





Abstract
The broad-spectrum mildew resistance genes RPW8.1 and RPW8.2 define a unique type of plant disease resistance (R) gene, and so far homologous sequences have been found in Arabidopsis thaliana only, which suggests a recent origin. In addition to RPW8.1 and RPW8.2, the RPW8 locus contains three homologs of RPW8, HR1, HR2, and HR3, which do not contribute to powdery mildew resistance. To investigate whether RPW8 has originated recently, and if so the processes involved, we have isolated and analyzed the syntenic RPW8 loci from Arabidopsis lyrata, and from Brassica rapa and B. oleracea. The A. lyrata locus contains four genes orthologous to HR1, HR2, HR3, and RPW8.2, respectively. Two syntenic loci have been characterized in Brassica; one locus contains three genes and is present in both B. oleracea and B. rapa, and the other locus contains a single gene and is detected in B. rapa only. The Brassica homologs have highest similarity to HR3. Sequence analyses suggested that the RPW8 gene family in Brassicaceae originated from an HR3-like ancestor gene through a series of duplications and that RPW8.1 and RPW8.2 evolved from functional diversification through positive selection several MYA. Examination of the sequence polymorphism of 32 A. thaliana accessions at the RPW8 locus and their disease reaction phenotypes revealed that the polymorphic RPW8 locus defines a major source of resistance to powdery mildew diseases. A possible evolutionary mechanism by which functional polymorphism at the AtRPW8 locus has been maintained in contemporary populations of A. thaliana is discussed.
Introduction
Members of plant species typically exhibit genetic variation in resistance and susceptibility to a particular disease. This variation is determined at polymorphic loci for disease resistance (R) genes (Staskawicz et al. 1995, Hammond Kosack and Jones 1997). In recent years, at least five different classes of R genes that confer resistance to a wide range of pathogens have been isolated (Dangl and Jones 2001). These genes provide an opportunity for plant biologists to address fundamental questions such as how plant R genes evolved and how they are maintained as polymorphic loci in natural populations.
The origin of a given R gene may be traced back to the earliest possible common ancestor that possessed it. The largest class of plant R genes encode proteins that contain a nucleotide binding site (NBS) and leucine-rich repeats (LRR). About 150 genes predicted to code for NBS and LRR motifs are found in the genome of Arabidopsis thaliana (Richly, Kurth, and Leister 2002; Meyers et al. 2003), and they have been detected in most other plant species (Cannon et al. 2002). The broad occurrence of NBS-LRR genes in plants indicates an ancient origin, and this hypothesis is further supported by the homology of NBS-LRR gene products to animal proteins involved in the regulation of apoptosis and/or innate immunity (van der Biezen and Jones 1998; Aravind, Dixit, and Koonin 1999; Dangl and Jones 2001; Staskawicz et al. 2001). However, it remains to be resolved more precisely when and how the NBS-LRR genes evolved into plant-specific R genes. In contrast, other classes of R genes have so far been functionally characterized only in a specific plant species or genus, even though homologous genes do exist in other plant species. For example, Pto of tomato (Martin et al. 1993) and Xa21 (Song et al. 1995) and a more recently cloned Xa21-like gene, Xa26 (Sun et al. 2004) of rice remain, respectively, as the only member(s) of the Pto-like kinase family and the Xa21-like receptor-like kinase family that have been characterized as naturally polymorphic R genes from the same or different plant species. This finding raises the possibility that some classes of R genes may have evolved recently in specific plant lineages via functional diversification of genes not originally specifying disease resistance.
The majority of previous evolutionary analyses of R genes have examined sequence divergence of gene family members within a genome (Cannon et al. 2002; Baumgarten et al. 2003; Meyers et al. 2003). Some analyses have focused on polymorphism at specific, characterized R loci to understand the evolutionary mechanisms by which the polymorphism is maintained in the contemporary populations (Caicedo, Schaal, and Kunkel 1999; Noël et al. 1999; Stahl et al. 1999). Two contrasting models for R gene evolution have arisen from these analyses. They are the frequency-dependent (balancing) selection model and the arms race model. According to the frequency-dependent selection model, polymorphism at R gene loci is maintained through a balance between selection for enhanced fitness in the presence of the pathogen and selection against a postulated reduced fitness in the absence of the pathogen. A feature of this model is that R genes could be ancient but present in only a proportion of the population. Stahl et al. (1999) reported that functional alleles of the RPM1 locus of A. thaliana have apparently coexisted with nonfunctional alleles for millions of years and that RPM1 is present in only 52% of accessions examined, which indicates that RPM1 is subject to frequency-dependent selection. More recently, RPM1 has been shown to be associated with a cost of resistance in the absence of the pathogen (Tian et al. 2003), which may explain the absence of RPM1 in some Arabidopsis accessions.
According to the arms race model, novel R genes arise in response to changes in the pathogen, and vice versa, in an interactive process. This model is supported by studies on many LRR-containing genes, such as Cf4/9, Xa21, RPP8, RPP5, Dm3, and L/M. (Anderson et al. 1997; Parniske et al. 1997; McDowell et al. 1998; Meyers et al. 1998; Wang et al. 1998; Noël et al. 1999). These studies indicate that the conserved LRR domain is under selection for novel forms that recognize variants of the pathogens. R gene clusters frequently reveal gene duplications and deletions, conversion by unequal crossing over, and intragenic and intergenic recombination, all of which may contribute to the rapid evolution of novel recognition specificities (Michelmore and Meyers 1998; Ellis, Dodds, and Pryor 2000; Holub 2001).
The broad-spectrum powdery mildew resistance genes RPW8.1 and RPW8.2 from A. thaliana accession Ms-0 encode novel proteins with a putative N-terminal transmembrane (TM) domain and a coiled-coil (CC) domain (Xiao et al. 2001). In susceptible accessions, the corresponding alleles specify different amino acid sequences, or the genes are deleted (Xiao et al. 2001). Resistance is expressed by activation of a salicylic acid–dependent and an EDS1-dependent pathway that is also used by a subset of the ancient NBS-LRR class of R genes that encode proteins that contain an N-terminal domain homologous to the Toll/interleukin receptor (Aarts et al. 1998; Xiao et al. 2001). Although sequence homologous to RPW8.1 and RPW8.2 has only been detected in A. thaliana, the ArabidopsisRPW8.1 and RPW8.2 genes also confer powdery mildew resistance in the unrelated genus Nicotiana (Xiao et al. 2003b), which indicates that the gene products can activate presumably conserved disease resistance signaling pathways. These observations raise the fundamental questions of how this locus arose and how it has been maintained in contemporary populations.
In this analysis, we aim to reconstruct the evolutionary history of the RPW8 locus by examining the sequence polymorphism at the syntenic RPW8 loci of four related species in Brassicaceae and the intraspecific sequence variation at RPW8.1 and RPW8.2 in A. thaliana. We show evidence that RPW8.1 and RPW8.2 evolved from a series of duplications of HR3, a candidate progenitor gene of the RPW8 gene family that is adjacent to RPW8.2, through diversifying selection in Arabidopsis after its separation from Brassica. We also reveal that the Ms-0 alleles of RPW8.1 and RPW8.2 constitute the major genetic resource of powdery mildew resistance in A. thaliana. We suggest that a likely mechanism for the maintenance of polymorphism for functional and nonfunctional genotypes at the RPW8 locus in contemporary populations of A. thaliana is balancing selection for a benefit of resistance against a cost of resistance.
Materials and Methods
Plant Materials, Pathogen Infection, and Phenotypic Examination
Seeds of Arabidopsis lyrata accessions were obtained from Charles Langley (University of California, Davis). Brassica oleracea ssp. Alboglabra cv. A12 and B. rapa ssp. Oleifera cv. R018 were used for BAC library construction and isolation of RPW8 homologs and for disease tests with powdery mildew isolates Erysiphe cruciferarum UEA1, E. cichoracearum UCSC1, E. orontii MGH, and Oidium lycopersici Oxford. Pathogen inoculation and disease scoring methods were the same as previously reported (Xiao et al. 2001).
Isolation of RPW8 Homologs from A. lyrata, B. oleracea, and B. rapa
Gene-specific primers (sequence available on request) were used to amplify the orthologous genes from A. lyrata by use of high-fidelity Taq polymerase (Roche). PCR products were sequenced. Intergenic regions of the AlRPW8 cluster of accession 99m9 were amplified by high-fidelity Taq polymerase with new primers designed against the A. lyrata homologs. PCR fragments were cloned into pGEM-T easy vector (Promega) and subsequently sequenced. The sequence of the A. lyrata orthologous genes were corrected by sequencing PCR products amplified with primers from flanking A. lyrata–specific sequence. For isolation of Brassica homologs, AtRPW8.1 and AtRPW8.2 genomic DNA were used as probes to screen a BAC library of B. oleracea ssp. Alboglabra var. A12 (O'Neill and Bancroft 2000) and a BAC library of B. rapa ssp. Oleifera var. R018 (unpublished). Filters of the two BAC libraries were incubated at 50°C for overnight and washed three times with 2 × SSC, 0.1% SDS. Positive BAC clones were fingerprinted by digestion of approximately 2 μg DNA from each clone with EcoRI and BamHI, followed by subsequent gel separation. The gel was also blotted and hybridized with 32P-labeled AtRPW8.1 and AtRPW8.2. Three positive fragments of a BAC clone (39H13), and one positive fragment of a different BAC clone (93M24) of B. rapa were recovered, cloned, and sequenced. The joining points of the three fragments were confirmed by PCR with specific primers and sequencing the PCR products. The orthologs of B. oleracea were amplified from positive BAC clone (25K1) of B. oleracea with primers specific to the B. rapa homologs. PCR products were then sequenced. Sequences have been submitted to GenBank under the following accession numbers: B. rapaRPW8 locus, AY225586; BrHRd, AY225590; BoHRa, AY225587; BoHRb, AY225588; BoHRc, AY225589; and A. lyrataRPW8 locus, AY225591.
