





Abstract
Molluscs in general, and bivalves in particular, exhibit an extraordinary degree of mitochondrial gene order variation when compared with other metazoans. Two factors inhibiting our understanding the evolution of gene rearrangement in bivalves are inadequate taxonomic sampling and failure to examine gene order in a phylogenetic framework. Here, we report the first complete nucleotide sequence (16,060 bp) of the mitochondrial (mt) genome of a North American freshwater bivalve, Lampsilis ornata (Mollusca: Paleoheterodonta: Unionidae). Gene order and mt genome content is examined in a comparative phylogenetic framework for Lampsilis and five other bivalves, representing five families. Mitochondrial genome content is shown to vary by gene duplication and loss among taxa and between male and female mitotypes within a species. Although mt gene arrangement is highly variable among bivalves, when optimized on an independently derived phylogenetic hypothesis, it allows for the reconstruction of ancestral gene order states and indicates the potential phylogenetic utility of the data. However, the interpretation of reconstructed ancestral gene order states must take in to account both the accuracy of the phylogenetic estimation and the probability of character state change across the topology, such as the presence/absence of atp8 in bivalve lineages. We discuss what role, if any, doubly uniparental inheritance (DUI) and recombination between sexual mitotypes may play in influencing gene rearrangement of the mt genome in some bivalve lineages.
Introduction
In the past decade, there has been an increased interest in gene arrangement of the mitochondrial (mt) genome. This is because the mt genome is a good model system to study genome evolution as it (1) has a relatively conserved gene content in most metazoans (but see Beagley, Okimoto, and Wolstenholme 1998), (2) is small in size, usually about 16 kilobases (kb), and (3) has well-studied processes including genetics and biochemistry (see Avise 2000 for review and other citations). Gene order of the mt genome can be used to investigate evolution of organisms and of their genomes by providing (1) characters that can be used in phylogenetic analysis of ancient lineages and (2) information that can be used to develop models for the mechanisms involved in gene rearrangement, replication, and regulation.
Until recently, studies on the gene order of the mt genome have been biased toward chordate taxa, which possess a similar gene arrangement; however in some invertebrate groups, gene order and genome content appears to be less conserved (see review in Boore 1999). The degree of invertebrate mt gene rearrangement appears to be variable across phyla and sometimes within a given phylum. In Arthropoda, the largest animal phylum, gene rearrangement generally appears to be limited to adjacent tRNA genes and areas surrounding noncoding (NC) regions (Boore 1999). For example, between the insect Drosophila and the chelicerate Limulus, gene order varies only in the placement of trnL (taa); there are no rearrangements among protein-coding or ribosomal genes (Staton, Daehler, and Brown 1997; Lavrov, Boore, and Brown 2000). Within specific arthropod lineages such as metastriate ticks (Black and Roehrdanz 1998), anomuran crustaceans (Morrison et al. 2001), and lice (Shao, Campbell, and Barker 2001), there is a greater amount of gene rearrangement that includes not only the translocation of tRNA genes but also protein-coding and occasionally rRNA genes. Although gene rearrangement is relatively limited in arthropods, gene order provides characters with sufficient phylogenetic signal to resolve relationships among distantly related lineages (Boore et al. 1995; Boore, Lavrov, and Brown 1998).
In contrast to the Arthropoda, Mollusca, the second largest animal phylum, displays an extraordinary amount of variation in gene arrangement. Currently, there are 12 complete and three incomplete mitochondrial genomes available on GenBank, representing four of the eight classes within the phylum (table 1). The two best-sampled classes are the gastropods and the bivalves. Four complete gastropod mt genomes are available, representing the pulmonates (Albinaria coerulea [Hatzoglou, Rodakis, and Lecanidou 1995] and Cepaea nemoralis [Terrett, Miles, and Thomas 1996]) and the opisthobranchs (Pupa strigosa [Kurabayashi and Ueshima 2000] and Roboastra europaea [Grande et al. 2002]), and one incomplete data set for the caenogastropod Littorina saxitilis (Wilding, Mill, and Grahame 1999). For bivalves, five complete mt genomes are available, including: Crassostrea gigas (S. H. Kim, E. Y. Je, and D. W. Park, personal communication; GenBank accession number NC_001276); the male and female mitotypes of Inversidens japanensis (M. Okazaki and R. Ueshima, personal communication; GenBank accession numbers AB055624 and AB055625); the male and female mitotypes of Venerupis philippinarum (M. Okazaki and R. Ueshima, personal communication; GenBank accession numbers AB065374 and NC_003354). In addition, there is a nearly complete mt genome for Mytilus edulis (Hoffman, Boore, and Brown 1992) and a partial mt genome for Pecten maximus (D. Sellos, M. Mommerot, and A. Rigaa, personal communication; GenBank accession number X92688).
Within the gastropods, the majority of gene rearrangement is restricted to tRNA genes among the pulmonates and opisthobranchs (Kurabayashi and Ueshima 2000; Grande et al. 2002). A more distinctive gene order is present in the partial mt genome sequence (8,022 bp) of the caenogastropod Littorina saxitilis (Wilding, Mill, and Grahame 1999), and gene translocation has be demonstrated to occur among five closely related caenogastropod species in the genus Dendropoma (Rawlings, Collins, and Bieler 2001). Even so, most gene boundaries are shared across the available gastropod sequences. Like arthropods, the relatively conserved gene order data in gastropods have been useful in phylogenetic reconstruction, providing evidence for a monophyletic Heterobranchia (Kurabayashi and Ueshima 2000; Grande et al. 2002), a monophyletic Cerithioidea (Lydeard et al. 2002), and phylogenetic relationships among closely related species in Dendropoma (Rawlings, Collins, and Bieler 2001).
Bivalves have the greatest amount of mt gene rearrangement, although this degree of variation may be an artifact of a wider taxonomic sampling in bivalves versus gastropods. Among the currently available taxa, there are very few shared gene boundaries (fig. 1) and gene translocation appears across all gene classes (protein-coding, tRNA, and rRNA). Genome content also varies among bivalve taxa due to apparent gene duplications and losses. Three of the four near-complete mt genome sequences (Crassostrea, Mytilus, and Venerupis) have duplicated tRNA genes. Crassostrea also has a duplicated rrnS, and Venerupis has a second tandem copy of cox2 of a different length.
In addition to the highly variable gene order and content of the mt genome, some bivalve lineages also have an unusual mode of inheritance for mtDNA, termed doubly uniparental inheritance (DUI) (Skibinski, Gallagher, and Beyon 1994; Zouros et al. 1994). Unlike most metazoans, these bivalves have two separate mtDNA lineages (two distinct and independent mt genomes) that appear to be segregated by sex and tissue type (but see Quesada, Skibinski, and Skibinski [1996] and Garrido-Ramos et al. [1998]). Males tend to be heteroplasmic, possessing both the female (F) and male (M) mitotypes, which are segregated between the somatic and gonadal tissues, respectively, whereas females usually are homoplasmic for the F-mitotype. Currently, DUI has been reported to occur within three disparate families: Mytilidae (Skibinski, Gallagher, and Beyon 1994; Zouros et al. 1994; Hoeh et al. 1996); Unionidae (Hoeh et al. 1996; Liu, Mitton, and Wu 1996); and Veneridae (Passamonti and Scali 2001), all of which are represented in this study. Comparison of F-mitotypes and M-mitotypes of Inversidens (Unionidae) and Venerupis (Veneridae) show limited differences in gene order and genome content and illustrate the independence of these sexual lineages.
Several models have been proposed to explain the mechanisms of gene translocation. The most familiar and accepted is the duplication and loss model, in which a portion of DNA is duplicated during replication, resulting in tandem copies of a gene or genome region. Once a disabling mutation arises in one of the gene copies, that copy is removed from the mt genome or reduced to noncoding sequence during subsequent replications. Recently, it has been proposed that gene loss may be a nonrandom process (Lavrov, Boore, and Brown 2002). There also may be an association between gene translocation and noncoding regions in the mt genome, especially the control region (CR) in vertebrates (Boore 1999). Two other less discussed models are the dimerization of two mt genomes and ‘illegitimate’ recombination between mt genomes (see description in Boore 2000). The general lack of knowledge for the mechanisms involved in gene translocation and genome rearrangement affects our interpretation of gene order data. Bivalves, with their great diversity, ancient lineages that span the Phanerozoic (Skelton and Benton 1993), and the observed variation in genome content should eventually provide a good system for modeling the mechanisms of gene movement.
In the present study, we determined the complete mtDNA sequence of the bivalve, Lampsilis ornata, a North American representative of the family Unionidae. In addition, we describe the notable features of the Lampsilis mt genome and compare these to other available bivalve taxa. Gene order of Lampsilis is examined in a comparative phylogenetic framework with five other bivalve sequences representing five families and four orders. Our examination of disparate gene arrangement indicates the potential phylogenetic utility of gene order and allows for the reconstruction of ancestral gene order states. Unique mechanisms that may be involved with gene translocation are discussed for Bivalvia.
