





Abstract
Viruses are notoriously capable of evolving resistance to drugs. However, if the endpoint of resistance evolution is only partial escape, a feasible strategy should be to stack drugs, so the combined effect of partial inhibition by several drugs results in net inhibition. Assessing the feasibility of this approach requires quantitative data on viral fitness before and after evolution of resistance to a drug, as done here with bacteriophage T7. An inhibitory gene expressed from a phage promoter aborts wild-type T7 infections. The effect is so severe that the phage population declines when exposed to the inhibitor but expands a billion-fold per hour in its absence. In prior work, T7 evolved modest resistance to this inhibitor, an expected result. Given the nature of the inhibitor, that it used the phage's own promoter to target the phage's destruction, we anticipated that resistance evolution would be limited as the phage may need to evolve a new regulatory system, with simultaneous changes in its RNA polymerase (RNAP) and many of its promoters to fully escape inhibition. We show here that further adaptation of the partially resistant phage led to complete resistance. Resistance evolution was due to three mutations in the RNAP gene and two other genes; unexpectedly, no changes were observed in promoters. Consideration of other mechanisms of T7 inhibition leaves hope that permanent inhibition of viral growth with drugs can in principle be achieved.
Introduction
Drugs are one of two main defenses against viral infections, but drug resistance seems ubiquitous (Richman 1996). Despite considerable effort to understand resistance evolution against some drugs, few insights to thwarting resistance evolution have been discovered. One of the biggest successes is combined drug therapy, now so widely used against HIV that it has its own acronym, HAART (Martinez-Cajas and Wainberg 2008), but which had its origins in the treatment of tuberculosis with antibiotics (Ryan 1993). Yet resistance to drugs continues despite combination therapy, so alternatives are needed. Instead of blocking viral evolution, it may be feasible to allow it. If drugs can be identified for which only partial resistance evolution is possible, then a combination of multiple inhibitors may collectively effect viral suppression even though resistance has evolved.
In using this approach, the critical issue is to employ inhibitors for which only partial resistance is attainable. For example, combined drug therapy with HIV does not lead to obvious viral suppression once resistance has evolved to the different drugs, so resistance evolution to those drugs is so complete as to render the drugs ineffective even in combination. Likewise, although bacterial resistance to antibiotics often involves a fitness cost, subsequent compensatory evolution largely overcomes that cost (Schrag et al. 1997). A priori, strong inhibitors should be good candidates, if only because the impact on the virus is large and hence offering more opportunity to trap viral fitness short of full recovery. Knowledge of how the inhibitor works and how resistance would be achieved may be useful in predicting the extent of recovery.
An inhibitor that a priori seems to possess suitable characteristics for preventing complete viral resistance is available for the bacteriophage T7. This inhibitor employs a key element of the phage regulatory system against the phage. Unlike most phages, T7 carries its own RNA polymerase (RNAP) and a set of 17 promoters recognized by the phage RNAP that are collectively essential for viability (Molineux 2006). Cell-lethal genes have been cloned into plasmid vectors that express the gene from a consensus T7 promoter (Bull et al. 2001), and infection by T7 leads to cell death before the phage produces progeny (abortive infection). These inhibitors are strong, and they present the virus with a “cruel bind” by using a promoter that is both abundant in and essential to the phage to target the phage's destruction. Viral escape thus should be difficult. In previous work, we observed that T7 evolved partial resistance to those inhibitors (Bull et al. 2001). The purpose of the present study was to assess the quantitative limit of viral resistance evolution during extended adaptation. The RNAP interacts with 17 promoters in the genome and at least 2 phage proteins, and it is the most connected protein in the phage genome network. It thus seemed likely that these constraints would limit resistance evolution to a level below that of a virus unaffected by the inhibitor. Alternatively, resistance may require so many changes that full recovery cannot be attained in a short-term adaptation.
