicomplexa). Comparison of CRMPs among apicomplexa similarly indicates that the equivalent CRMPs are represented by different numbers of paralogs in these different taxa. These observations are consistent with CRMPS having broad utility and being able to undergo expansion and specialization to meet the needs for pellicular complexity in different alveolates.
A glimpse into the microevolution of CRMPs within closely related taxa offers an even more intriguing view of the evolutionary behavior of these proteins. Toxoplasma gondii is most closely related to Neospora caninum and shares considerable gene synteny. Thus, conservation of sequence similarity and, in most cases, gene order enables unambiguous assignment of orthology. When two CRMP orthologs are compared, we observe a remarkable disparity between the evolution of the repetitive versus nonrepetitive regions of the proteins. For example, figure 5 shows two protein orthologs (T. gondii, TGME49_048570; N. caninum, NCLIV_064740) each with a repetitive region (shown in blue/pink) flanked by nonrepetitive termini. The N- and C-termini show strong primary sequence conservation, and the position of the repetitive region is also strongly conserved (fig. 5). Within the repetitive regions, however, the repeat consensuses show no primary sequence conservation between these orthologs. They are, however, conserved in: 1) repeat length, 2) approximate repeat number, 3) level of sequence identity shared among the repeats, and 4) amino acid composition of the repeats (fig. 5). Therefore, as the primary sequence of the repeats has evolved, all copies of the repeats have changed in unison (or within a short time). Furthermore, these changed repeats have not strayed outside of the conserved amino acid composition that we observe generally for CRMPs. This trend in evolution of the repetitive regions of CRMP homologs was observed for several proteins investigated. Although the mechanism for this very constrained form of molecular evolution is unclear, it implies strong selective forces acting on these repeat motifs.
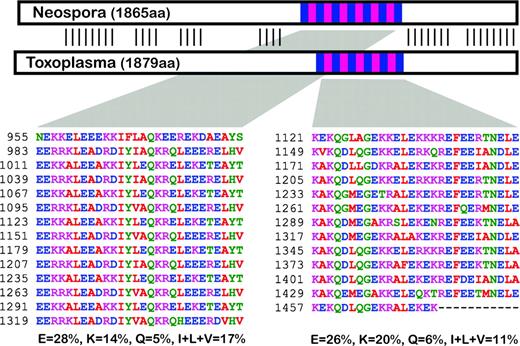
Comparison of two CRMP (Viral A-like) orthologs in closely related apicomplexans Neospora caninum (NCLIV_064740) and Toxoplasma gondii (TGME49_048570). Protein schematic shows repetitive regions (blue/pink) and nonrepetitive regions (white) with strong primary sequence similarity indicated by verticals lines. Repetitive sequence is shown for both proteins and the corresponding amino acid composition of these regions for residues E, K, Q, I, L, and V.
Conclusions: An Extended Molecular Nexus Unifying Alveolate Pellicles
The pellicle of alveolate organisms is a complex arrangement of three layers of membranes, subpellicular microtubules, many other specialized structures and compartments (e.g., trichocysts, oral apparatus, apical complex), and a membrane skeleton acting as structure. The first molecular nexus uniting the Infrakingdom Alveolata was the alveolin protein family that associates with the alveolar sacs of the pellicle and is characterized by short repeat elements (Gould et al. 2008). This study has extended this nexus identifying numerous further common proteins, many of which contain particular repeat motifs enriched in distinctive charged and hydrophobic amino acids. Localization of such newly identified proteins in both Tetrahymena and Toxoplasma confirmed that they reside exclusively within the pellicular structures in both phyla. It is intriguing that both Toxoplasma proteins tagged in this study associated with specialist pellicular structures involved in host cell invasion, namely, the apical complex, despite no obvious equivalent structure occurring in ciliates. The apical complex is a unique and defining feature of apicomplexa, and due to its role in parasitism and disease is the subject of intensive research. Here, two novel proteins of this apparatus have been identified solely by their similarity to CRMPs found associated with the pellicular structure in a ciliate. Moreover, the new Tetrahymena protein that is located exclusively in the oral apparatus is the homolog of one of the conoid proteins of apicomplexa. This suggests that through evolution of the different pellicular structures in the different phyla—for example, apical complexes in apicomplexans, oral apparatuses in ciliates—these common types of proteins have continued to be implicated in the divergent structures, and this might in turn point to common basic mechanisms of their organization and assembly. Our working hypothesis is that CRMPs are an adaptable group of structural proteins abundant in the pellicle of alveolates; their propensity for gene amplification and divergence in a lineage-specific manner, including hyperevolution of the repetitive motifs, is consistent with such a role.