Materials and Methods
Isolation and Amplification of the Mitochondrial Genome
High-molecular-weight mitochondrial DNA (mtDNA) was isolated (as described by Zardoya, Garrido-Pertierra, and Bautista 1995) from female Lampsilis ornata. To ensure that only the female mitotype was isolated, a single female was selected and only the somatic tissues were used for the procedure. After homogenization of fresh gill tissue, intact nuclei and cellular debris were removed by low-speed centrifugation. Mitochondria were then pelleted by high-speed centrifugation. mtDNA was removed from the organelle by alkaline lysis and treated with RNase, followed by a phenol/chloroform extraction. mtDNA was purified further with Microcon concentrators (Millipore) according to the manufacture's instructions.
The total length (∼16.0 kb) of L. ornata mtDNA was amplified by polymerase chain reaction (PCR). In brief, a small (∼500 bp) cox1 segment was amplified using primers from Folmer et al. (1994), following the protocol described in Roe and Lydeard (1998). New primers (COI-477F and COI-215R) were designed from sequence of the small cox1 segment, then used in long PCR (LA PCR kit TaKaRa) to amplify the remaining mtDNA region (∼16 kb). The resulting large fragment was purified with Microcon concentrators (Millipore) and used as template for cycle sequencing reactions. As a new sequence was generated, it was aligned to the original cox1 sequence fragment to create a continuous DNA strand (contig). New primers were designed from the ends of the contig. To quicken the process of sequencing, additional mitochondrial regions were utilized for primer design to create multiple origins of primer walking. Primers for three of these regions came from the following sources: rrnL (Lydeard, Mulvey, and Davis 1996), rrnS (White, Mcpheron, and Stauffer 1996), nad1 (Serb, Buhay, and Lydeard 2003), and cob (B. Bowen, personal communication). Original primers for this study were also designed from conserved sequence blocks within nad2, nad3, and nad4. Table 2 lists sequence and mt genome position of primers used in PCR and cycle-sequencing reactions. Sequence of the mt genome of Lampsilis ornata has been submitted to GenBank under the accession number AY365193. The voucher specimen is housed in the University of Alabama Unionid Collection under UAUC 3192.
Analysis of Sequence Data
Protein-coding genes were identified in the program MacVector version 7.0 (Oxford Molecular Group) using the similarity search option BlastX. The putative termini of protein-coding genes were inferred to be at the first in-frame start codon (L, I, M, or V) and the first in-frame stop codon. Nucleotide and amino acid sequences of the protein-coding genes were aligned with homologous genes from other molluscs, annelids, and arthropods using the alignment option ClustalW (Thompson, Higgins, and Gibson 1994) in MacVector to provide additional support for putative start and stop codons in protein-coding genes. The 5′ and 3′ ends of the rrnS and rrnL were assumed to be adjacent to the flanking tRNA genes. Transfer RNA genes (tRNAs) were identified using the program tRNAscan-SE (version 1.1, http://www.genetics.wustl.edu/eddy/tRNAscan-SE [Lowe and Eddy 1997]) using the invertebrate mitochondrial codon sequence and setting the cove cutoff score to 1 or manually when tRNA genes were not recognized by this program. Potential secondary structure for identified tRNAs were determined in tRNAscan, which uses a consensus model when predicting secondary structure. Nucleotides that do not fit tRNAscan consensus models are indicated by lowercase letters.
To examine phylogenetic histories of gene arrangement, gene order was coded and the character states optimized on an independently derived topology. The program MacClade version 4.0 (Maddison and Maddison 2000) was used to parsimoniously assign character states to internal nodes according to information from the topology. The DELTRAN tracing option was chosen to select among the most-parsimonious reconstructions. This tracing method delays changes away from the root, maximizing parallelisms and minimizing reversals. Gene arrangement for each of the molluscan taxa and outgroups was coded by gene boundaries following Boore et al. (1995) and Boore and Staton (2002), where the character was the gene X being either immediately upstream or downstream of the character state gene Y. This method of coding also provides information on transcriptional orientation for each gene relative to its neighbor. For this analysis, two data sets were generated, one including and the other excluding tRNA genes. tRNAs were omitted from one data matrix to determine whether there was any phylogenetic signal associated with mtDNA protein-coding and ribosomal genes, which might be masked by the apparently higher observed rate of translocations of tRNA genes. Genome sequence of taxa with unusual mt genome content, such as an incomplete mt genome sequence or duplication and/or loss of genes, were reevaluated using the methods described above to verify previous authors' findings. Several changes in gene annotation were made for the gene order analysis. In Pecten, the second copy of trnK was reassigned as trnQ, and trnV was newly identified downstream of trnQ. In Crassostrea, five additional tRNA genes were identified, including trnS1, trnS2, a second copy of trnQ, and two undetermined tRNAs that are most likely trnA and trnF. All new putative tRNA genes are illustrated in figure 2. In the F-mitotype of Inversidens, a portion of atp8 was identified between trnD and nad4L based on amino acid similarity (table 3). All tRNA duplications in Mytilus, Venerupis, and Crassostrea and the duplication of rrnS in Crassostrea and cox2 in Venerupis were independently confirmed during our reevaluation. Character states of duplicated, nonadjacent genes (such as trnM in Mytilus) were examined in an attempt to identify the original and duplicated copy of the gene. However, since the majority of duplication codings resulted in autapomorphic character states for the given taxon and did not resolve the question of gene homology, we decided on a conservative approach where nontandem gene duplications were assigned as unknown (“?”) in the gene order matrix. Gene arrangement from only the F-mitotype was compared across bivalve taxa under the assumption that gender-specific mitotypes represented orthologous sequences (but see Results and Discussion). A traditional phylogeny of the sampled molluscan classes was used for this study (Salvini-Plawen and Steiner 1996) (fig. 3). Non-molluscan taxa included the arthropod Drosophila melanogaster (Lewis, Farr, and Kaguni 1995) and the annelid Lumbricus terrestris (Boore and Brown 1995). Relationships within Gastropoda were based on Kurabayashi and Ueshima (2000) and Grande et al. (2002). Bivalve relationships in the topology were based on a hypothesis resulting from a total-evidence approach of DNA sequences and morphology (Giribet and Wheeler 2002). Character states were reconstructed using the DELTRAN tracing option in MacClade. Unambiguous reconstructions that support a particular node were noted for bivalve taxa as an estimation of phylogenetic signal in the gene order data. Using only unambiguously reconstructed character states, rather than reconstructing states at equivocal nodes, is a conservative approach of examining gene order data. It is understood that the reconstructed ancestral state for a character is only an estimate and has a level of uncertainty that depends on the phylogeny estimation.
Gene order data were also analyzed phylogenetically in PAUP* version 4.0b10 (Swofford 2002) in an effort to find the optimal topology. Data from the incomplete sequences of Littorina and Pecten were excluded from the analyses. Both gene order data sets, including and excluding tRNAs, were analyzed using parsimony criteria and employing a heuristic search of 100 random-order addition replicates and tree bisection-reconstruction (TBR) branch swapping. Only minimum-length trees were retained, and zero-length branches were collapsed. Support for the individual nodes of the resulting phylogenetic hypotheses was assessed by bootstrap values using the FAST stepwise addition option (1,000 replicates) in PAUP*.
Results and Discussion
Organization and Structural Features of Lampsilis ornata mt Genome
Mitochondrial genome of Lampsilis ornata is 16,060 base pairs (bp) in length and has an A+T composition of 62.4%. All 37 of the genes typically found in the metazoan mt genome were identified in Lampsilis. The gene arrangement of L. ornata is unique to all other molluscan and metazoan mt genomes. Genes are arranged in the following order (bolded genes are translated from the opposite strand): nad4, nad4L, atp8, trnD, atp6, cox3, cox1, cox2, nad3, trnH, trnA, trnS2, trnS1, trnE, nad2, trnM, trnW, trnR, rrnS, trnK, trnT, trnY, rrnL, trnL2, trnN, trnP, cob, trnF, nad5, trnQ, trnC, trnI, trnV, trnL1, nad1, trnG, and nad6 (fig. 4). The two leucine and two serine tRNA genes are differentiated by their anticodon sequence: trnL1 = (cua), trnL2 = (uaa), trnS1 = (aga), and trnS2 = (uca). Typically, start codons in the mitochondrial invertebrate code vary between L (UUG), I (AUU, AUC), M (AUA, AUG), and V (GUG) amino acid codons. In Lampsilis, no protein-coding genes begin with L, three begin with I (nad1, nad4, and nad6), eight begin with M (cob, cox2, cox3, atp6, atp8, nad2, nad3, and nad5), and two begin with V (cox1 and nad4L). The two stop codons were utilized in the Lampsilis mt genome (UAA and UAG). Interestingly, there was only one occurrence of gene overlap between nad4 and nad4L, where the beginning of nad4 overlapped nad4L by seven nucleotides. Although gene overlap is not uncommon in animal mitochondrial genomes (e.g., Yamazaki et al. 1997; Boore and Brown 2000; Helfenbein, Brown, and Boore 2001), most occurrences are between tRNA genes. An alternative interpretation for the spatial relationship between the nad4 and nad4L genes could be the presence of a truncated stop codon, represented as a single T or TA, thus removing the putative overlap region. The sequences of these codons are completed by polyadenylation after transcript processing of the truncated stop codons (Ojala, Montoya, and Attardi 1981). The remaining protein-coding genes in the Lampsilis mt genome had complete stop codons.