Transcripts Analysis
RNA was extracted by use of the RNeasy kit (QIAGEN) from leaves before and 8 days after inoculation with powdery mildew pathogens. For this procedure, 4-week-old plants of A. thaliana accessions Col-0, Ler, and Ms-0 and 6-week-old plants of A. lyrata accession 99m9 were inoculated with E. cichoracearum UCSC1 and 8-week-old plants of B. oleracea ssp. Alboglabra cv. A12 and B. rapa ssp. Oleifera cv. R018 were inoculated with E. crucifereaum UEA1. First-strand cDNA was synthesized by use of Superscript II (Gibco-BRL). A 10-fold dilution of the cDNA solution was used for PCR amplification (20 s at 94°C, 20 s at 60°C, and 40 s at 72°C; 32 cycles). The PCR products were cloned and sequenced.
Sequence Analysis
Homology analysis was done by application of the “gap” or “best-fit” function of the GCG package at http://www.hgmp.mrc.ac.uk. Nucleotide sequences of the RPW8 genes were aligned by ClustalW (Thompson, Higgins, and Gibson 1994) and were improved manually. Intron data were excluded from phylogenetic analyses. Synonymous and nonsynonymous substitution rates (Ka and Ks) between sequences were calculated for an alignment of 369 nucleotides after gapped sites were removed. Calculations were made by the yn00 program of the PAML version 3.11 software (Yang 1997), which implemented the method of Yang and Nielsen (Yang and Nielsen 2000) and allowed for both transition and codon usage biases. Subsequent to these analyses the CODEML program of the PAML software was used to test hypotheses of positive diversifying selection derived from the results of the yn00 analyses. This program enables one to construct several models that allow for different levels of heterogeneity in the Ka/Ks ratio among lineages. The simplest model assumes the same ratio for all branches in a phylogeny. The most general model assumes independent Ka/Ks ratio for each branch in a phylogeny. Here, we use the likelihood ratio test to compare models involving independent Ka/Ks ratios for branches believed to be evolving under positive diversifying selection against a series of null models for each lineage having a Ka/Ks ratio of 1 or less. The tree topology used for the CODEML analyses is that derived from our phylogenetic analyses.
The fit of the data to 56 models of base substitution was tested by application of Modeltest version 3.06 (Posada and Crandall 1998), which made use of parameter estimates and likelihood values obtained for each model by PAUP* (Swofford 1998). Modeltest uses the log-likelihood ratio tests and the Akaike information criterion (AIC) (Akaike 1973) to determine which of the models best describes the data. These parameters were then used in subsequent maximum-likelihood (ML) phylogenetic analyses. All phylogenetic analyses were performed by PAUP* version 4.0b10 (Swofford 1998) with gapped sites excluded by both ML and maximum parsimony (MP) with the branch and bound search option. All bootstrap analyses were performed with 1,000 replicates. To estimate the divergence time, we first calculated the synonymous substitution rate within the RPW8 gene family on the basis of the divergence time of 14 to 21MYA for the separation of Brassica and Arabidopsis lineages (Yang et al. 1999; Koch, Haubold, and Mitchell-Olds 2000). Synonymous mutation rate estimates were calculated as an average of all pairwise estimates for sequence comparisons that traverse the Brassica-Arabidopsis divergence by application of the formula ds/2T = synonymous mutation rate (T = divergence time). For a divergence time of 14 MYA, the synonymous substitution rate is 1.12 × 10−8 substitutions per site per year, and for a divergence time of 21 MYA the rate is 7.57 × 10−9 substitutions per site per year. One the basis of these substitution rates, we estimated the ages of key nodes within the phylogeny. For each node, the average of all pairwise synonymous divergences descending from the node was used to estimate the age of the divergence event.
Results
Gene Organization of the Syntenic RPW8 Loci of Arabidopsis and Brassica Species
The RPW8 locus of powdery mildew–resistant A. thaliana accession Ms-0 contains AtRPW8.1 and AtRPW8.2 and three homologs of RPW8, AtHR1, AtHR2, and AtHR3 (AF273059 [fig. 1]) (Xiao et al. 2001). The RPW8 locus of a mildew-susceptible A. thaliana accession Col-0 also contains AtHR1, AtHR2, AtHR3 but lacks AtRPW8.1 and AtRPW8.2, and in their place is a new homolog, AtHR4 (AL133363 [fig. 1]). In the two accessions, the RPW8 locus was flanked by a gene (At3g50440) that showed homology to rice pir7a and by a serine/threonine protein kinase gene, AtSPK-2 (At3g50500) (fig. 1). Sequence analysis of other A. thaliana accessions indicated that the Ms-0 and Col-0 genotypes represent two basic haplotypes at the RPW8 locus (see later text). Thus, the RPW8 gene family of A. thaliana contains at least six members.
Comparison by Blast2 and GCG gap revealed that Ms-0 and Col-0 differ only in the interval from 1,332 bp from the telomeric side of the AtHR3 stop codon to 234 bp centromeric to the AtSPK-2 stop codon and are otherwise nearly identical (>90% identity) in the other regions of the locus (fig. 1). In Ms-0, the divergent sequence is 5,518 bp and contains AtRPW8.1, AtRPW8.2, and an approximately 860-bp highly repetitive sequence (denoted “A” in figure 1). In Col-0, the divergent sequence is 10,077 bp and contains AtHR4 and a 632-bp sequence (At3g50490, “Retro” in figure 1) that encodes a pseudoreverse transcriptase highly homologous to the polyprotein-like retroelements BAB09878 and AAC32926 (E > 10−66), which belong to the long-terminal repeat (LTR)–type of retrotransposons. Blast searches showed that a fragment of 3,642 bp (denoted “B” in figure 1) containing the putative retrotransposon had at least 40 near-identical (E value > 10−100) sequences in the Arabidopsis genome, which indicates that this sequence is related to an active or past-active LTR-type retrotransposon (Le et al. 2000). Within the divergent 5,518-bp sequence of Ms-0 and the 10,077-bp sequence of Col-0, there are also six 220-bp to 460-bp segments that have 75% to 95% homology between Ms-0 and Col-0 (fig. 1).
Dot plots revealed two imperfect direct repeats (95% identity), 266 bp and 268 bp, respectively, in the intergenic region of AtHR1 and AtHR2 in Ms-0 and Col-0 (denoted “C” in figure 1). A Blast search identified 10 sequences with significant homology (E > 10−70) to this repeat, two of which (AC007178 from chromosome 2 and AC023279 from chromosome 1) also contain the approximately 860-bp repetitive sequence in Ms-0 (A in figure 1) with only a 40-bp to 109-bp gap, but not the retrotransposon identified in Col-0 (B in fig. 1). This finding suggests that these two types of repeats (A and C in figure 1) may have been associated with each other before the occurrence of the retrotransposon in the Col-0 haplotype.
To examine the origin of AtRPW8.1 and AtRPW8.2 at the RPW8 locus, we identified and determined the sequences of the syntenic RPW8 loci in A. thaliana and the related species, A. lyrata, Brassica oleracea, and B. rapa. In each species, the RPW8 locus is flanked by homologs of the genes pir7a and SPK-2 (fig. 1).
The RPW8 locus from A. lyrata contains four genes that are apparently orthologous to AtHR1, AtHR2, AtHR3, and AtRPW8.2 and is more compact in size (fig. 1). The orthologs are more similar (81.2% to 92.9% identity at nucleotide level) between A. thaliana and A. lyrata than are the paralogs (48.7% to 74.3%) within each species (see table 1 in Supplementary Material online), and the intergenic regions from A. thaliana and A. lyrata have little or no homology.
An ortholog of AtRPW8.1 is absent from the AlRPW8 locus. Whether this condition indicates the deletion of RPW8.1 from A. lyrata or the later formation of RPW8.1 in A. thaliana after the separation of these two Arabidopsis species cannot be inferred from these sequence data. However, RPW8.2 had clearly evolved before the separation of A. thaliana from A. lyrata. This finding indicates a deletion of AtRPW8.2 in the Col-0 haplotype in a later event, which provides direct evidence that the Ms-0 haplotype is older than the Col-0 haplotype. Moreover, the AtRPW8.2 gene in Ms-0 is flanked by small duplicated sequences, which are contiguous in Col-0 (fig. 1). This finding suggests that AtRPW8.2 may have been deleted by an unequal recombination event during the emergence of Col-0. Thus, it seems likely that the present AtRPW8 locus of the Col-0 haplotype evolved from an Ms-0-like progenitor haplotype type after a series of sequence rearrangements that included an insertion of a retrotransposon that eventually resulted in the deletion of AtRPW8.2 and transformation of AtRPW8.1 into AtHR4.
The RPW8 loci of B. rapa and B. oleracea each contain three RPW8 homologs without clear orthology to the Arabidopsis genes, and we named these homologs HRa, HRb, and HRc. BrHRa, BrHRb, and BrHRc are highly homologous (≥92%) to each other and to the corresponding orthologs at the locus in B. oleracea (96.4% to 99.7%). The intergenic regions were also similar (85.8%), which indicates a recent origin of this gene cluster. These six BrassicaRPW8 genes have 79% to 80% nucleotide sequence identity to AtHR3 and AlHR3, but their sequence identity to other ArabidopsisRPW8 homologs is lower and ranges from 50% to 74%, which suggests that the BrassicaRPW8 locus arose from duplications of an HR3-like gene. The B. rapa genome also contains an additional locus that consists of a single homolog of RPW8, which we named BrHRd (fig. 1). HRd was not detected in the B. oleracea genome by Southern analysis (data not shown). Interestingly, BrHRd is also most similar to Al/AtHR3 (73.8%) (see table 1 in Supplementary Material online). These results suggested that the RPW8 locus in Brassica and Arabidopsis resulted from independent duplications of an HR3-like progenitor gene flanked by SPK-2 and pir7a.
The RPW8 Gene Family Members Are Divergent in Sequence but Conserved in Structure, and Most of Them Are Expressed
DNA sequence analysis, confirmed by sequencing of the reverse transcriptase-polymerase chain reaction (RT-PCR) products (see below), revealed that the 17 members of the RPW8 gene family in A. thaliana, A. lyrata, B. oleraceae, and B. rapa vary in size, from 644 bp (AtRPW8.1) to 1158 bp (BrHRd), but each contains two exons split by a phase 2 intron where an arginine codon (AG¦G) is completely conserved (table 1). Fourteen of the 17 genes have a 296-bp coding sequence for the first exon (CD1 in table 1). The size of the second exon (CD2) varies considerably from 166 bp (AtRPW8.1) to 355 bp (BoHRb and BrHRb). The size of the intron also varies from 104 bp (BoHRc) to 543 bp (BrHRd).