Materials and Methods
Strains
The bacterial host was IJ1133 (E. coli K-12 ΔlacX74 thiDΔ[mcrC-mrr]102::Tn10), a strain lacking type I and other restriction loci (Garcia and Molineux 1996). Inhibition of phage T7 was effected by plasmid pSB2, containing the EcoKI restriction enzyme subunit hsdR transcribed from the strong T7 promoter ø10 in vector pT7-7 (Webb et al. 1996). The HsdR subunit of EcoKI carries the nuclease and DNA translocase activities of EcoK1 but is not known to possess any enzyme activity in the absence of the HsdM and HsdS subunits. Nevertheless, pSB2 inhibits T7 growth in restriction-minus hosts lacking the hsd locus such as IJ1133. The mechanism is unknown. A second inhibitory plasmid was used to assay fitness of the evolved phages. Plasmid pS105a52g (Smith et al. 1998) carries a modified version of the phage λ holin gene (S) under control of the T7 ø10 promoter. In previous work, this plasmid was shown to inhibit growth of wild-type T7. The mechanism is presumed to be premature disruption of the cell's membrane potential, different from that underlying the lethality of pSB2 (Bull et al. 2001).
T7 Biology
One of the early genes expressed after T7 infection is a phage genome-specific RNAP. This enzyme is responsible for all late gene expression, priming of origin-specific DNA replication, and initiation of DNA packaging (Molineux 2006). It therefore clearly provides essential functions that cannot be circumvented by alternative pathways. The enzyme recognizes specific 23-bp sequences on the phage genome that are classified as replication, class II and class III promoters. The five class III promoters (including ø10, the promoter controlling expression of the plasmid-borne inhibitory genes used here) and the rightmost replication promoter are completely conserved (identical) in sequence and are considered the strongest. The class III promoters direct expression of all the structural and morphogenetic genes of the phage, whose products are overall those required in largest amounts for phage propagation. In fact, a deletion of ø10 prevents phage growth unless a downstream terminator is also removed (Molineux IJ, unpublished observation).
T3 wild-type (T3+), T7+, and T7groR1 were the three phage strains used. T3+ is a relative of T7, with similar genome organization and homologs of all essential genes as well as of most nonessential genes. However, its RNAP is only weakly active on T7 promoters, so T3 can serve as a control phage that is not inhibited by pSB2. T7+ is highly sensitive to pSB2; T7groR1 is a phage exhibiting partial resistance to pSB2, having been grown on this inhibitor in a prior study (Bull et al. 2001). The descendent of T7groR1 that was adapted to the inhibitor in the present work is designated T7groR1E, for “evolved.”
Passage Conditions and Fitness Assays
Phage were propagated on IJ1133(pSB2) grown in Luria-Bertani broth (10 g NaCl, 10 g Bacto Tryptone, and 5 g Bacto yeast extract per liter) supplemented with 100 mg/ml ampicillin to maintain the plasmid. Cells from frozen aliquots were inoculated into 10-ml cultures in 125-ml flasks, grown for 1 h at 37 °C to a density of approximately 108 cells/ml. Typically, 105 phage were added, grown 30–60 min (until the phage density had reached 107/ml), then transferred directly to the next flask. The phage density was occasionally allowed to accumulate until the entire cell culture lysed, resulting in a period with high multiplicity of infection (MOI) that facilitates recombination between different phage infecting the same host. The transfer volume was adjusted to contain approximately 105 phage, but higher phage numbers were transferred when phage fitness was low. A sample from the finished flask was treated with chloroform and stored. Transfers from one culture to the next on the same day were not treated with chloroform, but the chloroform-treated sample from the last transfer on 1 day was used to start the first transfer on the subsequent day. The accumulated adaptation of T7groR1E conducted for this study involved 49 transfers for a cumulative time of 30 h beyond T7groR1. T3+ was grown for a cumulative time of 10.5 h (21 transfers); although it was not expected to be affected by pSB2, it was allowed to adapt to IJ1133(pSB2) and the passage conditions to overcome other factors that might affect its fitness. Cells with the pSB2 plasmid were grown without induction, whereas those with pS105a52g were induced with 1 mM isopropyl β-D-1-thiogalactopyranoside 10 min before phage addition.
Fitness is the log2 change in phage density per hour under the conditions used for passage and was measured across multiple, consecutive transfers; a single fitness estimate was determined from the titer at the end of the first infected culture to the end of at least one other, to avoid effects of life-cycle synchronization that occur when phage are added to the first culture. Phage density in fitness assays was usually much lower than during passage conditions (never exceeding 0.1 MOI), thus minimizing coinfection of the same host by multiple phages.