Codon Usage and Codon Bias
A total of 3,717 amino acids are encoded by the Lampsilis mt genome (table 4). There is a codon bias for TTT (F) in Lampsilis, which is also present in Mytilus (Hoffman, Boore, and Brown 1992) but not in the gastropods Pupa (Kurabayashi and Ueshima 2000), Roboastra (Grande et al. 2002), and Albinaria (Hatzoglou, Rodakis, and Lecanidou 1995), where TTA (L) is the most abundant (comparisons only made with published data). Interestingly, in the most basal molluscan representative, Katharina, TTT (F) and TTA (L) are used equally frequently (Boore and Brown 1994). As more molluscan mt genomes are determined and analyzed, there may be evidence for historical constraint of codon usage across the phylum. All codons in each codon family occur in the Lampsilis mt genome. Excluding termination codons, TGC (C) and CGC (R) are used less than 10 times. This differs somewhat from Mytilus, where TGC (C) is more frequent. In Lampsilis mtDNA, codons ending in A or T are more frequent (67%) than those ending in G or C and reflect A+T base compositional bias (table 4).
Noncoding Regions in Lampsilis
There are as many as 28 noncoding (NC) regions found throughout the Lampsilis mt genome, ranging in size from 2 to 282 bp (fig. 4). The three largest NC regions are found between nad2-trnE (282 bp), trnQ-nad5 (230 bp), and nad5-trnF (136 bp). Of these, only the 136-bp NC region has an increased A+T composition (A+T = 76.8%), which is one characteristic typically used to identify origins of replication. The control region (CR), a noncoding region of DNA involved in replication of the mt genome, has not been identified in Lampsilis. Unlike vertebrates, the CR in invertebrates is not well characterized and lacks discrete, conserved sequence blocks used in identification (see Mytilus [Hoffman, Boore, and Brown 1992]). Multiple copies of the CR also have been documented in vertebrates (Kumazawa et al. 1998), which may further confound the identification of the origin of replication of the light (OL) and heavy (OH) strands in less well-studied organisms such as many invertebrate taxa. All bivalve sequences examined for this study had multiple NC regions throughout their mt genomes.
tRNA Secondary Structure
Twenty-two sequences that folded into tRNA-like secondary structures and possessed correct anticodon sequences were identified in Lampsilis (fig. 5). Nearly all of the putative Lampsilis tRNAs have a seven-member aminoacyl acceptor arm, a five-member anticodon stem (but see trnA, trnE, and trnG), and a seven-member anticodon loop. The anticodon loop of trnR is unusual, having only six members. A “?” marks the place in the loop that may be missing a base (fig. 5). In Lampsilis, both trnS1 and trnS2 have shortened DHU arm stems, where potential pairing consists of only one (G-T) or two (G-C, T-A) base pairs, respectively. These configurations for trnS are not shown in figure 5. This is not atypical, as the DHU arm of the trnS1 (AGN) is unpaired in many metazoan taxa (Garey and Wolstenholme 1989; Hoffman, Boore, and Brown 1992; Yamazaki et al. 1997; Tomita et al. 2002). The TψC arm loop is missing in the LampsilistrnC, but is present in Mytilus (Hoffman, Boore, and Brown 1992) and Katharina (Boore and Brown 1994); however, the length of TψC arm is variable in gastropods, where either the stem is missing or reduced (Yamazaki et al. 1997). Mispairing between bases in stems is consistent across several taxa. For example, the second base pair in the anticodon stem of trnW has a T-T mispairing in Lampsilis, Mytilus, and Katharina and a T-G pairing in several gastropods (Yamazaki et al. 1997).
Content Variation of Bivalve mt Genomes
Bivalves have much more variation in mt genome content than other sampled molluscs. Gene duplication and/or loss is present in almost every taxon in which a complete mt genome is available. For example, rrnS is duplicated in Crassostrea, and tRNA genes have been duplicated in Mytilus (trnM), Venerupis (trnM and trnV), and Crassostrea (trnQ). Hoffman, Boore, and Brown (1992) were the first to report that the protein-coding gene atp8 is absent in Mytilus, a condition that was only previously known to occur in nematodes (Okimoto et al. 1991). Subsequent studies have shown that other bivalves also lack this gene, including Crassostrea (S. H. Kim,E. Y. Je, and D. W. Park, personal communication) and Venerupis (M. Okazaki and R. Ueshima, personal communication). Although atp8 is present in the Inversidens F-mitotype (identified during this study from M. Okazaki and R. Ueshima, personal communication [see table 3]), it appears to be nonfunctional due to the presence of several stop codons after the first 31 amino acids. Interestingly, there does not appear to be an identifiable remnant of this gene in the male mitotype (M. Okazaki and R. Ueshima, personal communication). In contrast, the other unionid, Lampsilis, is the only bivalve in this study that possesses a complete copy of atp8; however, this copy encodes a protein that is longer than the “typical” ATP8 by 13 extra amino acids on the C-terminus. At this time, it cannot be determined whether this gene copy is functional in Lampsilis.
Phylogenetic Implications of Mitochondrial Gene Arrangement in Bivalvia
Two gene order matrices, one including and one excluding tRNA genes, were constructed for the 13 molluscan and two distantly related outgroup taxa. There were 74 characters in the complete matrix and 30 characters when tRNA genes were excluded. Examination of gene arrangement when mapped onto the independently derived molluscan topology (fig. 3) showed several unambiguously reconstructed character states that support the Bivalvia and relationships within the class. The absence of atp8 was reconstructed as the ancestral condition of Bivalvia in both gene order data sets. Another putative ancestral state for Bivalvia was a shared gene boundary between of rrnL-cob when excluding tRNAs from the data matrix. Adjacent trnG and trnN genes are reconstructed as the ancestral character state for the clade containing Crassostrea, Pecten, and Mytilus. This clade was not supported unambiguously when excluding tRNA genes from the analysis. The Unionidae (Lampsilis + Inversidens) was supported by multiple synapomorphies and unambiguous character state reconstructions in both data sets (see below for description), including the presence of atp8 (fig. 6). The placement of Venerupis on the independently derived topology was not supported with either gene order data set. Each of the bivalve taxa has many autapomorphic characters, illustrating the number of unique gene boundaries. Figure 6 lists unambiguously reconstructed ancestral gene order conditions within Bivalvia.
More unambiguously reconstructed changes were observed in the gene order data when tRNAs were included, which was surprising since it has been suggested that the secondary structure of tRNA genes allow them to translocate more frequently (Moritz and Brown 1987; Staton et al. 1994). It would be expected that if the rate of tRNA gene translocation was much higher than protein-coding and ribosomal gene translocation, that deeper divergences would not have unambiguous support from gene order changes involving tRNAs due to a higher level of homoplasy. Although there is little unambiguous support for Bivalvia in either data set, tRNA gene order data does support clades of Unionidae and Mytilus (Pecten + Crassostrea).
Maximum-parsimony analysis was conducted on both gene order data sets to generate a topology based solely on gene boundaries. Analysis of the gene order data, including tRNAs, provided a total of 67 parsimony informative characters. The MP analysis resulted in 12 most-parsimonious trees (488 steps). The Mollusca was not recovered as monophyletic, and higher-order relationships among the classes were unresolved (not shown). Both Bivalvia and Gastropoda were recovered as monophyletic groups. Within Bivalvia, relationships differed from the inferred topology (Giribet and Wheeler 2002) by the placement of Venerupis in a polytomy with Crassostrea and Mytilus. Bootstrap values were greater than 50% for three clades: Gastropoda (91%), Pupa+Roboastea (76%), and Lampsilis+Inversidens (96%). Analysis of gene order data, excluding tRNAs, resulted in 25 equally parsimonious trees of 159 steps (not shown). The strict consensus indicates less resolution overall and a monophyletic Bivalvia was not recovered.