Except for AlHR1, which contains a G to T substitution at 37 bp after the start codon and results in an early stop codon, the remaining 16 members were predicted to encode peptides of different size ranging from 148 amino acids (aa) (AtRPW8.1) to 216 aa (BoHRb and BrHR). Alignments of the nucleotide sequences (not shown) and predicted peptide sequences (fig. 2) showed that the degree of homology between the 17 genes varied considerably, particularly at the C-termini. The N-terminal amino acids encoded by the CD1 of all the family members contained a putative TM domain at positions of approximately 1 to 30 aa, and all except for AtRPW8.1 and AtHR4 also contained a CC domain (at positions of approximately 63 to 97 aa). AtHR4 is the most divergent member of the family, and it has five 42-bp imperfect tandem repeats (GATACAAGTCC/GACCAATGGAC/TCGATATCAAAGAAATGAAGGC, encoding IQVH/DQWT/IDIKEMKA) at its 3′ end, indicative of intragenic duplications (fig. 2).
Expressed sequence tag (EST) databases were searched for homologous sequences of the 17 members of the RPW8 family, but only three ESTs, all representing transcripts of AtHR4, were found (GenBank accession numbers C99860, C99861, and C99864). To determine whether other RPW8 family members were expressed, RNA was extracted from leaves of A. thaliana accessions Ms-0 and Col-0, and Ler (which contains divergent AtRPW8.1 and AtRPW8.2 alleles [see table 2]), both before and 8 days after inoculation with E. cichoracearum UCSC1. cDNA products were isolated by RT-PCR with gene-specific primers. cDNAs corresponding to each of the RPW8 family members were detected in samples before and after inoculation with the pathogen, with the expected exception that cDNAs for AtRPW8.1 and AtRPW8.2 could not be detected in Col-0, and cDNA for AtHR4 could not be detected in Ms-0 and Ler (fig. 3A). Similar RT-PCR experiments with A. lyrata plants detected amplified cDNA products from AlHR3 and AlRPW8.2 but not from AlHR1 (which contains an early stop codon and may be a pseudogene) and from AlHR2 under our experimental conditions (fig. 3B). The cDNA products amplified with primers based on the sequences of either HRa or HRb or HRc from B. oleracea or B. rapa contained a mixture of cDNA of these three genes that were indistinguishable in size (fig. 3C). Cloning and sequencing of the cDNA products revealed that all three genes were expressed in B. oleracea and B. rapa plants, before and after inoculation with E. cruciferarum UEA1. Similarly, cDNA of BrHRd was detected in B. rapa, before and after challenge with E. cruciferarum UEA1 (data not shown). Sequence analysis of the amplified cDNAs together with their corresponding genomic DNA confirmed the exon-intron splicing sites as predicted in table 1.
RPW8.1 and RPW8.2 Evolved from Gene Duplication and Subsequent Functional Diversification via Positive Selection in Arabidopsis
A phylogenetic tree of the 17 members of the RPW8 gene family was constructed, and divergence times were estimated (fig. 4) on the basis of (1) the rate of synonymous substitution, (2) the split of the Arabidopsis and Brassica lineages, which has been estimated to have happened between 14 and 21 MYA (Yang et al. 1999; Koch, Haubold, and Mitchell-Olds 2000). The age of the first gene duplication event within this gene family (node A in figure 4) was estimated to be 23 to 34 MYA. This indicates the antiquity of the gene family, the origin of which predates the evolutionary split of the plant lineages leading to Brassica and Arabidopsis. From the topology of the tree and information on gene organization (fig. 1) it can be concluded that HR1 and HR2 orthologs were deleted from the locus within the Brassica lineage after the split of Arabidopsis and Brassica. The origin of the functional R genes AtRPW8.1 and AtRPW8.2 is estimated to be 10 to 15 MYA, when the lineage giving rise to AtRPW8.1, AtRPW8.2, AlRPW8.2, and AtHR4 first emerged (node C in figure 4). RPW8.1 was probably deleted in A. lyrata after the split that formed the two Arabidopsis species. The subsequent emergence of AtHR4 (and probably the deletion of AtRPW8.2) in A. thaliana accession Col-0(node F in figure 4) is estimated to have happened 6 to 10 MYA.
The types of forces that drive natural selection can be inferred from the types of nucleotide substitutions in coding sequences. An informative parameter of the evolution of genes under strong selection, such as those at vertebrate MHC loci and plant LRR-containing R genes (Hughes and Nei 1988; Nei, Gu, and Sitnikova 1997; Parniske et al. 1997; Michelmore and Meyers 1998; Bergelson et al. 2001), has been the ratio of the rate of nonsynonymous substitution (Ka) that causes an amino acid change to the rate of synonymous substitution (Ks) that does not. We, therefore, used the parameter Ka/Ks to assess whether duplicated genes at the RPW8 locus may have evolved by purifying selection (Ka/Ks < 1) that conserved the amino acid sequence, or neutral selection (Ka/Ks ≈ 1) that had no constraint for sequence divergence, or diversifying selection (Ka/Ks > 1) that led to different peptides.
The Ka/Ks ratio estimated by CODEML under the one-ratio model for all branches on the tree (see Materials and Methods) was 0.81, which indicates close to neutral selection (see table 1 in Supplementary Material online). The log-likelihood value in this model is −595.14. However, the individual pairwise Ka/Ks ratios among the 17 genes calculated by yn00 (see Materials and Methods) varied from 0.17 (AlHR3/AtHR3) to 2.81 (AtRPW8.1/AlRPW8.2). We do not include the Ka/Ks ratios for the Brassica homologs here, because these sequences have very low nucleotide divergences, and the ratios may, therefore, be unreliable (Yang and Neilson 2000). The average Ka/Ks ratio between all combinations of HR1, HR2, and HR3 pairs was 0.36 ± 0.18, whereas the average Ka/Ks ratio between HR3, RPW8.1, RPW8.2, and HR4 was 1.88 ± 0.54 (table 3, and table 1 in Supplementary Material online). On the basis of the Ka/Ks data, HR1, HR2, and HR3 have been under purifying selection that has preserved the amino acid sequences of these genes. This finding suggests that these genes may have a conserved function. AlHR3/AtHR3 has the lowest Ka/Ks ratio (0.17), which indicates that HR3 has been subject to the strongest purifying selection. Further supporting this conclusion, analysis of the nucleotide sequences of AtHR3 from nine A. thaliana accessions showed that the predicted amino acid sequences were identical (table 2). In contrast, RPW8.1, RPW8.2, and HR4 appear to have been subject to diversifying selection that resulted in novel amino acid sequences with respect to each other and to HR3. To test this hypothesis, we first estimated Ka/Ks ratios under an eight-ratio model with a different Ka/Ks ratio for each branch between node C (fig. 4) and the genes AtRPW8.1, AtHR4, AtRPW8.2, and AlRPW8.2 and another ratio for all remaining branches. The log-likelihood under this model is −2563.99. We then carried out separate analyses with each of the free-to-vary branches constrained so that Ka/Ks ≤ 1. The likelihood values under the eight-ratio model with and without the constraint Ka/Ks ≤ 1 were then compared, to test whether the branches of interest have a Ka/Ks ratio that is significantly greater than 1. Three branches were identified with Ka/Ks ratios significantly greater than 1 (P < 1% [fig. 4]).
To identify regions of the coding sequence in AtRPW8.1, AtRPW8.2, and AtHR4 that diverged from AtHR3 for novel amino acids, we conducted a sliding-window analysis to show the distribution of the nonsynonymous substitutions along the 369-bp alignable coding sequence among the four genes. As shown in figure 5, although the distribution was quite even, there appeared to have been three regions (I, II, and III in figure 5) where nonsynonymous substitutions were more frequent. The second region approximately corresponds to the site that encodes the CC domain. Relatively high nonsynonymous substitutions in this region might affect the normal function of the CC domain. In fact, extensive mutations abolished the CC domains of AtRPW8.1 and AtHR4. Interestingly, AtRPW8.1 had a different putative CC domain at its C-terminus. We also conducted a sliding-window analysis for AtHR1, AtHR2, and AtHR3 that are suggested to have been under purifying selection. As expected, the overall nonsynonymous substitutions are much less frequent than between AtRPW8.1, AtRPW8.2, AtHR4, and AtHR3 (fig. 5). The region of the CC domain in these three genes was particularly conserved. Interestingly, a region that approximately corresponds to amino acids 45 to 60 in AtHR3 had much higher nonsynonymous substitutions compared with the rest of the coding sequence. Possibly, this local divergence reflects subtle functional differences between AtHR1, AtHR2, and AtHR3.
As was reflected by the protein sequence alignment (fig. 2), the C-termini of the gene family are highly variable compared with the other parts of the genes. We were unable to align the sequences in this region and could not, therefore, incorporate them in Ka/Ks analyses, and yet, these variations might have a major contribution to the functional diversification of this gene family.
RPW8 Defines a Major Powdery Mildew Resistance Locus in Arabidopsis thaliana
Approximately 20% of A. thaliana accessions are resistant to powdery mildews (Adam et al. 1999). To examine the role of the RPW8 locus in this resistance, we determined the nucleotide sequences of AtRPW8.1, AtRPW8.2, and AtHR4 in 32 A. thaliana accessions, and results of the amino acid sequence analysis are summarized in table 2. Thirteen accessions that contain the Ms-0 alleles of either AtRPW8.1 or AtRPW8.2 or both (sequence identical in amino acids) were resistant to powdery mildew. Seventeen out of 19 accessions with AtRPW8.1 and AtRPW8.2 alleles that differed in amino acid sequence from the Ms-0 alleles, or in which these genes were deleted, were susceptible. This finding indicated that the Ms-0 alleles of AtRPW8.1 and AtRPW8.2 are the main source of powdery mildew resistance in A. thaliana. It was also noted that eight accessions, including Col-0, that lack AtRPW8.1 and AtRPW8.2 but possess AtHR4 were generally more susceptible than those accessions that contained divergent RPW8.1 and RPW8.2 alleles. Some divergent alleles may, therefore, be partially functional in disease resistance.