Identification of Resistance-Conferring Changes
Nucleotide changes were determined with Sanger sequencing on an ABI3100 from an isolate of the final phage population of T7groR1E and compared with that of wild-type T7. To determine which mutations conferred resistance to pSB2, as opposed to enhancing growth independently of the inhibitor, we used a recombination-outgrowth assay (Rokyta et al. 2002; Bull et al. 2007): An isolate of the adapted T7groR1E was recombined with wild-type T7+ and the mix grown on IJ1133 lacking pSB2 for 10 h. By starting with approximately 50% frequency of each evolved mutation, selection should rapidly resolve the beneficial from the nonbeneficial. The outgrowth environment lacked the inhibitory plasmid, so any mutation ascending under these conditions does not specifically incur resistance to pSB2 and could have evolved in the absence of the plasmid. Those mutations are thus not considered to be strictly resistance mutations. Note that this assay tends to resolve resistance mutations as those with a deleterious effect in the absence of the inhibitor; mutations with weak effects in the absence of the inhibitor (and perhaps those closely linked to a mutation beneficial in the absence of the plasmid) can be misclassified. This outgrowth population was sequenced over the sites of changes.
Results and Discussion
Our initial T7groR1 was a descendant of T7+, adapted to pSB2 in the same E. coli K12 host used here. Prior to the present study, no attempt was made to formally quantify the level of resistance of either phage or to evolve it to maximum resistance (Bull et al. 2001 reported efficiency of plating as the only fitness measure), so the present study provides the first formal quantification of fitness. As measured here, T7+ is so inhibited by pSB2 that it exhibits slight negative growth (−2.1 doublings/h) compared with the T3 control, which grows at 30 doublings/h (230 or a billion-fold increase in titer per hour; fig. 1). The previously adapted T7groR1 had evolved substantial resistance at 10 doublings/h (1,000-fold increase per hour above wild type) but remained a million-fold per hour below the T3 upper limit.
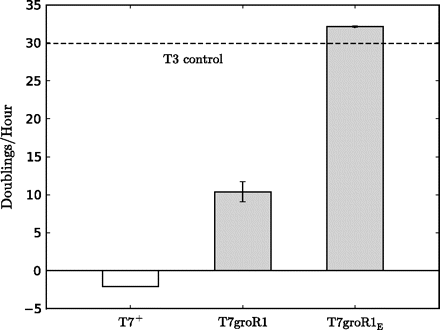
Fitness (doublings/hour) of T7+, T7groR1, and T7groR1E on the inhibitory host. The latter achieved a fitness maximum similar to the control, phage T3 adapted to this host. T3 should not be inhibited by the plasmid because T3 transcribes only weakly from T7 promoters, but some T3 adaptation to host and growth conditions is feasible. Bars indicate 95% confidence intervals.
Here, we further adapted T7groR1 to pSB2. Our final T7groR1E completely escaped the inhibitory effects of pSB2, with a fitness slightly exceeding that of the T3+ control. The gain in fitness of T7groR1E over its partially resistant ancestor T7groR1 is profound at 20 doublings/h. The fact that the fitness of T7groR1E slightly exceeded that of T3 is not necessarily surprising, as a moderate superiority of T7 has been observed in a study in which both T3+ and T7+ were adapted to the same host (Bull et al. 2004). Thus, T3 (or any other phage) should be considered a semiquantitative control.
The course of this evolution might have followed any of a few pathways. As one possible mechanism for the initial evolution, the resistance observed in groR1 might have operated by detoxifying or otherwise blocking synthesis of the inhibitory protein encoded by the plasmid. Instead it reduced transcription of the plasmid-borne inhibitor (Bull et al. 2001). From this intermediate, there are several possibilities for the evolution of groR1E, depending largely on the genetic basis of the intermediate fitness of groR1. Two theoretical pathways are those proposed above: detoxification or preventing synthesis of the inhibitor.
One different idea borrows from the standard model for the evolution of bacterial antibiotic resistance (Schrag et al. 1997; Lenski 1998): groR1 might have already achieved complete avoidance of expression from the plasmid promoter but had low fitness because the resistance mutations had pleiotropic effects that impaired the phage RNAP and thus reduced expression from phage promoters. Evolution of higher fitness in groR1E would then have been compensatory, restoring RNAP activity on phage promoters while maintaining avoidance of expression from the plasmid. The possibility that groR1 had achieved full suppression of the inhibitory plasmid was not supported by reporter gene expression data, which showed only a moderate decrease (Bull et al. 2001), but the resolution of that assay is limited at low expression levels. However, fitnesses of groR1 and groR1E are virtually the same (28.9 ± 0.6, 29.7 ± 0.5, respectively) on IJ1133 lacking inhibitory plasmids (“intrinsic” fitness). This result indicates that the substantial, further evolution of groR1E fitness on pSB2 was not directly compensatory for a low intrinsic fitness of groR1. We note, however, that the intrinsic fitness of both groR1 and groR1E is much lower than T7+ (35.6) before any adaptation to IJ1133 (T7+ can evolve to a fitness of 47.9, Heineman and Bull 2007).