Variation Within Unionidae
Gene arrangement between female mt genomes of the North American Lampsilis ornata and Asian Inversidens japanensis are identical except for the region between rrnS and cox1. This region includes three protein-coding (nad2, nad3, and cox2) and eight tRNA (A, E, W, R, M, S1, S2, and H) genes (fig. 7). Interestingly, this region is also variable between the M- mitotypes and F-mitotypes of Inversidens (M. Okazaki and R. Ueshima, personal communication). In all three mt genomes, this variable region contains the largest NC sequence: Lampsilis, 282 bp between trnE and nad2; Inversidens-F, 287 bp between trnW and trnE; and Inversidens-M, 316 bp between trnH and cox1. The association of an NC region with an area of gene translocation has been observed in arthropod taxa (Boore 1999) and may provide a starting point in the identification and characterization of a CR in the Unionidae. Although gene order is more similar between Lampsilis-F and Inversidens-M, phylogenetic analysis of nucleotide sequence variation among the mitotypes recovers the F-mitotypes of Lampsilis and Inversidens as sister taxa supporting the homology of these two genomes under the criteria described by Hoeh et al. (1996) (J. M. Serb, unpublished data). Homology of the F-mitotypes suggest that there has not been a mitotype reversal in the lineage for Lampsilis and Inversidens, and our limited taxonomic sample agrees with the conclusions of Hoeh et al. (1996) that there is a single origin of DUI in the Unionidae lineage.
Once homology was established for the unionid mt genomes, ancestral character states were reconstructed from the gene order data. When including tRNAs, six unambiguous character states (gene boundaries) support the Unionidae and are putative synapomorphies: trnA-trnS2, trnG-nad6, trnL2-trnN, nad2-trnM, trnQ-nad5, and atp8-nad4L. When excluding tRNAs from the gene order data, three unambiguous character states support the Unionidae: atp6-atp8-nad4L and cob-nad5 (fig. 6).
What Makes Bivalve Gene Order So Variable?
At this time, the available mt gene order data do not support a particular model of gene rearrangement for bivalves, but it does provide tantalizing clues. Multiple copies of genes exist in extant taxa, but the majority of gene duplications are not tandem. This may be because either the duplication did not form in tandem or the gene translocation occurred subsequently to the duplication event. Our data also show an association of large NC regions within a region of gene rearrangement in unionids and between the rrnS duplication in Crassostrea. Sampling mt genome sequences of more closely related bivalves may provide insight to the mechanism of gene movement by limiting the number of gene translocations while at the same time utilizing gene duplication and loss that frequently occurs in Bivalvia. Bivalves provide a unique opportunity to formulate models by providing historical “snapshots” of gene movement, as duplications are predicted to be intermediate steps in mt genome rearrangements. Also, because of their unique mode of mtDNA inheritance, bivalves may have mechanisms for gene translocation not present in other molluscs (see below).
It is important when examining gene rearrangement to consider the likelihood of the change of character states across the topology. Loss and subsequent regain of a gene would appear to be unlikely, whereas gene duplication and loss of one copy would be more probable with our current understanding of gene translocation models. For example, it is unlikely that atp8 was lost in the ancestor of Bivalvia then regained in the Unionidae lineage. Instead, it is possible that the estimation of phylogeny used in our analysis was incorrect or that taxon sampling for gene order was poor. If Venerupis is actually more closely related to Mytilus (Crassostrea + Pecten) than Unionidae, as indicated in the phylogenetic analysis of the gene order data, then the loss of atp8 would not be a putative ancestral state for the Bivalvia but a synapomorphy for a more limited clade, including Venerupis, Mytilus, Crassostrea, and Pecten. When the topology reflects this relationship, two new unambiguously reconstructed ancestral states (trnW-trnR and trnP-cob) are designated for the Bivalvia. Adding taxa may also affect our estimation of character transformation by filling taxonomic gaps in gene order data for class members. However, if the topology of Giribet and Wheeler (2002) is correct, an alternative hypothesis to explain the distribution of atp8 across Bivalvia is multiple and independent losses of atp8 in various lineages. Again, only after a more thorough taxonomic sampling can this hypothesis be addressed.
The occurrence of doubly uniparental inheritance found in some bivalve lineages may also be involved in gene translocation, gene duplication/loss events, and recombination. The data show that gene content and order can vary between M-mitotypes and F-mitotypes of a species (Inversidens japanensis, M. Okazaki andR. Ueshima, personal communication; GenBank accession numbers AB055624 and AB055625), and it has been previously demonstrated that the mechanism that separates the two mitotype lineages may lapse or has multiple origins in Bivalvia. Phylogenetic evidence from Hoeh et al. (1996) and the existence of heteroplasmic females and homoplasmic males (Quesada, Skibinski, and Skibinski 1996; Garrido-Ramos et al. 1998) suggests that the DUI mechanism may occasionally experience breakdowns where a new M-mitotype lineage may be derived from an F-mitotype (mitotype role reversal or “masculinization”). A reversal can also occur in the opposite direction, where an M-mitotype is “feminized” (Garrido-Ramos et al. 1998). Using the example of atp8, if atp8 is deleted from the F-mitotype early in the bivalve evolutionary history, it could be subsequently “gained” in a later lineage (e.g., Unionidae) if that lineage experiences a role-reversal of the mitotypes. In other words, the M-mitotype, which contains a copy of atp8, would replace the F-mitotype that lacked a copy of that gene. This would result in the presence of atp8 in the newly created F-mitotype and in that bivalve lineage. Hoeh et al. (1996) recognizes at least three independent mitotype reversals across Mytilidae and Unionidae. Although controversial, another possible mechanism associated with changes of gene order and genome content is recombination between sexual mitotypes. Ladoukakis and Zouros (2001) found evidence for homologous recombination between a newly masculinized F-mitotype and a maternally inherited F-mitotype and in the gonadal tissue of a male individual of Mytilus galloprovincialis. A second study by Burzynski et al. (2003) provides additional evidence of recombination in M. trossulus at a different location of the mt genome. Further study will be necessary to establish whether mtDNA recombination is a general phenomenon in bivalves or limited to Mytilus, whether recombination is restricted to particular portions of the mt genome, and whether there is a relationship between recombination and DUI. However, if portions of the mt genome can undergo recombination between mitotypes, then this may be another mechanism for gene rearrangement that may not result in tandem gene duplication.
This study represents the first examination of mitochondrial gene rearrangement in bivalves using a phylogenetic framework. One advantage of this method is the ability to infer ancestral character states for mt gene order in Bivalvia. In addition, the ability to map gene order changes on the phylogeny has the potential to elucidate what mechanisms may be responsible for creating the diversity of gene order variation. Several considerations must be made when attempting to reconstruct gene translocation, including the accuracy of the phylogenetic estimation and the probability of the change of character states across the topology. To better understand and model gene rearrangement in the mitochondrial genomes of bivalves, it will be necessary to include the sequences of additional bivalve mt genomes. Increased taxonomic sampling will allow a more accurate estimation of ancestral character states for mt gene order. It also will be paramount to determine how widespread DUI is within the class and the number of times this mode of inheritance has been compromised. Unrecognized mitotype reversals in the evolution of Bivalvia will confound analysis of gene order data if nonhomologous mt genomes are compared.
Present address: Department of Ecology, Evolution, and Marine Biology, University of California, Santa Barbara.