Discussion
We describe here the origin of a novel type of R gene in Arabidopsis that has become the main source of resistance to the powdery mildew pathogens. This development of resistance has apparently involved duplications of an ancestral gene similar to HR3. HR3 appears to have been maintained by purifying selection but is not involved in specifying resistance. Functional diversification after gene duplication events gave rise to RPW8.1 and RPW8.2, and the Ms-0 alleles of these confer resistance to powdery mildew pathogens.
The Origin and Maintenance of RPW8 in Arabidopsis
The inability to detect homologs of RPW8 in databases may reflect the incompleteness of sequence data from other genomes. Alternatively, both AtRPW8.1 and AtRPW8.2 evolved recently in the plant lineage that contains Arabidopsis. Recently, an RPW8 homolog has been claimed to be present in the rice genome (Goff et al. 2002). However, no sequence data was given to the claimed rice RPW8 homolog, and we failed to detect any rice sequences with significant homology to RPW8 by Blast search of the public databases.
To examine whether homologs of RPW8 exist in other species, we searched for RPW8 homologs from three close relatives of A. thaliana. We isolated the syntenic RPW8 loci from A. lyrata, B. oleracea, and B. rapa. Our data from gene organization and phylogenetic analyses indicated that AtRPW8.2 indeed evolved in an ancestor of A. lyrata and A. thaliana after divergence from the Brassica lineage and that AtRPW8.1 most likely evolved in Arabidopsis before the speciation of A. thaliana and A. lyrata. AtRPW8.1 was subsequently deleted in the latter. We estimated, on the basis of synonymous substitutions of the RPW8 gene family members, that a duplication of an HR3-like gene between 10 to 15 MYA was the first event in the origin of the RPW8.1 and RPW8.2 genes (node C in figure 4). The discovery of this critical period for the evolution of RPW8.1 and RPW8.2, together with the gene organization data, has enabled us to infer that HR3, the paralogous gene next to RPW8.2, most probably is the progenitor gene of the small RPW8 gene family.
Our data support a model of “gene duplication and diversification” for the birth of Al/AtRPW8.2 and Al/AtRPW8.1, with fortuitous duplication of an HR3-like progenitor gene providing the molecular basis for selection. We presume that one copy (HR3) retained the original function of the progenitor gene, whereas other copy diverged for novel function. Our Ka/Ks analysis showed that HR3 has been subject to purifying selection, whereas Al/AtRPW8.2 and AtRPW8.1 have been subject to diversifying selection, which provides molecular evidence for this model. Preliminary transgenic analysis showed that AlRPW8.2 conferred enhanced resistance in A. thaliana Col-0 to E. cichoracearum UCSC1 (S. Xiao and J. Turner, unpublished data), which suggests that AlRPW8.2 may also be a functional R gene in A. lyrata. Thus, RPW8.2 may have gained resistance function several MYA. It seems likely that selection pressure imposed on Arabidopsis plants by powdery mildew pathogens may have been the main driving force for the sequence diversification of AlRPW8.2, AtRPW8.2, and AtRPW8.1.
We assume that after the genes AlRPW8.2, AtRPW8.2, and AtRPW8.1 had gained resistance function, they must have diversified in the Arabidopsis populations. Our sequence and phenotypic data from 32 A. thaliana accessions (table 2) indicated that polymorphism at the RPW8 locus probably accounts for a large proportion of the genetic variation of powdery mildew resistance among the A. thaliana accessions. This finding is consistent with the results from previous genetic analyses of powdery mildew resistance in three A. thaliana accessions (Xiao et al. 1997; Schiff, Wilson, and Somerville 2001; Wilson et al. 2001). One may reason that these functional R genes have been maintained in the population because they render plants a “benefit-of-resistance” in the presence of the powdery mildew pathogens. However, we found that nine of 11 accessions that contained AtRPW8.1 and AtRPW8.2 alleles divergent from those of Ms-0 at the protein level were susceptible to powdery mildews. Furthermore, seven of eight Col-0–like accessions that lacked AtRPW8.1 and AtRPW8.2 were more susceptible to powdery mildew (table 2). These observations have two implications: (1) some of the divergent alleles may be partially functional, and (2) there has been selection against the functional RPW8 alleles. It is also possible that some of the nonfunctional genes may have simply resulted from drift in the absence of selection for and against RPW8 in situations where the pathogen was absent and the level of RPW8 expression was low. Genetic variation in disease resistance has led to speculation of a “cost-of-resistance” that may be associated with R genes (Bergelson et al. 1996; Purrington 2000; Heil and Baldwin 2002). A cost of resistance has recently been revealed for RPM1 that confers resistance in A. thaliana to Pseudomonas syringae (Tian et al. 2003). Preliminary experiments that compared the fitness of Col-0 wild-type with Col-0 plants that carry a single copy of RPW8.1 and RPW8.2 under control of the native promoters showed that in the absence of powdery mildew pathogens, Col-0 transgenic plants had lower (∼94%) seed yield than those of Col-0 wild-type plants (100%) (Xiao et al. unpublished data). This result suggests that a cost-of-resistance may be associated with expression of AtRPW8.1 and AtRPW8.2. Additional evidence comes from our recent observation that over 30% of the Col-0transgenic plants carrying AtRPW8.1 and AtRPW8.2 under control of the native promoters exhibited reduced stature (compared with Col-0 wild-type) and spontaneous cell death in the absence of the pathogens (Xiao et al. 2003a). Thus, it is conceivable that both a benefit and a cost of resistance are associated with AtRPW8.1 and AtRPW8.2, and it is the balance between these two aspects of the RPW8 phenotypes that forms the driving force in maintaining these two R genes at a polymorphic locus in A. thaliana. This hypothesis conforms to the balancing selection model suggested for evolution of RPM1 and Pto (Stahl et al. 1999; Riely and Martin 2001). However, more comprehensive experiments on cost of resistance are needed to confirm this speculation.
All Col-0–like accessions contain AtHR4, a gene that appears to have arisen from AtRPW8.1, at the position corresponding to AtRPW8.1 and AtRPW8.2 in Ms-0 (fig. 1). Phylogenetic and gene organization data (figs. 1 and 4) indicate that AtHR4 is probably of most recent origin. Although it is the least similar to other members of the RPW8 gene family, it still encodes a full-length ORF and has been subject to diversifying selection (table 3). Interestingly, transcription of AtHR4 is induced by powdery mildew (S. Somerville, personal communication) and Peronospora parasitica (E. Holub, personal communication). Identification of its function in relation to disease resistance would be revealing.
Molecular Mechanisms of DNA Sequence Divergence at the RPW8 Locus
R genes of different types are found in complex loci where multiple homologous genes are clustered together (Hulbert 1997; Parniske et al. 1997; Song et al. 1997; Botella et al. 1998; Meyers et al. 1998; Noël et al. 1999; Riely and Martin 2001; Wei, Wing, and Wise 2002). It has, therefore, been suggested that gene duplication and subsequent sequence diversification plays an important role in evolution of plant R genes (Michelmore and Meyers 1998; Holub 2001). The evolutionary history of the RPW8 locus revealed here reiterates this theme.
Unequal recombination and transposon-mediated duplication events have been implicated for the duplications at some R loci (Song et al. 1997; Sun et al. 2001). The lack of sequence exchanges between the RPW8 homologs at the Al/AtRPW8 locus indicates that unequal crossing over has not contributed significantly to the sequence diversification of these genes. At the Al/AtRPW8 locus, the orthologs are more similar (81% to 93% nucleotide identity) and the paralogs are more divergent (49% to 73%), which indicates the relative stability of this locus. A similar situation has been found at the R loci Dm3 in lettuce and Pto in tomato (Meyers et al. 1998; Riely and Martin 2001).
Repetitive elements at the RPW8 locus are found in other tandem arrays in Arabidopsis. The approximately 860-bp repetitive sequence at the AtRPW8 locus of Ms-0 (denoted “A” in fig. 1) has 53 nearly identical matches (E value = 0), 42 highly homologous sequences (E > 10−100), and 160 homologous sequences (E > 10−30) in the Arabidopsis genome. These matches vary in size from 100 to approximately 860 bp, of which up to 220 bp have 86% identity to Atg2g12440, a non-LTR retrotransposon. Thus, the approximately 860-bp repetitive sequences may be footprints of transposons or generated from unknown genome duplication mechanisms. This finding suggests that the formation of the RPW8 cluster in Ms-0 may have been initiated by duplication of an HR3-like ancestral gene, possibly mediated by a transposon insertion and subsequent excision. In Col-0, the absence of the approximately 860-bp repetitive sequence and the presence of an LTR retrotransposon-like sequence indicate that this latter element may have originated from another, later, transposition event at the RPW8 locus in the Ms-0 haplotype, coincident with the emergence of the Col-0 haplotype. Transposon-related sequences were also found in other R gene loci such as Xa21 (Song et al. 1997), RPP5 (Noël et al. 1999), and Mla (Wei, Wing, and Wise 2002).
The age of AtRPW8.2, AtRPW8.1, and AtHR4 at the AtRPW8 locus was estimated to be no more than 10 to 15 Myr (fig. 4). However, the average sequence identity (nucleotide) between them and their presumed progenitor gene HR3 is only 65.3%, which indicates an accelerated sequence diversification. The high Ka/Ks ratio associated with AtRPW8.1, AtRPW8.2, and AtHR4 suggests that point mutation had played a major role in functional diversification of this small gene family. Point mutations in individual genes are also the primary source of novel polymorphisms in other plant R genes and in vertebrate MHC genes (Nei, Gu, and Sitnikova 1997; Michelmore and Meyers 1998). Transposon-mediated gene deletion (AtRPW8.1 and AtRPW8.2), and intragenic duplications (AtHR4) were probably associated with the formation of the Col-0 haplotype for the AtRPW8 locus. Small deletions and insertions (indels) might also have played an important role, because AtRPW8.1 and AtRPW8.2 are the smallest two members of this family, each of which had deletions of 180 bp and 117 bp, respectively, in the 3′ part of the CD2 compared with AtHR3. This observation implies that the structural changes at the C-termini of these proteins might have contributed to the resistance function of AtRPW8.1 and AtRPW8.2.