Second, the fitness increase from groR1 to groR1E was tested on a second inhibitory plasmid (pS105a52g). If evolution of groR1E was specific to the toxic gene in pSB2, rather than to the expression vector, then the fitness of groR1E should show little gain on this second inhibitor. Instead, the fitness gain on pS105a52g is profound and is nearly the same as on pSB2 (11.9 ± 1.04 for groR1, 27.4 ± 0.9 for groR1E). These data thus suggest that the evolution of groR1E from groR1 further reduced gene expression from the T7 promoter on the plasmid. Furthermore, the data argue against the detoxification or prevention of synthesis ideas, as these are expected to be gene specific.
Sequencing of the complete genome of a T7groR1E isolate revealed several substitutions, five of which appear by the recombination-outgrowth assay to be specific to growth in the presence of pSB2 and seven that confer a general advantage to phage growth in the absence of the inhibitor (table 1). Of the five resistance substitutions, two had been present in T7groR1. Three of the five occurred in the RNAP gene, two of them new to T7groR1E. One present in both T7groR1 and T7groR1E but discovered here occurred in an internal core protein important for genome entry. A substitution unique to T7groR1E was found in the tail fiber gene. Perhaps surprisingly, and despite three changes in the RNAP gene that presumably affected polymerase expression from the plasmid-borne promoter, there were no substitutions in any of the 17 phage promoters. Likewise, two T7 genes degrade host DNA and might have been expected to evolve to hasten plasmid degradation, but no resistance mutations were observed in them.
Locations and Genetic Characteristics of Five Adaptive Changes in T7groR1E That Were Specifically Beneficial to the Inhibitory Plasmid
| Gene (Function) | Position | Changea | groR1 | groR1E |
| 1 (RNAP) | 4,417 | T→C F416S | + | + |
| 5,479 | A→G D770G | + | ||
| 5,485 | A→G H772R | + | ||
| 16 (Core protein) | 30,719 | C→A P619H | + | + |
| 17 (Tail fiber) | 35,411 | C→T A263V | + |
| Gene (Function) | Position | Changea | groR1 | groR1E |
| 1 (RNAP) | 4,417 | T→C F416S | + | + |
| 5,479 | A→G D770G | + | ||
| 5,485 | A→G H772R | + | ||
| 16 (Core protein) | 30,719 | C→A P619H | + | + |
| 17 (Tail fiber) | 35,411 | C→T A263V | + |
In addition, seven changes in T7groR1E were not specific to the inhibitor pSB2 and thus are not considered resistance mutations: 1,855* A→G (0.6B), 2,515* T→G (0.7), 2,664 T→C (0.7), 2,708–2,993 deletion (0.7), 8,750 T→C (1.8), 36,389* A→G (17.5), and 36,462* T→C (17.5); an asterisk indicates that the mutation was also present in the immediate ancestor T7groR1.
Locations and Genetic Characteristics of Five Adaptive Changes in T7groR1E That Were Specifically Beneficial to the Inhibitory Plasmid
| Gene (Function) | Position | Changea | groR1 | groR1E |
| 1 (RNAP) | 4,417 | T→C F416S | + | + |
| 5,479 | A→G D770G | + | ||
| 5,485 | A→G H772R | + | ||
| 16 (Core protein) | 30,719 | C→A P619H | + | + |
| 17 (Tail fiber) | 35,411 | C→T A263V | + |
| Gene (Function) | Position | Changea | groR1 | groR1E |
| 1 (RNAP) | 4,417 | T→C F416S | + | + |
| 5,479 | A→G D770G | + | ||
| 5,485 | A→G H772R | + | ||
| 16 (Core protein) | 30,719 | C→A P619H | + | + |
| 17 (Tail fiber) | 35,411 | C→T A263V | + |
In addition, seven changes in T7groR1E were not specific to the inhibitor pSB2 and thus are not considered resistance mutations: 1,855* A→G (0.6B), 2,515* T→G (0.7), 2,664 T→C (0.7), 2,708–2,993 deletion (0.7), 8,750 T→C (1.8), 36,389* A→G (17.5), and 36,462* T→C (17.5); an asterisk indicates that the mutation was also present in the immediate ancestor T7groR1.