Richard Thomas, Associate Editor
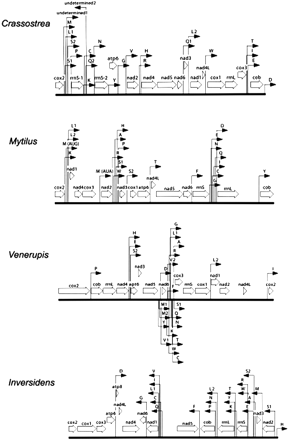
Mitochondrial gene order of four bivalve representatives (Crassostrea, Mytilus, Venerupis, and Inversidens). Full citations for these sequences can be found in table 1. Protein and rRNA genes are abbreviated as in the text. tRNA genes are identified by the one-letter code for the corresponding amino acid. The two leucine and two serine tRNA genes are differentiated by their anticodon sequence: L1 = trnL(cua), L2 = trnL(uaa), S1 = trnS(aga), and S2 = trnS(uca). Other tRNA duplications in Crassostrea, Mytilus, and Venerupis are listed by copy number 1 or 2, but this designation does not presume homology across taxa. The direction of transcription is depicted by arrows
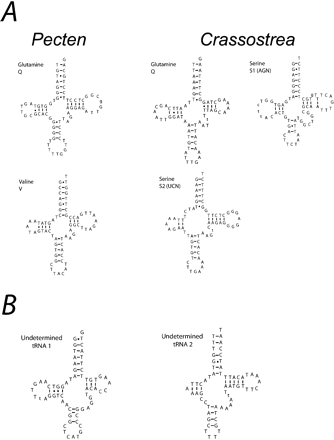
The potential secondary structure for newly identified tRNAs in Pecten and Crassostrea using tRNAscan-SE. Two and three putative tRNAs are illustrated for Pecten and Crassostrea, respectively (A) and two undetermined tRNAs from Crassostrea (B). Bars between nucleotide pairs indicate base pairing; G-T pairing is shown with dots. Lowercase letters indicate base positions that did not fit tRNAscan models
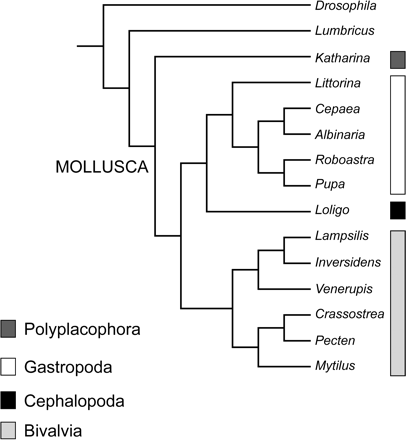
Topology illustrating the traditional relationships among molluscan classes (Salvini-Plawen and Steiner 1996). Gene order data is optimized on this topology. Relationships within gastropods and bivalves were determined by independent data sets from Kurabayashi and Ueshima (2000) and Grande et al. (2002) (gastropods) and Giribet and Wheeler (2002) (bivalves)
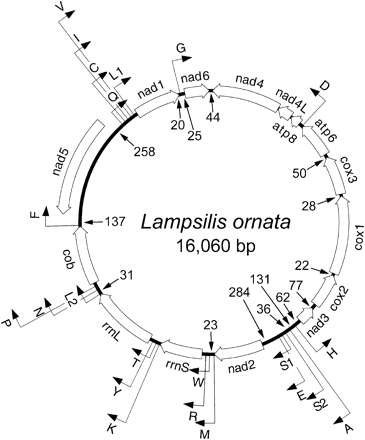
Gene map of the mitochondrial genome of Lampsilis ornata. Protein and rRNA genes are abbreviated as in the text. tRNA genes are identified by the one-letter code for the corresponding amino acid. The two leucine and two serine tRNA genes are differentiated by their anticodon sequence: L1 = trnL(cua), L2 = trnL(uaa), S1 = trnS(aga), and S2 = trnS(uca). The direction of transcription is depicted by arrows. Noncoding regions greater than 20 nucleotides are listed by size, and their genome position and transcriptional orientation are indicated with arrows
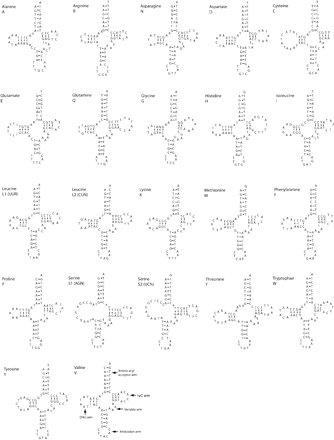
The potential secondary structures for 22 inferred tRNAs of Lampsilis ornata. Amino acid identities are given above each sequence. Bars between nucleotide pairs indicate base pairing; G-T pairing is shown with dots. Lowercase letters indicate base positions that did not fit tRNAscan models; “?” indicates a possible missing base. Features of tRNA secondary structure are illustrated on tRNA (V)
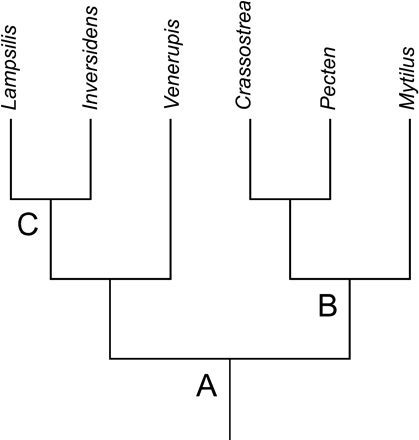
Unambiguous character state changes optimized on an independently derived topology for bivalves. Nodes are specified by letters A–C. Bolded genes designate opposite transcriptional orientation. The following are unambiguous character state reconstructions that support a particular node; character state reconstructions at a given node when excluding tRNA genes are listed in ( ). Adjacent genes are separated by a “–.” A: atp8 absent; (atp8 absent, rrnL-cob). B: trnG-trnN; (none). C:trnA-trnS2, trnG-nad6, trnL2-trnN, nad2-trnM, trnQ-nad5, atp8-nad4L; (atp6-atp8-nad4L, cob-nad5). The number of unambiguous autapomorphic character state changes for the bivalve taxa are as follows, where the number in ( ) is from the gene order data set excluding tRNAs: Lampsilis 2 (0); Inversidens 1 (1); Venerupis 11 (8); Crassostrea 2 (0); Pecten 1 (0); Mytilus 4 (2)
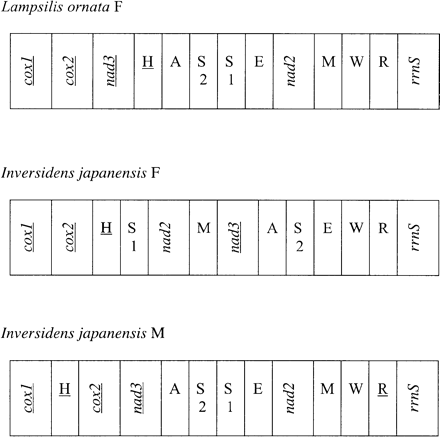
Gene map of the variable gene order portion in the unionid taxa Lampsilis ornata-F (female mitotype) and Inversidens japanensis-F and Inversidens japanensis-M (male mitotype). Genes transcribed by the opposite strand are underlined. Transfer RNA genes are designated by a single letter for the corresponding amino acid. There are two tRNAs for serine that are differentiated by numerals: S1 and S2 recognize the codons AGN and UCN, respectively. Notation for all other genes is standard and gene size is not to scale
Mitochondrial Genomes of Molluscan Taxa Available on GenBank Used in this Study.
| Taxon | GenBank Accession Numbers | Authors | ||
|---|---|---|---|---|
| Bivalvia | ||||
| Mytilus edulisa | M83756–M83762 | Hoffman, Boore, and Brown 1992 | ||
| Crassostrea gigas | NC_001276 | S. H. Kim, E. Y. Je, and D. W. Park, personal communication | ||
| Venerupis philippinarum-Fb | NC_003354 | M. Okazaki and R. Ueshima, personal communication | ||
| Inversidens japanensis-F | AB055625 | M. Okazaki and R. Ueshima, personal communication | ||
| Inversidens japanensis-Mc | AB055624 | M. Okazaki and R. Ueshima personal communication | ||
| Pecten maximusa | X92688 | D. Sellos, M. Mommerot, and A. Rigaa, personal communication | ||
| Gastropoda | ||||
| Albinaria coerulea | NC_001761 | Hatzoglou, Rodakis, and Lecanidou 1995 | ||
| Cepaea nemoralis | NC_001816 | Terrett, Miles, and Thomas 1996 | ||
| Littorina saxitilisa | AJ132137 | Wilding, Mill, and Grahame 1999 | ||
| Pupa strigosa | NC_002176 | Kurabayashi and Ueshima 2000 | ||
| Roboastra europaea | AY083457 | Grande et al. 2002 | ||
| Polyplacophora | ||||
| Katharina tunicata | U09810 | Boore and Brown 1994 | ||
| Cephalopoda | ||||
| Loligo bleekeri | NC_002507 | Tomita et al. 2002 | ||
| Taxon | GenBank Accession Numbers | Authors | ||
|---|---|---|---|---|
| Bivalvia | ||||
| Mytilus edulisa | M83756–M83762 | Hoffman, Boore, and Brown 1992 | ||
| Crassostrea gigas | NC_001276 | S. H. Kim, E. Y. Je, and D. W. Park, personal communication | ||
| Venerupis philippinarum-Fb | NC_003354 | M. Okazaki and R. Ueshima, personal communication | ||
| Inversidens japanensis-F | AB055625 | M. Okazaki and R. Ueshima, personal communication | ||
| Inversidens japanensis-Mc | AB055624 | M. Okazaki and R. Ueshima personal communication | ||
| Pecten maximusa | X92688 | D. Sellos, M. Mommerot, and A. Rigaa, personal communication | ||
| Gastropoda | ||||
| Albinaria coerulea | NC_001761 | Hatzoglou, Rodakis, and Lecanidou 1995 | ||
| Cepaea nemoralis | NC_001816 | Terrett, Miles, and Thomas 1996 | ||
| Littorina saxitilisa | AJ132137 | Wilding, Mill, and Grahame 1999 | ||
| Pupa strigosa | NC_002176 | Kurabayashi and Ueshima 2000 | ||
| Roboastra europaea | AY083457 | Grande et al. 2002 | ||
| Polyplacophora | ||||
| Katharina tunicata | U09810 | Boore and Brown 1994 | ||
| Cephalopoda | ||||
| Loligo bleekeri | NC_002507 | Tomita et al. 2002 | ||
Note.—Bold terms are molluscan classes.
aIncomplete data.
bF = female mitotype.
cM = male mitotype.