Although the nucleotide and predicted protein sequences of the Arabidopsis paralogous members of the RPW8 gene family are highly divergent, their gene structure is conserved. In particular, the phase and position, but not the size of the singular introns, is conserved, which suggests possible selection for the “split” amino acid—a conserved arginine in all RPW8 family members—or the integrity of the splice site (Fichant 1992). In members of other R gene families, intron positions are conserved in N, L6, and RPP5, each of which belongs to TIR-NBS-LRR class of R genes (Parker et al. 1997). Intron position and number are conserved among RGC2 family members at the Dm3 locus (Meyers et al. 1998). Significantly, except for AlHR1, all the RPW8 gene family members encode full-length ORFs and most are also expressed. Thus, they could be active genes maintained by selection. Future work to determine the possible functions of HR1, HR2, HR3, and HR4 may, therefore, reveal the original function of the RPW8 resistance genes.
Formation of the RPW8 Loci in Arabidopsis and Brassica via Independent Duplication Events
Gene deletion was revealed in complex loci such as Cf-4/9 and Pto in different tomato species (Parniske et al. 1997; Riely and Martin 2001). Our data suggest that B. rapa contains two syntenic RPW8 loci, whereas B. oleracea contains one. The common one that contains three HR3-like genes apparently evolved from more recent duplication events within the Brassica evolutionary lineage (figs. 1 and 4). The other locus of B. rapa contains only one HR3-like gene. The formation of Al/AtHR1 and Al/AtHR2 in Arabidopsis was estimated to have resulted from the earliest duplication event of the RPW8 gene family around 23 to 34 MYA, before the Arabidopsis-Brassica split (14 to 21 MYA) (Yang et al. 1999; Koch, Haubold, and Mitchell-Olds 2000). Thus, the HR1 and HR2 must have been deleted from these two loci in the Brassica lineage after the Arabidopsis-Brassica split. It has been hypothesized that one Brassica genome (i.e., B. nigra) evolved from triplication of an Arabidopsis-like progenitor genome (Lagercrantz 1998). An implication from this hypothesis is that the B. rapa and B. oleracea genomes may have contained three syntenic RPW8 loci, one of which has been completely deleted. A precedent for this event is provided by the deletion of the RPM1 ortholog in four of six syntenic loci detected in the B. napus genome (AACC) that is an amphidiploid hybrid of B. rapa (AA) and B. oleracea (CC) (Grant et al. 1998). An additional screening of the Brassica genomic libraries by using the two genes flanking RPW8 locus as probe may help to consolidate this speculation.
Through mutually informative gene organization and molecular phylogenetic data, we have been able to gain an understanding of the origin of RPW8 genes involved in conferring broad-spectrum resistance to powdery mildew in A. thaliana. Future work that investigates the RPW8 locus in other close relatives of Arabidopsis and characterizes the fitness consequences of individual alleles should be illuminating.
Present address: Center for Biosystems Research, University of Maryland Biotechnology Institute, College Park, Maryland.
Geoffrey McFadden, Associate Editor
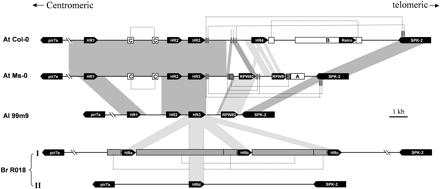
Gene organization of the syntenic RPW8 loci of A. thaliana (At Col-0, AAT20E23; At Ms-0, AF273059), A. lyrata (Al99m9, AY225591), and B. rapa ssp. Oleifera (Br R018, AY225586). Sequences with greater than 90% identity between different genotypes are linked by gray shading and sequences exhibiting 75% to 90% identity are linked by stippled gray. Small stretches of direct duplication (>75% identity) within each cluster are represented by dark gray bars, or by open rectangles if they are repetitive sequences also found in other part of the A. thaliana genome. The duplicated segments are linked by dashed lines. Size differences between duplicated segments are caused by indels. The coding sequences are black rectangles, and the arrows indicate the direction of transcription. SPK-2 is serine-threonine protein kinase 2, identified in A. thaliana (At3g50500). pir7a is a hypothetical gene (At3g50450) that shows homology to rice pir7a. Retro indicates sequence encoding for a pseudoretrotransposon. A indicates an approximately 860-bp repetitive sequence in Ms-0. B indicates a 3,642-bp repetitive sequence in Col-0. C indicates an approximately 266-bp repetitive sequence in Ms-0 and Col-0. The regions beyond “\\” were not sequenced, but the presence of the genes and the physical distance was determined by PCR with specific primers

Alignment of predicted amino acid sequences of the RPW8 gene family. Protein sequences of 14 RPW8 family members (three members of B. oleracea were not included, as they are very similar to their orthologs of B. rapa) are aligned with ClustalW and the alignment is shaded by use of the BOXSHADE WWW server (http://www.ch.embnet.org/software/BOX_form.html). White type on a black background indicates a residue that is identical in at least half of the proteins shown, whereas a gray background indicates conservative substitutions as defined by the BOXSHADE 3.21 default parameters. A short dash (-) indicates a gap. Arrowhead indicates the arginine residue coded by the conserved intron split codon (AG¦G) (see text). Single thick line indicates the putative transmembrane domain. Doubled line indicates the putative coiled-coil domain (except for AtRPW8.1 and AtHR4). Arrows indicate residues identical among the 14 members. Divided line indicates five intragenic duplications in AtHR4 only. Ψ indicates that AlHR1 is a pseudogene, which has a stop codon after the 13th amino acid (indicated by *). Orthologs are linked by solid lines
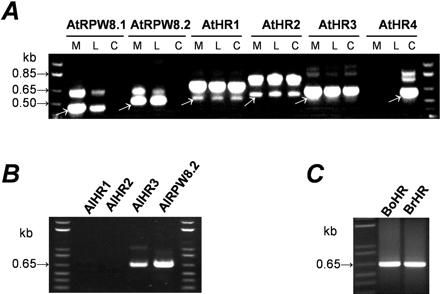
Transcripts analysis of the RPW8 gene family members. RNA was extracted from leaves of plants before and 8 days postinoculation (dpi) of powdery mildews and used for first-strand cDNA synthesis. Ten-fold dilutions of the cDNA were used as template for PCR amplification with gene-specific primers of the indicated RPW8 homologs. Results from pathogen-infected plants were represented. Data for BrHRd was not shown. (A) Transcripts of the A. thaliana family members. Plants were inoculated with E. cichoracearum UCSC1. M indicates Ms-0, L indicates Ler, and C indicates Col-0. White arrows indicate cDNA products. (B) Transcripts of the A. lyrata family members. Plants were inoculated with E. cichoracearum UCSC1. (C) Transcripts of the Brassica family members. Plants were inoculated with E. cruciferarum UEA1
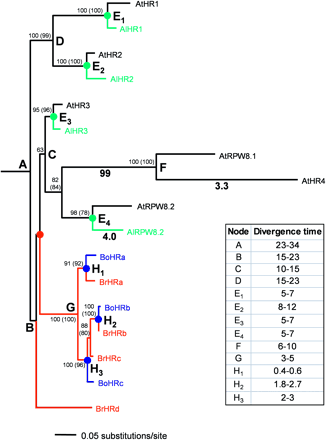
Phylogenetic analysis of RPW8 gene family. A maximum-likelihood tree constructed from 369 alignable coding nucleotides of the RPW8 gene family, with bootstrap support values (100 replicates). Maximum-parsimony analysis arrived at exactly the same topology and comparable branch lengths (bootstrap values shown in brackets). In the absence of an outgroup sequence, the root position of the tree was defined from the mid-point root of a neighbor-joining analysis of synonymous distances. This method removes the problem of substitution rate differences among branches caused by differing selective pressures for amino acid change that could lead to incorrect rooting. Branches and nodes are color-coded to represent genetic divergences arising from speciation events. Red lines and red node denote the divergence between Brassica and Arabidopsis, blue lines and nodes denote speciation between B. rapa and B. oleracea, and green lines and nodes denote speciation between A. thaliana and A. lyrata. Letters at nodes correspond to the divergence time estimates (see Materials and Methods) in the table beside the tree. Numbers under branches are Ka/Ks ratios found to be significantly above 1
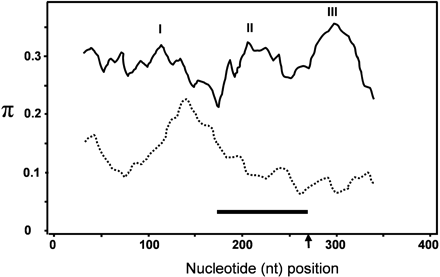
Distribution of nonsynonymous substitutions along the 369 alignable coding sequence of RPW8 family members by application of a sliding window of 50 nt with 5-nt steps (DnaSP3.51). Solid line indicates the estimated nucleotide diversity (π) between AtRPW8.1, AtRPW8.2, AtHR4, and AtHR3, and three regions with higher π values are indicated by I, II, and III. Dashed line indicates the estimated π between AtHR1, AtHR2, and AtHR3. Arrow indicates the split site of the two exons. Bar indicates the approximate nt position of the coiled-coil motif
Size of the RPW8 Gene Family Members.