The largest gain in fitness (from T7groR1 to T7groR1E) was thus due to three resistance mutations—a pair of mutations in the RNAP gene, a single change in the tail fiber gene—and possibly changes in two genes that were not specifically resistant to the inhibitor (from table 1). There is no a priori basis for expecting resistance changes in either the internal core protein or the tail fiber. The change in gp16 was present in T7groR1 but had not been detected in our earlier study, but the efficiency of plating assays with recombinant genomes suggested that the major fitness gains were associated with the portion of the genome that carries the RNAP gene, rather than with the portion carrying late genes, coding for virion structural proteins.
Additional changes to the RNAP gene between T7groR1 and T7groR1E were perhaps expected, based on the discovery of gene 1 changes in several independent adaptations from the earlier study (Bull et al. 2001) and given that the RNAP physically binds to phage promoter sequences. In our previous study, where growth in the presence of pSB2 and pS105a52g was selected, we isolated several different mutations affecting T7 RNAP. Two of these, D770G (present in groR1, which was used in this work) and E775G both lie close to the promoter recognition helix but distal to its binding region. Nevertheless, we had suggested that the altered RNAPs, perhaps in conjunction with other mutant proteins, may be able to discriminate between promoters present on supercoiled plasmid DNAs and promoters on relaxed, linear phage DNA. Adaptation of groR1 to yield groR1E revealed a second change in RNAP: H772R. H772 also lies very close to DNA downstream of the enzyme's active site; the clustering of different mutations in this region strongly suggests that transcription is affected, but perhaps more at the stage of elongation than initiation.
We considered the possibility that the speed of transcription by T7 RNAP might be reduced by the D770G and H772R substitutions, hypothesizing that a slower RNAP could reduce the rate of synthesis of the inhibitor and thereby allow phage escape. The cost to the phage would be a reduced rate of synthesis of its own proteins, which could extend the latent period and reduce intrinsic fitness of groR1E. Lysis of groR1E- infected IJ1133 is indeed slightly longer than after T7+ infection (data not shown), but the lack of a more appropriate control phage or host prevents us from making this a strong conclusion.
Strategies for Permanent Viral Inhibition
The present study raises the specter that T7 can evolve to substantially overcome many if not most types of inhibitors. In light of this foreboding result, we can expand our perspective and search for generalities among other experimental adaptations using T7, studies that include inhibition by modifications of the host as well as modifications of the phage genome. A big advantage of T7 is that the mechanistic basis of inhibition and of viral escape is often understood. Such an understanding may be essential to the design or at least choice of better inhibitors, although the present study illustrates the difficulty of prediction even when a priori information is available.
A naive evolutionary perspective provides one foundation for predicting the ease or difficulty of resistance evolution: the phylogenetic breadth of inhibition. A priori, an inhibitor that is effective against a group of related viruses (hence is phylogenetically broad) should be less prone to viral resistance evolution than one specific to one or a few viruses. On a molecular level, we expect that more mutations will be required to overcome a phylogenetically broad inhibitor than to overcome a phylogenetically narrow one, and perhaps that the level of resistance attainable will decline as the phylogenetic breadth of the inhibitor increases.
Studies of or otherwise relevant to inhibition of T7 may be grouped into several categories based on the genetic nature of the inhibition.
1. Viral-driven suicide gene expression. The present study falls into this category. Prior studies with a Lactoccus phage and with T7 established that phage evolution could at least partly overcome this type of inhibition with single mutations (Djordjevic and Klaenhammer 1997; Bull et al. 2001). Resistance evolution led to complete recovery here. This result is surprising given that the inhibitor uses the consensus phage promoter sequence, whose expression in the phage genome is absolutely essential. The inhibitor should thus have confronted the phage with a “cruel bind.” The ability of T7 to evolve resistance might be argued from the fact that inhibition is phylogenetically narrow—the inhibition does not operate against relatives of T7. However, this logic was not thought to apply with this type of inhibitor, because the RNAP should not be able to evolve without simultaneously changing promoters. Given that promoter changes were not required, evolution of complete resistance is not necessarily surprising, but we could not have anticipated that promoter changes were not required.