Mitochondrial Genomes of Molluscan Taxa Available on GenBank Used in this Study.
| Taxon | GenBank Accession Numbers | Authors | ||
|---|---|---|---|---|
| Bivalvia | ||||
| Mytilus edulisa | M83756–M83762 | Hoffman, Boore, and Brown 1992 | ||
| Crassostrea gigas | NC_001276 | S. H. Kim, E. Y. Je, and D. W. Park, personal communication | ||
| Venerupis philippinarum-Fb | NC_003354 | M. Okazaki and R. Ueshima, personal communication | ||
| Inversidens japanensis-F | AB055625 | M. Okazaki and R. Ueshima, personal communication | ||
| Inversidens japanensis-Mc | AB055624 | M. Okazaki and R. Ueshima personal communication | ||
| Pecten maximusa | X92688 | D. Sellos, M. Mommerot, and A. Rigaa, personal communication | ||
| Gastropoda | ||||
| Albinaria coerulea | NC_001761 | Hatzoglou, Rodakis, and Lecanidou 1995 | ||
| Cepaea nemoralis | NC_001816 | Terrett, Miles, and Thomas 1996 | ||
| Littorina saxitilisa | AJ132137 | Wilding, Mill, and Grahame 1999 | ||
| Pupa strigosa | NC_002176 | Kurabayashi and Ueshima 2000 | ||
| Roboastra europaea | AY083457 | Grande et al. 2002 | ||
| Polyplacophora | ||||
| Katharina tunicata | U09810 | Boore and Brown 1994 | ||
| Cephalopoda | ||||
| Loligo bleekeri | NC_002507 | Tomita et al. 2002 | ||
| Taxon | GenBank Accession Numbers | Authors | ||
|---|---|---|---|---|
| Bivalvia | ||||
| Mytilus edulisa | M83756–M83762 | Hoffman, Boore, and Brown 1992 | ||
| Crassostrea gigas | NC_001276 | S. H. Kim, E. Y. Je, and D. W. Park, personal communication | ||
| Venerupis philippinarum-Fb | NC_003354 | M. Okazaki and R. Ueshima, personal communication | ||
| Inversidens japanensis-F | AB055625 | M. Okazaki and R. Ueshima, personal communication | ||
| Inversidens japanensis-Mc | AB055624 | M. Okazaki and R. Ueshima personal communication | ||
| Pecten maximusa | X92688 | D. Sellos, M. Mommerot, and A. Rigaa, personal communication | ||
| Gastropoda | ||||
| Albinaria coerulea | NC_001761 | Hatzoglou, Rodakis, and Lecanidou 1995 | ||
| Cepaea nemoralis | NC_001816 | Terrett, Miles, and Thomas 1996 | ||
| Littorina saxitilisa | AJ132137 | Wilding, Mill, and Grahame 1999 | ||
| Pupa strigosa | NC_002176 | Kurabayashi and Ueshima 2000 | ||
| Roboastra europaea | AY083457 | Grande et al. 2002 | ||
| Polyplacophora | ||||
| Katharina tunicata | U09810 | Boore and Brown 1994 | ||
| Cephalopoda | ||||
| Loligo bleekeri | NC_002507 | Tomita et al. 2002 | ||
Note.—Bold terms are molluscan classes.
aIncomplete data.
bF = female mitotype.
cM = male mitotype.
Primers Used in the Amplification and Sequencing of the Complete Mitochondrial Genome of Lampsilis ornata.
| Primer | Primer Sequence (5′ to 3′) | Base Position Range |
|---|---|---|
| ND4-1084R | GGAGCTGCCATGTTCATTGATCTTA | 263–287 |
| aND1-end | ATAGCACACATTAAAAACCAACACAAAGC | 278–306 |
| ND4-630F | TCGCCAAAGCTCATGTTGAGG | 705–725 |
| aAsp-end | GGTGCTTATTTTTTCTTTTAGGGTTGGTTC | 1013–1042 |
| aAsp-196F | GAAGCAGCGTTAGGGTTGAGTGTATTAG | 1402–1429 |
| aAsp-150Fc | GTGAAGTGGGGTTTTGGTAAGAAGTTTAG | 1668–1696 |
| bAsp-54F | GCAGGCTTATGTGTTTGTTTTGTTGG | 1972–1997 |
| aCO3-350F | CTGTCTTTGACTTTTGTGTTGGCTAG | 2310–2335 |
| aCO3-60F | TCATCTTTGGATGCTGGGTTAAGCTA | 2592–2617 |
| CO3-730R | TCAAACTACATCTACAAAATGTCAG | 2724–2748 |
| CO3-312F | GTATGGGTTAGGTTTAACGGTCTTATTGG | 2963–2991 |
| CO3-176R | CACAACCCAACTCCACAGTTGGAACAAG | 3128–3155 |
| CO3-180F | TGGTGGCGAGATGTAATTCGTGAAG | 3278–3302 |
| CO3-60R | GGTCTTTCCTGCGTCATTTCACTCG | 3528–3552 |
| bCO3-486R | GTTAGACAATACAATACCCGTCAATCC | 3963–3989 |
| HCO2189a | TTAACTTCAGGGTGACCAAAAAATCA | 4312–4338 |
| COI-477F | GATTCCGTTGTTTGTATGGGCTGTTACTG | 4475–4503 |
| J2160-F | CTTTACATCTTGCTGGTGCCTCTTCTAT | 4570–4597 |
| COI-215R | CCATATCAGGAGCCCCAATCATAAGAG | 4766–4792 |
| LCO1490a | GGTCAACAAATCATAAAGATATTGG | 4996–5020 |
| J408-R | TTGGGCTCTTCCTTCAGTCGGTGT | 5224–5247 |
| J190-R | CCTNACAAGAACTCGCCTATAAACCCCA | 5268–5295 |
| aHis-580R | ATTTGTTCTTGTCTTGGTTTCGTCTTTTG | 5624–5652 |
| J420/190R | GCCACTCATGAATAACCCCACCAAA | 5841–5865 |
| Ala-F | GCAATCAACCATTGAGTCCAACTCTAGAAC | 6460–6489 |
| aAla-540F | GTCCTATCTTCCTCAAACTGATTAACAACC | 7086–7115 |
| Met-R | GAAGAGTGATCATTTTGGGTTATGAGCC | 8023–8052 |
| L1091b | TAATAAAAAACTAGGATTAGATACCCTATTAT | 8603–8636 |
| L1091-R | GTAGTCGGGTATCTAATCCGAGTTTTGAGGTTGT | 8603–8638 |
| 12S-100R | GATGAGGAAGTAAAGCACCGCCACA | 8689–8713 |
| 12S-360F | TTAGCACACCAAGCAAATCTGAACAAGAACT | 8952–8982 |