| Gene | Accession Number | CD1 (nt) | Intron (nt) | CD2 (nt) | Protein (aa) |
|---|---|---|---|---|---|
| AtHR1 | At3g50450 | 296 | 151 | 274 | 189 |
| AtHR2 | At3g50460 | 296 | 191 | 322 | 205 |
| AtHR3 | At3g50470 | 296 | 375 | 346 | 213 |
| AtRPW8.2 | AF273059a | 296 | 128 | 229 | 174 |
| AtRPW8.1 | AF273059a | 281 | 197 | 166 | 148 |
| AtHR4 | At3g50480 | 311 | 291 | 292 | 200 |
| AlHR1b | AY22591a | 296 | 165 | 274 | — |
| AlHR2 | AY22591a | 296 | 204 | 325 | 206 |
| AlHR3 | AY22591a | 296 | 387 | 346 | 213 |
| AlRPW8.2 | AY22591a | 296 | 269 | 349 | 214 |
| BoHRa | AY225587 | 296 | 106 | 340 | 211 |
| BoHRb | AY225588 | 296 | 105 | 355 | 216 |
| BoHRc | AY225589 | 296 | 104 | 352 | 215 |
| BrHRa | AY225586a | 296 | 107 | 343 | 212 |
| BrHRb | AY225586a | 296 | 105 | 355 | 216 |
| BrHRc | AY225586a | 296 | 105 | 352 | 215 |
| BrHRd | AY225590 | 293 | 543 | 322 | 204 |
| Gene | Accession Number | CD1 (nt) | Intron (nt) | CD2 (nt) | Protein (aa) |
|---|---|---|---|---|---|
| AtHR1 | At3g50450 | 296 | 151 | 274 | 189 |
| AtHR2 | At3g50460 | 296 | 191 | 322 | 205 |
| AtHR3 | At3g50470 | 296 | 375 | 346 | 213 |
| AtRPW8.2 | AF273059a | 296 | 128 | 229 | 174 |
| AtRPW8.1 | AF273059a | 281 | 197 | 166 | 148 |
| AtHR4 | At3g50480 | 311 | 291 | 292 | 200 |
| AlHR1b | AY22591a | 296 | 165 | 274 | — |
| AlHR2 | AY22591a | 296 | 204 | 325 | 206 |
| AlHR3 | AY22591a | 296 | 387 | 346 | 213 |
| AlRPW8.2 | AY22591a | 296 | 269 | 349 | 214 |
| BoHRa | AY225587 | 296 | 106 | 340 | 211 |
| BoHRb | AY225588 | 296 | 105 | 355 | 216 |
| BoHRc | AY225589 | 296 | 104 | 352 | 215 |
| BrHRa | AY225586a | 296 | 107 | 343 | 212 |
| BrHRb | AY225586a | 296 | 105 | 355 | 216 |
| BrHRc | AY225586a | 296 | 105 | 352 | 215 |
| BrHRd | AY225590 | 293 | 543 | 322 | 204 |
Note.—Except for the A. thaliana genes, all A. lyrata, B. oleracea, and B. rapa sequences were determined in this analysis. nt = nucleotide; aa = amino acids.
a Accession number assigned to a gene cluster.
b AlHR1 contains a stop codon at the 37-bp position and is pseudogene.
Size of the RPW8 Gene Family Members.
| Gene | Accession Number | CD1 (nt) | Intron (nt) | CD2 (nt) | Protein (aa) |
|---|---|---|---|---|---|
| AtHR1 | At3g50450 | 296 | 151 | 274 | 189 |
| AtHR2 | At3g50460 | 296 | 191 | 322 | 205 |
| AtHR3 | At3g50470 | 296 | 375 | 346 | 213 |
| AtRPW8.2 | AF273059a | 296 | 128 | 229 | 174 |
| AtRPW8.1 | AF273059a | 281 | 197 | 166 | 148 |
| AtHR4 | At3g50480 | 311 | 291 | 292 | 200 |
| AlHR1b | AY22591a | 296 | 165 | 274 | — |
| AlHR2 | AY22591a | 296 | 204 | 325 | 206 |
| AlHR3 | AY22591a | 296 | 387 | 346 | 213 |
| AlRPW8.2 | AY22591a | 296 | 269 | 349 | 214 |
| BoHRa | AY225587 | 296 | 106 | 340 | 211 |
| BoHRb | AY225588 | 296 | 105 | 355 | 216 |
| BoHRc | AY225589 | 296 | 104 | 352 | 215 |
| BrHRa | AY225586a | 296 | 107 | 343 | 212 |
| BrHRb | AY225586a | 296 | 105 | 355 | 216 |
| BrHRc | AY225586a | 296 | 105 | 352 | 215 |
| BrHRd | AY225590 | 293 | 543 | 322 | 204 |
| Gene | Accession Number | CD1 (nt) | Intron (nt) | CD2 (nt) | Protein (aa) |
|---|---|---|---|---|---|
| AtHR1 | At3g50450 | 296 | 151 | 274 | 189 |
| AtHR2 | At3g50460 | 296 | 191 | 322 | 205 |
| AtHR3 | At3g50470 | 296 | 375 | 346 | 213 |
| AtRPW8.2 | AF273059a | 296 | 128 | 229 | 174 |
| AtRPW8.1 | AF273059a | 281 | 197 | 166 | 148 |
| AtHR4 | At3g50480 | 311 | 291 | 292 | 200 |
| AlHR1b | AY22591a | 296 | 165 | 274 | — |
| AlHR2 | AY22591a | 296 | 204 | 325 | 206 |
| AlHR3 | AY22591a | 296 | 387 | 346 | 213 |
| AlRPW8.2 | AY22591a | 296 | 269 | 349 | 214 |
| BoHRa | AY225587 | 296 | 106 | 340 | 211 |
| BoHRb | AY225588 | 296 | 105 | 355 | 216 |
| BoHRc | AY225589 | 296 | 104 | 352 | 215 |
| BrHRa | AY225586a | 296 | 107 | 343 | 212 |
| BrHRb | AY225586a | 296 | 105 | 355 | 216 |
| BrHRc | AY225586a | 296 | 105 | 352 | 215 |
| BrHRd | AY225590 | 293 | 543 | 322 | 204 |
Note.—Except for the A. thaliana genes, all A. lyrata, B. oleracea, and B. rapa sequences were determined in this analysis. nt = nucleotide; aa = amino acids.
a Accession number assigned to a gene cluster.
b AlHR1 contains a stop codon at the 37-bp position and is pseudogene.
Genotypes of A. thaliana Accessions at AtRPW8 and Their Disease Phenotypes.
| Group | Accessiona | AtRPW8.1b | AtRPW8.2b | AtHR4c | AtHR3c | Phenotyped |
|---|---|---|---|---|---|---|
| 1 | Ms-0 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | HR3/Ms-0 | R |
| Kas-1 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | HR3/Ms-0 | R | |
| Wa-1 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | HR3/Ms-0 | R | |
| Ei-5 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| Nw-0 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| Shahdara | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| Sorbo | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| 2 | C24 | RPW8.1/Ms-0 | RPW8.2/C24 | n | — | R |
| Ksk-1 | RPW8.1/Ms-0 | RPW8.2/Ksk-1 | n | — | R | |
| Nok-0 | RPW8.1/Ms-0 | RPW8.2/Nok-1 | n | — | R | |
| Sah-0 | RPW8.1/Ms-0 | RPW8.2/Sah-0 | n | — | R | |
| 3 | Ts-7 | RPW8.1/Ts-7 | RPW8.2/Ms-0 | n | — | R |
| Uk-1 | RPW8.1/Uk-1 | RPW8.2/Ms-0 | n | — | R | |
| 4 | Can-0 | RPW8.1/Can-0 | RPW8.2/Can-0 | n | HR3/Ms-0 | R |
| Fl-1 | RPW8.1/Fl-1 | RPW8.2/Fl-1 | n | — | R | |
| Bu-06 | RPW8.1/Bu-06 | RPW8.2/Sah-0 | n | — | S | |
| Is-0 | RPW8.1/Ler | RPW8.2/Is-0 | n | — | S+ | |
| Kl-0 | RPW8.1/Kl-0 | RPW8.2/Kl-0 | n | — | S | |
| Ler | RPW8.1/Ler | RPW8.2/Ler | n | HR3/Ms-0 | S | |
| Na-1 | RPW8.1/Na-1 | RPW8.2/Fl-1 | n | — | S | |
| Nd-0 | RPW8.1/Can-0 | RPW8.2/Fl-1 | n | HR3/Ms-0 | S | |
| Sy-0 | RPW8.1/Sy-0 | RPW8.2/Sy-0 | n | HR3/Ms-0 | S | |
| Ws-0 | RPW8.1/Bu-06 | RPW8.2/Sy-0 | n | HR3/Ms-0 | S+ | |
| Xx-0 | RPW8.1/Ler | RPW8.2/Is-0 | n | — | S | |
| 5 | Col-0 | n | n | HR4/Col-0 | HR3/Ms-0 | S+ |
| Fr-3 | n | n | HR4/Fr3 | — | S+ | |
| Is-1 | n | n | HR4/Fr3 | — | S+ | |
| Nie-0 | n | n | HR4/Col-0 | — | S | |
| Sg-2 | n | n | HR4/Fr3 | — | S+ | |
| Sh-0 | n | n | HR4/Fr3 | — | S+ | |
| Wc-1 | n | n | HR4/Fr3 | — | S+ | |
| Wt-4 | n | n | HR4/Fr3 | — | S+ |
| Group | Accessiona | AtRPW8.1b | AtRPW8.2b | AtHR4c | AtHR3c | Phenotyped |
|---|---|---|---|---|---|---|
| 1 | Ms-0 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | HR3/Ms-0 | R |
| Kas-1 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | HR3/Ms-0 | R | |
| Wa-1 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | HR3/Ms-0 | R | |
| Ei-5 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| Nw-0 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| Shahdara | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| Sorbo | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| 2 | C24 | RPW8.1/Ms-0 | RPW8.2/C24 | n | — | R |
| Ksk-1 | RPW8.1/Ms-0 | RPW8.2/Ksk-1 | n | — | R | |
| Nok-0 | RPW8.1/Ms-0 | RPW8.2/Nok-1 | n | — | R | |
| Sah-0 | RPW8.1/Ms-0 | RPW8.2/Sah-0 | n | — | R | |
| 3 | Ts-7 | RPW8.1/Ts-7 | RPW8.2/Ms-0 | n | — | R |
| Uk-1 | RPW8.1/Uk-1 | RPW8.2/Ms-0 | n | — | R | |
| 4 | Can-0 | RPW8.1/Can-0 | RPW8.2/Can-0 | n | HR3/Ms-0 | R |
| Fl-1 | RPW8.1/Fl-1 | RPW8.2/Fl-1 | n | — | R | |
| Bu-06 | RPW8.1/Bu-06 | RPW8.2/Sah-0 | n | — | S | |
| Is-0 | RPW8.1/Ler | RPW8.2/Is-0 | n | — | S+ | |
| Kl-0 | RPW8.1/Kl-0 | RPW8.2/Kl-0 | n | — | S | |
| Ler | RPW8.1/Ler | RPW8.2/Ler | n | HR3/Ms-0 | S | |
| Na-1 | RPW8.1/Na-1 | RPW8.2/Fl-1 | n | — | S | |
| Nd-0 | RPW8.1/Can-0 | RPW8.2/Fl-1 | n | HR3/Ms-0 | S | |
| Sy-0 | RPW8.1/Sy-0 | RPW8.2/Sy-0 | n | HR3/Ms-0 | S | |
| Ws-0 | RPW8.1/Bu-06 | RPW8.2/Sy-0 | n | HR3/Ms-0 | S+ | |
| Xx-0 | RPW8.1/Ler | RPW8.2/Is-0 | n | — | S | |
| 5 | Col-0 | n | n | HR4/Col-0 | HR3/Ms-0 | S+ |
| Fr-3 | n | n | HR4/Fr3 | — | S+ | |
| Is-1 | n | n | HR4/Fr3 | — | S+ | |
| Nie-0 | n | n | HR4/Col-0 | — | S | |
| Sg-2 | n | n | HR4/Fr3 | — | S+ | |
| Sh-0 | n | n | HR4/Fr3 | — | S+ | |
| Wc-1 | n | n | HR4/Fr3 | — | S+ | |
| Wt-4 | n | n | HR4/Fr3 | — | S+ |
a All accessions, except for Can-0, were chosen from a previous survey (Adam et al. 1999), and their origins can be found at http://nasc.life.nott.ac.uk/catalogue.html.