2. Essential host factors. Some host factors are required by the phage but not by the host, and elimination of them from the host inhibits the virus. With respect to T7 and relatives, the host protein thioredoxin is required by the phage to render its DNA polymerase processive (Qimron et al. 2006). This requirement is absolute—no phage progeny are produced in the absence of thioredoxin. Furthermore, neither T7 nor its relatives have been observed to escape this requirement (Molineux IJ, unpublished observations). Thus, phylogenetic breadth coincides with lack of resistance evolution in this case. We might thus anticipate that blocking viral access to such an essential host factor would lead to permanent inhibition, but this possibility remains to be tested.
3. Restriction-Modification (RM) systems. The classic mechanism of phage inhibition is RM, in which cellular enzymes bind to unmethylated, specific-recognition sequences in the DNA and cleave the DNA. The first T7 gene introduced into the cell protects against type I RM systems, but the phage has no defense against type II RM systems, except that its genome lacks common 6-base type II recognition sequences (a close relative does carry a defense against type I and type II systems, however). Phage genomes with some but few RM sites occasionally escape restriction and become methylated, so that they epigenetically “evolve” resistance (they immediately lose this methylation-based resistance when grown in a host lacking the RM system). Genomes with many RM sites remain vulnerable and should not typically evolve resistance, because many changes are required simultaneously (Korona et al. 1993; Korona and Levin 1993). The long-term evolution of T7 in response to RM has not been studied in detail, but the opportunity is provided by the RM system of phage P1 lysogens, because T7 carries many sites restricted by that RM system.
The fate of viral resistance evolution against RM systems may defy any rule of phylogenetic breadth. Because of redundancy in the genetic code, genomes of related phages may differ greatly in the number of sites recognized by a specific restriction enzyme without having much impact on peptide sequences. For example, although T7 and T3 each have many EcoP15 sites, in T7 but not T3, they are oriented in the same direction, so the genome would not be cleaved. T7 has only 6 GATC sites, whereas the relative SP6 has 53.
4. Viral traps. Dennehy et al. (2007) proposed adding sensitive but nonpermissive hosts to a culture of permissive cells. The nonpermissive hosts act as traps, because they can be infected but produce no progeny. If the traps are sufficiently common, the viral population will decline even though permissive hosts are present. One advantage of this system is that no inhibitor per se need be designed, rather any nonpermissive host that can be infected will do, and one could potentially chose from a wide range of nonpermissive cell types. However, a vast excess of the trap may be required: If the two hosts have the same adsorption rates to the virus, viral eradication requires that trap cells must outnumber permissive cells by a factor of nearly the burst size (equations in Bull 2006). Furthermore, there is strong selection for viral variants that avoid the nonpermissive host (Bull 2006; Heineman et al. 2008), and this avoidance—a form of resistance—has been shown to evolve in T7 under much less extreme conditions than nonpermissive hosts (Heineman et al. 2008) although not in isometric phages (Guyader and Burch 2008). This form of inhibition will thus usually be phylogenetically narrow, and resistance evolution is expected to be easy.
A variety of naturally occurring antiphage mechanisms are known from bacteria, and several have been introduced into industrial bacteria to control phage growth (Sturino and Klaenhammer 2006). In other cases, inhibitory cassettes have been engineered from natural components. Some appear to offer at least temporary avoidance of phage resistance evolution, possibly permanent resistance, although efforts to evolve resistant phages have not been heroic (Sturino and Klaenhammer 2006).
In the last couple of decades, it has become apparent that evolutionary principles are relevant to medicine, especially in the treatment of infectious diseases (Williams and Nesse 1991; Nesse and Williams 1998; Bull and Wichman 2001). It is now to the point that evolutionary principles are being used to propose drug design strategies (e.g., Pepper 2008). The present study is a caution that predictions based solely on principles may be naive. In our case, the rational design of an inhibitor, based on detailed biochemical information about a virus, resulted in only temporary inhibition in which a far simpler mechanism of resistance resulted in complete escape than was deemed possible. This is not to suggest that failure is assured, and indeed, the review of cases for T7 offers hope that permanent inhibition is attainable. Rather the lesson is that empirical tests are still required to discover how evolution works in individual cases.
We thank Rachael Springman for assistance with various technical aspects of the study and Whitney Yin for suggestions on the likely effects of the mutations isolated in this work. We are grateful to Eric Davidson for the gift of a plasmid and Andy Ellington for use of some equipment. This work was supported by NIH GM57756 to J.J.B., G.M. 32095 to I.J.M.; J.J.B. also receives support as the Miescher Regents Professor.
References
Author notes
Manolo Gouy, Associate Editor

{kind=link}