| H1478b | TAATAATAAGAGCGACGGGCGATGTGT | 8992–9019 |
| 16S-70R | CTGTGGTGGGCGTAGAGGTAGTGAC | 9369–9393 |
| 16Sar-L-mytc | CGACTGTTTAACAAAAACAT | 9925–9943 |
| 16S-780F | CTAAATAATAAAGGAAGACGAAAAGACC | 10127–10154 |
| 16S-78R | CGGGGTCTTTTCGTCTTCCTTTATTAT | 10131–10157 |
| 16Sbr-H-mytc | CCGTTCTGAACTCAGCTCATGT | 10432–10452 |
| Pro-10R | CTATTTTCTGGGGTGAGTATGTGTAGG | 10747–10773 |
| unicytbFd | TGTGGGGCGACTGTTATTACTAA | 11253–11275 |
| cytB-150R | GCAATAGCAAAAGGAAGGACAAAGTGAAG | 11373–11401 |
| cytB-390F | GTACTAACTCCCACTTGCTTGTTCTTCC | 11544–11571 |
| unicytbRd | AAGAAGTATCATTCGGGTTG | 11637–11656 |
| Phe-47F | GCCATTGCCCTTCTCCATTTTCCCATAC | 12034–12061 |
| aPhe-334F | CAATACCAGAAATCAACCTTCCAGACAC | 12387–12414 |
| aPhe-768Fc | TACACTCCTTAGCACCAAACACCCAC | 12871–12896 |
| bGln-210Rc | CTACTCTTGTTGAGTGTAGATCACATTGC | 13942–13970 |
| ND1-350R | GGTTGTTGGAAATAAAGGTAGATGGGGTAAGAT | 14015–14045 |
| Gln-47R | GCAAATTGGTTGAAAGCGGAAATATTGG | 14220–14249 |
| ND1-110R | AGGTAGATAGTTTGAAGAGGTTGGTGTTA | 14707–14735 |
| ND1-240F | CTAGCCGTCTACACAACCCTCATAACAG | 14856–14883 |
| NIJ-12073e | TCGGATTCTCCTTCTGCAAAGTC | 15120–15142 |
| ND1-1750F | TACCATAATCTAACCTCAACTCTAAACA | 15410–15437 |
| aND1-385F | ACTCTCCCTCAAACTATCAAACAAATC | 15823–15849 |
| Primer | Primer Sequence (5′ to 3′) | Base Position Range |
|---|---|---|
| ND4-1084R | GGAGCTGCCATGTTCATTGATCTTA | 263–287 |
| aND1-end | ATAGCACACATTAAAAACCAACACAAAGC | 278–306 |
| ND4-630F | TCGCCAAAGCTCATGTTGAGG | 705–725 |
| aAsp-end | GGTGCTTATTTTTTCTTTTAGGGTTGGTTC | 1013–1042 |
| aAsp-196F | GAAGCAGCGTTAGGGTTGAGTGTATTAG | 1402–1429 |
| aAsp-150Fc | GTGAAGTGGGGTTTTGGTAAGAAGTTTAG | 1668–1696 |
| bAsp-54F | GCAGGCTTATGTGTTTGTTTTGTTGG | 1972–1997 |
| aCO3-350F | CTGTCTTTGACTTTTGTGTTGGCTAG | 2310–2335 |
| aCO3-60F | TCATCTTTGGATGCTGGGTTAAGCTA | 2592–2617 |
| CO3-730R | TCAAACTACATCTACAAAATGTCAG | 2724–2748 |
| CO3-312F | GTATGGGTTAGGTTTAACGGTCTTATTGG | 2963–2991 |
| CO3-176R | CACAACCCAACTCCACAGTTGGAACAAG | 3128–3155 |
| CO3-180F | TGGTGGCGAGATGTAATTCGTGAAG | 3278–3302 |
| CO3-60R | GGTCTTTCCTGCGTCATTTCACTCG | 3528–3552 |
| bCO3-486R | GTTAGACAATACAATACCCGTCAATCC | 3963–3989 |
| HCO2189a | TTAACTTCAGGGTGACCAAAAAATCA | 4312–4338 |
| COI-477F | GATTCCGTTGTTTGTATGGGCTGTTACTG | 4475–4503 |
| J2160-F | CTTTACATCTTGCTGGTGCCTCTTCTAT | 4570–4597 |
| COI-215R | CCATATCAGGAGCCCCAATCATAAGAG | 4766–4792 |
| LCO1490a | GGTCAACAAATCATAAAGATATTGG | 4996–5020 |
| J408-R | TTGGGCTCTTCCTTCAGTCGGTGT | 5224–5247 |
| J190-R | CCTNACAAGAACTCGCCTATAAACCCCA | 5268–5295 |
| aHis-580R | ATTTGTTCTTGTCTTGGTTTCGTCTTTTG | 5624–5652 |
| J420/190R | GCCACTCATGAATAACCCCACCAAA | 5841–5865 |
| Ala-F | GCAATCAACCATTGAGTCCAACTCTAGAAC | 6460–6489 |
| aAla-540F | GTCCTATCTTCCTCAAACTGATTAACAACC | 7086–7115 |
| Met-R | GAAGAGTGATCATTTTGGGTTATGAGCC | 8023–8052 |
| L1091b | TAATAAAAAACTAGGATTAGATACCCTATTAT | 8603–8636 |
| L1091-R | GTAGTCGGGTATCTAATCCGAGTTTTGAGGTTGT | 8603–8638 |
| 12S-100R | GATGAGGAAGTAAAGCACCGCCACA | 8689–8713 |
| 12S-360F | TTAGCACACCAAGCAAATCTGAACAAGAACT | 8952–8982 |
| H1478b | TAATAATAAGAGCGACGGGCGATGTGT | 8992–9019 |
| 16S-70R | CTGTGGTGGGCGTAGAGGTAGTGAC | 9369–9393 |
| 16Sar-L-mytc | CGACTGTTTAACAAAAACAT | 9925–9943 |
| 16S-780F | CTAAATAATAAAGGAAGACGAAAAGACC | 10127–10154 |
| 16S-78R | CGGGGTCTTTTCGTCTTCCTTTATTAT | 10131–10157 |
| 16Sbr-H-mytc | CCGTTCTGAACTCAGCTCATGT | 10432–10452 |
| Pro-10R | CTATTTTCTGGGGTGAGTATGTGTAGG | 10747–10773 |
| unicytbFd | TGTGGGGCGACTGTTATTACTAA | 11253–11275 |
| cytB-150R | GCAATAGCAAAAGGAAGGACAAAGTGAAG | 11373–11401 |
| cytB-390F | GTACTAACTCCCACTTGCTTGTTCTTCC | 11544–11571 |
| unicytbRd | AAGAAGTATCATTCGGGTTG | 11637–11656 |
| Phe-47F | GCCATTGCCCTTCTCCATTTTCCCATAC | 12034–12061 |
| aPhe-334F | CAATACCAGAAATCAACCTTCCAGACAC | 12387–12414 |
| aPhe-768Fc | TACACTCCTTAGCACCAAACACCCAC | 12871–12896 |
| bGln-210Rc | CTACTCTTGTTGAGTGTAGATCACATTGC | 13942–13970 |
| ND1-350R | GGTTGTTGGAAATAAAGGTAGATGGGGTAAGAT | 14015–14045 |
| Gln-47R | GCAAATTGGTTGAAAGCGGAAATATTGG | 14220–14249 |
| ND1-110R | AGGTAGATAGTTTGAAGAGGTTGGTGTTA | 14707–14735 |
| ND1-240F | CTAGCCGTCTACACAACCCTCATAACAG | 14856–14883 |
| NIJ-12073e | TCGGATTCTCCTTCTGCAAAGTC | 15120–15142 |
| ND1-1750F | TACCATAATCTAACCTCAACTCTAAACA | 15410–15437 |
| aND1-385F | ACTCTCCCTCAAACTATCAAACAAATC | 15823–15849 |
dBonnie Bowen, unpublished data.
Primers Used in the Amplification and Sequencing of the Complete Mitochondrial Genome of Lampsilis ornata.