b Alleles identical to that of Ms-0 accession at amino acid level are denoted RPW8.1(2)/Ms-0; alleles diverged from that of Ms-0 are denoted with the names of the accessions if unique, or with the name of the accession that appears first in the table if identical at amino acid level. n indicates not present.
c For HR4, the genotypes are either Col-0 or Fr-3 alleles at amino acid level, or not present (n). For HR3, all are identical at amino acid level. Dashes (—) indicate not determined.
d All the accessions (at least five plants for each) were tested with Erysiphe cruciferaum UEA1, E. cichoracearum UCSC1, E. orontii MGH, and Oidium lycopersici Oxford in this study. The phenotypes were scored as either resistant (R), susceptible (S), or very susceptible (S+). As the disease phenotypes of all the accessions to the four isolates were similar, only the phenotypes to E. cichoracearum UCSC1 were presented here.
Genotypes of A. thaliana Accessions at AtRPW8 and Their Disease Phenotypes.
| Group | Accessiona | AtRPW8.1b | AtRPW8.2b | AtHR4c | AtHR3c | Phenotyped |
|---|---|---|---|---|---|---|
| 1 | Ms-0 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | HR3/Ms-0 | R |
| Kas-1 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | HR3/Ms-0 | R | |
| Wa-1 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | HR3/Ms-0 | R | |
| Ei-5 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| Nw-0 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| Shahdara | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| Sorbo | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| 2 | C24 | RPW8.1/Ms-0 | RPW8.2/C24 | n | — | R |
| Ksk-1 | RPW8.1/Ms-0 | RPW8.2/Ksk-1 | n | — | R | |
| Nok-0 | RPW8.1/Ms-0 | RPW8.2/Nok-1 | n | — | R | |
| Sah-0 | RPW8.1/Ms-0 | RPW8.2/Sah-0 | n | — | R | |
| 3 | Ts-7 | RPW8.1/Ts-7 | RPW8.2/Ms-0 | n | — | R |
| Uk-1 | RPW8.1/Uk-1 | RPW8.2/Ms-0 | n | — | R | |
| 4 | Can-0 | RPW8.1/Can-0 | RPW8.2/Can-0 | n | HR3/Ms-0 | R |
| Fl-1 | RPW8.1/Fl-1 | RPW8.2/Fl-1 | n | — | R | |
| Bu-06 | RPW8.1/Bu-06 | RPW8.2/Sah-0 | n | — | S | |
| Is-0 | RPW8.1/Ler | RPW8.2/Is-0 | n | — | S+ | |
| Kl-0 | RPW8.1/Kl-0 | RPW8.2/Kl-0 | n | — | S | |
| Ler | RPW8.1/Ler | RPW8.2/Ler | n | HR3/Ms-0 | S | |
| Na-1 | RPW8.1/Na-1 | RPW8.2/Fl-1 | n | — | S | |
| Nd-0 | RPW8.1/Can-0 | RPW8.2/Fl-1 | n | HR3/Ms-0 | S | |
| Sy-0 | RPW8.1/Sy-0 | RPW8.2/Sy-0 | n | HR3/Ms-0 | S | |
| Ws-0 | RPW8.1/Bu-06 | RPW8.2/Sy-0 | n | HR3/Ms-0 | S+ | |
| Xx-0 | RPW8.1/Ler | RPW8.2/Is-0 | n | — | S | |
| 5 | Col-0 | n | n | HR4/Col-0 | HR3/Ms-0 | S+ |
| Fr-3 | n | n | HR4/Fr3 | — | S+ | |
| Is-1 | n | n | HR4/Fr3 | — | S+ | |
| Nie-0 | n | n | HR4/Col-0 | — | S | |
| Sg-2 | n | n | HR4/Fr3 | — | S+ | |
| Sh-0 | n | n | HR4/Fr3 | — | S+ | |
| Wc-1 | n | n | HR4/Fr3 | — | S+ | |
| Wt-4 | n | n | HR4/Fr3 | — | S+ |
| Group | Accessiona | AtRPW8.1b | AtRPW8.2b | AtHR4c | AtHR3c | Phenotyped |
|---|---|---|---|---|---|---|
| 1 | Ms-0 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | HR3/Ms-0 | R |
| Kas-1 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | HR3/Ms-0 | R | |
| Wa-1 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | HR3/Ms-0 | R | |
| Ei-5 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| Nw-0 | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| Shahdara | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| Sorbo | RPW8.1/Ms-0 | RPW8.2/Ms-0 | n | — | R | |
| 2 | C24 | RPW8.1/Ms-0 | RPW8.2/C24 | n | — | R |
| Ksk-1 | RPW8.1/Ms-0 | RPW8.2/Ksk-1 | n | — | R | |
| Nok-0 | RPW8.1/Ms-0 | RPW8.2/Nok-1 | n | — | R | |
| Sah-0 | RPW8.1/Ms-0 | RPW8.2/Sah-0 | n | — | R | |
| 3 | Ts-7 | RPW8.1/Ts-7 | RPW8.2/Ms-0 | n | — | R |
| Uk-1 | RPW8.1/Uk-1 | RPW8.2/Ms-0 | n | — | R | |
| 4 | Can-0 | RPW8.1/Can-0 | RPW8.2/Can-0 | n | HR3/Ms-0 | R |
| Fl-1 | RPW8.1/Fl-1 | RPW8.2/Fl-1 | n | — | R | |
| Bu-06 | RPW8.1/Bu-06 | RPW8.2/Sah-0 | n | — | S | |
| Is-0 | RPW8.1/Ler | RPW8.2/Is-0 | n | — | S+ | |
| Kl-0 | RPW8.1/Kl-0 | RPW8.2/Kl-0 | n | — | S | |
| Ler | RPW8.1/Ler | RPW8.2/Ler | n | HR3/Ms-0 | S | |
| Na-1 | RPW8.1/Na-1 | RPW8.2/Fl-1 | n | — | S | |
| Nd-0 | RPW8.1/Can-0 | RPW8.2/Fl-1 | n | HR3/Ms-0 | S | |
| Sy-0 | RPW8.1/Sy-0 | RPW8.2/Sy-0 | n | HR3/Ms-0 | S | |
| Ws-0 | RPW8.1/Bu-06 | RPW8.2/Sy-0 | n | HR3/Ms-0 | S+ | |
| Xx-0 | RPW8.1/Ler | RPW8.2/Is-0 | n | — | S | |
| 5 | Col-0 | n | n | HR4/Col-0 | HR3/Ms-0 | S+ |
| Fr-3 | n | n | HR4/Fr3 | — | S+ | |
| Is-1 | n | n | HR4/Fr3 | — | S+ | |
| Nie-0 | n | n | HR4/Col-0 | — | S | |
| Sg-2 | n | n | HR4/Fr3 | — | S+ | |
| Sh-0 | n | n | HR4/Fr3 | — | S+ | |
| Wc-1 | n | n | HR4/Fr3 | — | S+ | |
| Wt-4 | n | n | HR4/Fr3 | — | S+ |
a All accessions, except for Can-0, were chosen from a previous survey (Adam et al. 1999), and their origins can be found at http://nasc.life.nott.ac.uk/catalogue.html.
b Alleles identical to that of Ms-0 accession at amino acid level are denoted RPW8.1(2)/Ms-0; alleles diverged from that of Ms-0 are denoted with the names of the accessions if unique, or with the name of the accession that appears first in the table if identical at amino acid level. n indicates not present.
c For HR4, the genotypes are either Col-0 or Fr-3 alleles at amino acid level, or not present (n). For HR3, all are identical at amino acid level. Dashes (—) indicate not determined.
d All the accessions (at least five plants for each) were tested with Erysiphe cruciferaum UEA1, E. cichoracearum UCSC1, E. orontii MGH, and Oidium lycopersici Oxford in this study. The phenotypes were scored as either resistant (R), susceptible (S), or very susceptible (S+). As the disease phenotypes of all the accessions to the four isolates were similar, only the phenotypes to E. cichoracearum UCSC1 were presented here.