The structural and functional complexity of the pellicle observed throughout alveolate phyla and the potential broad role for charged repeat motifs within these structures suggested that CRMPs might be a very conspicuous category of proteins in this important taxonomic group. To determine if alveolates are enriched in these types of proteins, we applied the CRMP search algorithm (exactly as applied to the T. thermophila proteome) to alveolate genomes and nonalveolate single-celled eukaryote genomes. Although gene number varies considerably among different eukaryotes, ciliates and apicomplexans consistently have more CRMPs and/or a higher percentage of CRMPs as a portion of the genome (table 1). Although it cannot be predicted what the expression pattern might be for these gene sets, this analysis does suggest that CRMPs are a conspicuously enlarged feature of alveolates. Other protists, such as Trichomonas vaginalis, also apparently share many of these types of proteins. This is consistent with the annotation of numerous T. vaginalis proteins as viral A-type inclusion proteins and the characterization of at least one such protein associating with cytoskeletal structures in this cell. The articulins of euglenoids are a further example of CRMPs that occur in other eukaryotic groups (Marrs and Bouck 1992). Thus, these data suggest that CRMPs as a general feature are not unique to alveolates; however, their expansion in this group might indicate that they have been instrumental in the establishment of the common pellicular architecture and the further development of complexity.
CRMPs Found in Complete Genome Sequence Data for Diverse Single-Celled Eukaryotes.
 |
|
CRMPs Found in Complete Genome Sequence Data for Diverse Single-Celled Eukaryotes.
|
|
At this point, the function of the individual repeats remains uncertain, but we hypothesize that the repeats are involved in extending interacting surface areas that mediate specific protein–protein interactions. The enriched residues of the repeats E, K, and Q are implicated in promoting intrinsically disordered protein structures capable of dynamically responding to interaction partners (Radivojac et al. 2007). Furthermore, the repeat regions are strongly predicted to form coiled coils (http://www.ch.embnet.org/software/COILS_form.html). Thus, the formation of induced coil-based interactions could form the rigid, detergent, and alkali-resistant protein structures recovered in the pellicle preparations (this study and [Mann and Beckers 2001]) and provide a dynamic flexible network required for these complex cells. We note that, in this study, CRMP motifs have been identified not only in structural proteins (e.g., alveolins, flagellar spoke proteins, epiplasmins, and TTHERM_00128280 from this study that might itself forms filaments [fig. 2L]) but also on metabolic proteins, such as molecular motors (e.g., kinesins and dyneins). We also observe that some CRMPs occur in structures early and sometimes transiently in their development (e.g., TTHERM_00188980 in nascent oral apparatus and TgME49_044470 in establishing the growing pellicle of daughter cells) and therefore might perform important roles in recruiting and coordinating other proteins to these structures. Thus, these repeats may not be confined only to structural proteins but underpin the coordination of many pellicular functions associated with these diverse eukaryotes.
Supplementary Material
Supplementary figure S1 and Supplementary Data–Supplementary Data are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
This project was supported by a Discovery Project Grant (DP0664097) from the Australian Research Council (ARC). S.B.G. was supported by a German Research Foundation fellowship. We are grateful to Jacek Gaertig for assistance with Tetrahymena transformation. K.L.F., A.M.C., and A.B. acknowledge support of funding from the Australian Centre for Plant Functional Genomics. G.I.M. is an ARC Federation Fellow and a HHMI International Scholar. Program grant support from the National Health and Research Council is acknowledged.
References
Author notes
Present address: Heinrich Heine University, Düsseldorf, Germany.
Associate editor: Martin Embley

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}