Ka/Ks Ratios Among Selected Members of the RPW8 Gene Familya.
| Gene Pairs | Ka/Ks |
|---|---|
| AtHR1–AlHR1 | 0.55 |
| AtHR2–AlHR2 | 0.23 |
| AtHR3–AlHR3 | 0.17 |
| Al/tHR1–Al/tHR2 | 0.61* |
| Al/tHR3–Al/tHR1 | 0.31* |
| Al/tHR3–Al/tHR2 | 0.29* |
| Group averageb | 0.36 ± 0.18 |
| AtRPW8.2–AlRPW8.2 | 1.32 |
| AtRPW8.1–AtHR4 | 2.24 |
| Al/tHR3–Al/tRPW8.2 | 0.95* |
| Al/tHR3–AtRPW8.1 | 1.88* |
| Al/tHR3–AtHR4 | 2.15* |
| Al/tRPW8.2–AtRPW8.1 | 2.45* |
| Al/tRPW8.2–AtHR4 | 2.15* |
| Group averageb | 1.88 ± 0.54 |
| Gene Pairs | Ka/Ks |
|---|---|
| AtHR1–AlHR1 | 0.55 |
| AtHR2–AlHR2 | 0.23 |
| AtHR3–AlHR3 | 0.17 |
| Al/tHR1–Al/tHR2 | 0.61* |
| Al/tHR3–Al/tHR1 | 0.31* |
| Al/tHR3–Al/tHR2 | 0.29* |
| Group averageb | 0.36 ± 0.18 |
| AtRPW8.2–AlRPW8.2 | 1.32 |
| AtRPW8.1–AtHR4 | 2.24 |
| Al/tHR3–Al/tRPW8.2 | 0.95* |
| Al/tHR3–AtRPW8.1 | 1.88* |
| Al/tHR3–AtHR4 | 2.15* |
| Al/tRPW8.2–AtRPW8.1 | 2.45* |
| Al/tRPW8.2–AtHR4 | 2.15* |
| Group averageb | 1.88 ± 0.54 |
Note.—asterisks (*) indicate average value.
a Ka/Ks ratios were calculated based on 369-bp alignable coding sequences of the RPW8 family members.
b t-test between the two groups of data showed t = 6.50, P < 0.0001.
Ka/Ks Ratios Among Selected Members of the RPW8 Gene Familya.
| Gene Pairs | Ka/Ks |
|---|---|
| AtHR1–AlHR1 | 0.55 |
| AtHR2–AlHR2 | 0.23 |
| AtHR3–AlHR3 | 0.17 |
| Al/tHR1–Al/tHR2 | 0.61* |
| Al/tHR3–Al/tHR1 | 0.31* |
| Al/tHR3–Al/tHR2 | 0.29* |
| Group averageb | 0.36 ± 0.18 |
| AtRPW8.2–AlRPW8.2 | 1.32 |
| AtRPW8.1–AtHR4 | 2.24 |
| Al/tHR3–Al/tRPW8.2 | 0.95* |
| Al/tHR3–AtRPW8.1 | 1.88* |
| Al/tHR3–AtHR4 | 2.15* |
| Al/tRPW8.2–AtRPW8.1 | 2.45* |
| Al/tRPW8.2–AtHR4 | 2.15* |
| Group averageb | 1.88 ± 0.54 |
| Gene Pairs | Ka/Ks |
|---|---|
| AtHR1–AlHR1 | 0.55 |
| AtHR2–AlHR2 | 0.23 |
| AtHR3–AlHR3 | 0.17 |
| Al/tHR1–Al/tHR2 | 0.61* |
| Al/tHR3–Al/tHR1 | 0.31* |
| Al/tHR3–Al/tHR2 | 0.29* |
| Group averageb | 0.36 ± 0.18 |
| AtRPW8.2–AlRPW8.2 | 1.32 |
| AtRPW8.1–AtHR4 | 2.24 |
| Al/tHR3–Al/tRPW8.2 | 0.95* |
| Al/tHR3–AtRPW8.1 | 1.88* |
| Al/tHR3–AtHR4 | 2.15* |
| Al/tRPW8.2–AtRPW8.1 | 2.45* |
| Al/tRPW8.2–AtHR4 | 2.15* |
| Group averageb | 1.88 ± 0.54 |
Note.—asterisks (*) indicate average value.
a Ka/Ks ratios were calculated based on 369-bp alignable coding sequences of the RPW8 family members.
b t-test between the two groups of data showed t = 6.50, P < 0.0001.
We thank Charles Langley (University of California, Davis) for A. lyrata seeds, Kathy Faulkner and Samantha Johnson (John Innes Center, UK) for help with Brassica BAC library screening, David Alden and Geoff Eagles for assistance in plant care, Nottingham Arabidopsis Stock Centre for Arabidopsis seeds. We also thank Piyavadee Charoenwattana, Daniel Jaggard, and Mark Coleman for assistance in sequence determination of some A. thaliana accessions, Andy Johnston and Charles Langley for helpful discussion and critical reading of the manuscripts, and three anonymous reviewers for comments on earlier versions of the manuscript. This work is supported by BBSRC grant P15697.
Literature Cited
Aarts, N., M. Metz, E. Holub, B. J. Staskawicz, M. J. Daniels, and J. E. Parker.
Adam, L., S. Ellwood, I. Wilson, G. Saenz, S. Xiao, R. P. Oliver, J. G. Turner, and S. Somerville.
Akaike, H.
Anderson, P. A., G. J. Lawrence, B. C. Morrish, M. A. Ayliffe, E. J. Finnegan, and J. G. Ellis.
Aravind, L., V. M. Dixit, and E. V. Koonin.
Baumgarten, A., S. Cannon, R. Spangler, and G. May.
Bergelson, J., M. Kreitman, E. A. Stahl, and D. C. Tian.
Bergelson, J., C. B. Purrington, C. J. Palm, and J. C. Lopez-Gutierrez.
Botella, M. A., J. E. Parker, L. N. Frost, P. D. Bittner-Eddy, J. L. Beynon, M. J. Daniels, E. B. Holub, and J. D. G. Jones.
Caicedo, A. L., B. A. Schaal, and B. N. Kunkel.
Cannon, S. B., H. Zhu, A. M. Baumgarten, R. Spangler, G. May, D. R. Cook, and N. D. Young.
Dangl, J. L., and J. D. G. Jones.
Ellis, J., P. Dodds, and T. Pryor.
Fichant, G. A.
Goff, S. A., D. Ricke, and T. H. Lan, et al. (55 co-authors).
Grant, M. R., J. M. McDowell, A. G. Sharpe, M. D. T. Zabala, D. J. Lydiate, and J. L. Dangl.
Hammond Kosack, K. E., and J. D. G. Jones.
Heil, M., and I. T. Baldwin.
Holub, E. B.
Hughes, A. L., and M. Nei.
Hulbert, S. H.
Koch, M. A., B. Haubold, and T. Mitchell-Olds.
Lagercrantz, U.
Le, Q. H., S. Wright, Z. H. Yu, and T. Bureau.
Martin, G. B., S. H. Brommonschenkel, J. Chunwongse, A. Frary, M. W. Ganal, R. Spivey, T. Wu, E. D. Earle, and S. D. Tanksley.
McDowell, J. M., M. Dhandaydham, T. A. Long, M. G. M. Aarts, S. Goff, E. B. Holub, and J. L. Dangl.
Meyers, B. C., A. Kozik, A. Griego, H. Kuang, and R. W. Michelmore.
Meyers, B. C., K. A. Shen, P. Rohani, B. S. Gaut, and R. W. Michelmore.
Michelmore, R. W., and B. C. Meyers.
Nei, M., X. Gu, and T. Sitnikova.
Noël, L., T. L. Moores, E. A. van der Biezen, M. Parniske, M. J. Daniels, J. E. Parker, and J. D. G. Jones.
O'Neill, C. M., and I. Bancroft.
Parker, J. E., M. J. Coleman, V. Szabo, L. N. Frost, R. Schmidt, E. A. van der Biezen, T. Moores, C. Dean, M. J. Daniels, and J. D. G. Jones.
Parniske, M., K. E. Hammond Kosack, C. Golstein, C. M. Thomas, D. A. Jones, K. Harrison, B. B. H. Wulff, and J. D. G. Jones.
Posada, D., and K. A. Crandall.
Richly, E., J. Kurth, and D. Leister.
Riely, B. K., and G. B. Martin.
Schiff, C. L., I. W. Wilson, and S. C. Somerville.
Song, W. Y., L. Y. Pi, G. L. Wang, J. Gardner, T. Holsten, and P. C. Ronald.
Song, W. Y., G. L. Wang, and L. L. Chen, et al. (12 co-authors).
Stahl, E. A., G. Dwyer, R. Mauricio, M. Kreitman, and J. Bergelson.
Staskawicz, B. J., F. M. Ausubel, B. J. Baker, J. G. Ellis, and J. D. G. Jones.
Staskawicz, B. J., M. B. Mudgett, J. L. Dangl, and J. E. Galan.
Sun, Q., N. C. Collins, M. Ayliffe, S. M. Smith, J. Drake, T. Pryor, and S. H. Hulbert.
Sun, X., Y. Cao, Z. Yang, C. Xu, X. Li, S. Wang, and Q. Zhang.
Swofford, D. L.
Thompson, J. D., D. G. Higgins, and T. J. Gibson.
Tian, D., M. B. Traw, J. Q. Chen, M. Kreitman, and J. Bergelson.
van der Biezen, E. A., and J. D. Jones.
Wang, G. L., D. L. Ruan, and W. Y. Song, et al. (12 co-authors).
Wei, F., R. A. Wing, and R. P. Wise.
Wilson, I. W., C. L. Schiff, D. E. Hughes, and S. C. Somerville.
Xiao, S., S. Brown, E. Patrick, C. Brearley, and J. G. Turner.
Xiao, S., P. Charoenwattana, L. Holcombe, and J. G. Turner.
Xiao, S. Y., S. Ellwood, O. Calis, E. Patrick, T. X. Li, M. Coleman, and J. G. Turner.
Xiao, S. Y., S. Ellwood, K. Findlay, R. P. Oliver, and J. G. Turner.
Yang, Y. W., K. N. Lai, P. Y. Tai, and W. H. Li.
Yang, Z. H.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}