| Primer | Primer Sequence (5′ to 3′) | Base Position Range |
|---|---|---|
| ND4-1084R | GGAGCTGCCATGTTCATTGATCTTA | 263–287 |
| aND1-end | ATAGCACACATTAAAAACCAACACAAAGC | 278–306 |
| ND4-630F | TCGCCAAAGCTCATGTTGAGG | 705–725 |
| aAsp-end | GGTGCTTATTTTTTCTTTTAGGGTTGGTTC | 1013–1042 |
| aAsp-196F | GAAGCAGCGTTAGGGTTGAGTGTATTAG | 1402–1429 |
| aAsp-150Fc | GTGAAGTGGGGTTTTGGTAAGAAGTTTAG | 1668–1696 |
| bAsp-54F | GCAGGCTTATGTGTTTGTTTTGTTGG | 1972–1997 |
| aCO3-350F | CTGTCTTTGACTTTTGTGTTGGCTAG | 2310–2335 |
| aCO3-60F | TCATCTTTGGATGCTGGGTTAAGCTA | 2592–2617 |
| CO3-730R | TCAAACTACATCTACAAAATGTCAG | 2724–2748 |
| CO3-312F | GTATGGGTTAGGTTTAACGGTCTTATTGG | 2963–2991 |
| CO3-176R | CACAACCCAACTCCACAGTTGGAACAAG | 3128–3155 |
| CO3-180F | TGGTGGCGAGATGTAATTCGTGAAG | 3278–3302 |
| CO3-60R | GGTCTTTCCTGCGTCATTTCACTCG | 3528–3552 |
| bCO3-486R | GTTAGACAATACAATACCCGTCAATCC | 3963–3989 |
| HCO2189a | TTAACTTCAGGGTGACCAAAAAATCA | 4312–4338 |
| COI-477F | GATTCCGTTGTTTGTATGGGCTGTTACTG | 4475–4503 |
| J2160-F | CTTTACATCTTGCTGGTGCCTCTTCTAT | 4570–4597 |
| COI-215R | CCATATCAGGAGCCCCAATCATAAGAG | 4766–4792 |
| LCO1490a | GGTCAACAAATCATAAAGATATTGG | 4996–5020 |
| J408-R | TTGGGCTCTTCCTTCAGTCGGTGT | 5224–5247 |
| J190-R | CCTNACAAGAACTCGCCTATAAACCCCA | 5268–5295 |
| aHis-580R | ATTTGTTCTTGTCTTGGTTTCGTCTTTTG | 5624–5652 |
| J420/190R | GCCACTCATGAATAACCCCACCAAA | 5841–5865 |
| Ala-F | GCAATCAACCATTGAGTCCAACTCTAGAAC | 6460–6489 |
| aAla-540F | GTCCTATCTTCCTCAAACTGATTAACAACC | 7086–7115 |
| Met-R | GAAGAGTGATCATTTTGGGTTATGAGCC | 8023–8052 |
| L1091b | TAATAAAAAACTAGGATTAGATACCCTATTAT | 8603–8636 |
| L1091-R | GTAGTCGGGTATCTAATCCGAGTTTTGAGGTTGT | 8603–8638 |
| 12S-100R | GATGAGGAAGTAAAGCACCGCCACA | 8689–8713 |
| 12S-360F | TTAGCACACCAAGCAAATCTGAACAAGAACT | 8952–8982 |
| H1478b | TAATAATAAGAGCGACGGGCGATGTGT | 8992–9019 |
| 16S-70R | CTGTGGTGGGCGTAGAGGTAGTGAC | 9369–9393 |
| 16Sar-L-mytc | CGACTGTTTAACAAAAACAT | 9925–9943 |
| 16S-780F | CTAAATAATAAAGGAAGACGAAAAGACC | 10127–10154 |
| 16S-78R | CGGGGTCTTTTCGTCTTCCTTTATTAT | 10131–10157 |
| 16Sbr-H-mytc | CCGTTCTGAACTCAGCTCATGT | 10432–10452 |
| Pro-10R | CTATTTTCTGGGGTGAGTATGTGTAGG | 10747–10773 |
| unicytbFd | TGTGGGGCGACTGTTATTACTAA | 11253–11275 |
| cytB-150R | GCAATAGCAAAAGGAAGGACAAAGTGAAG | 11373–11401 |
| cytB-390F | GTACTAACTCCCACTTGCTTGTTCTTCC | 11544–11571 |
| unicytbRd | AAGAAGTATCATTCGGGTTG | 11637–11656 |
| Phe-47F | GCCATTGCCCTTCTCCATTTTCCCATAC | 12034–12061 |
| aPhe-334F | CAATACCAGAAATCAACCTTCCAGACAC | 12387–12414 |
| aPhe-768Fc | TACACTCCTTAGCACCAAACACCCAC | 12871–12896 |
| bGln-210Rc | CTACTCTTGTTGAGTGTAGATCACATTGC | 13942–13970 |
| ND1-350R | GGTTGTTGGAAATAAAGGTAGATGGGGTAAGAT | 14015–14045 |
| Gln-47R | GCAAATTGGTTGAAAGCGGAAATATTGG | 14220–14249 |
| ND1-110R | AGGTAGATAGTTTGAAGAGGTTGGTGTTA | 14707–14735 |
| ND1-240F | CTAGCCGTCTACACAACCCTCATAACAG | 14856–14883 |
| NIJ-12073e | TCGGATTCTCCTTCTGCAAAGTC | 15120–15142 |
| ND1-1750F | TACCATAATCTAACCTCAACTCTAAACA | 15410–15437 |
| aND1-385F | ACTCTCCCTCAAACTATCAAACAAATC | 15823–15849 |
| Primer | Primer Sequence (5′ to 3′) | Base Position Range |
|---|---|---|
| ND4-1084R | GGAGCTGCCATGTTCATTGATCTTA | 263–287 |
| aND1-end | ATAGCACACATTAAAAACCAACACAAAGC | 278–306 |
| ND4-630F | TCGCCAAAGCTCATGTTGAGG | 705–725 |
| aAsp-end | GGTGCTTATTTTTTCTTTTAGGGTTGGTTC | 1013–1042 |
| aAsp-196F | GAAGCAGCGTTAGGGTTGAGTGTATTAG | 1402–1429 |
| aAsp-150Fc | GTGAAGTGGGGTTTTGGTAAGAAGTTTAG | 1668–1696 |
| bAsp-54F | GCAGGCTTATGTGTTTGTTTTGTTGG | 1972–1997 |
| aCO3-350F | CTGTCTTTGACTTTTGTGTTGGCTAG | 2310–2335 |
| aCO3-60F | TCATCTTTGGATGCTGGGTTAAGCTA | 2592–2617 |
| CO3-730R | TCAAACTACATCTACAAAATGTCAG | 2724–2748 |
| CO3-312F | GTATGGGTTAGGTTTAACGGTCTTATTGG | 2963–2991 |
| CO3-176R | CACAACCCAACTCCACAGTTGGAACAAG | 3128–3155 |
| CO3-180F | TGGTGGCGAGATGTAATTCGTGAAG | 3278–3302 |
| CO3-60R | GGTCTTTCCTGCGTCATTTCACTCG | 3528–3552 |
| bCO3-486R | GTTAGACAATACAATACCCGTCAATCC | 3963–3989 |
| HCO2189a | TTAACTTCAGGGTGACCAAAAAATCA | 4312–4338 |
| COI-477F | GATTCCGTTGTTTGTATGGGCTGTTACTG | 4475–4503 |
| J2160-F | CTTTACATCTTGCTGGTGCCTCTTCTAT | 4570–4597 |
| COI-215R | CCATATCAGGAGCCCCAATCATAAGAG | 4766–4792 |
| LCO1490a | GGTCAACAAATCATAAAGATATTGG | 4996–5020 |
| J408-R | TTGGGCTCTTCCTTCAGTCGGTGT | 5224–5247 |
| J190-R | CCTNACAAGAACTCGCCTATAAACCCCA | 5268–5295 |
| aHis-580R | ATTTGTTCTTGTCTTGGTTTCGTCTTTTG | 5624–5652 |
| J420/190R | GCCACTCATGAATAACCCCACCAAA | 5841–5865 |
| Ala-F | GCAATCAACCATTGAGTCCAACTCTAGAAC | 6460–6489 |
| aAla-540F | GTCCTATCTTCCTCAAACTGATTAACAACC | 7086–7115 |
| Met-R | GAAGAGTGATCATTTTGGGTTATGAGCC | 8023–8052 |
| L1091b | TAATAAAAAACTAGGATTAGATACCCTATTAT | 8603–8636 |
| L1091-R | GTAGTCGGGTATCTAATCCGAGTTTTGAGGTTGT | 8603–8638 |
| 12S-100R | GATGAGGAAGTAAAGCACCGCCACA | 8689–8713 |
| 12S-360F | TTAGCACACCAAGCAAATCTGAACAAGAACT | 8952–8982 |
| H1478b | TAATAATAAGAGCGACGGGCGATGTGT | 8992–9019 |
| 16S-70R | CTGTGGTGGGCGTAGAGGTAGTGAC | 9369–9393 |
| 16Sar-L-mytc | CGACTGTTTAACAAAAACAT | 9925–9943 |
| 16S-780F | CTAAATAATAAAGGAAGACGAAAAGACC | 10127–10154 |
| 16S-78R | CGGGGTCTTTTCGTCTTCCTTTATTAT | 10131–10157 |
| 16Sbr-H-mytc | CCGTTCTGAACTCAGCTCATGT | 10432–10452 |
| Pro-10R | CTATTTTCTGGGGTGAGTATGTGTAGG | 10747–10773 |
| unicytbFd | TGTGGGGCGACTGTTATTACTAA | 11253–11275 |
| cytB-150R | GCAATAGCAAAAGGAAGGACAAAGTGAAG | 11373–11401 |
| cytB-390F | GTACTAACTCCCACTTGCTTGTTCTTCC | 11544–11571 |
| unicytbRd | AAGAAGTATCATTCGGGTTG | 11637–11656 |
| Phe-47F | GCCATTGCCCTTCTCCATTTTCCCATAC | 12034–12061 |
| aPhe-334F | CAATACCAGAAATCAACCTTCCAGACAC | 12387–12414 |
| aPhe-768Fc | TACACTCCTTAGCACCAAACACCCAC | 12871–12896 |
| bGln-210Rc | CTACTCTTGTTGAGTGTAGATCACATTGC | 13942–13970 |
| ND1-350R | GGTTGTTGGAAATAAAGGTAGATGGGGTAAGAT | 14015–14045 |
| Gln-47R | GCAAATTGGTTGAAAGCGGAAATATTGG | 14220–14249 |
| ND1-110R | AGGTAGATAGTTTGAAGAGGTTGGTGTTA | 14707–14735 |
| ND1-240F | CTAGCCGTCTACACAACCCTCATAACAG | 14856–14883 |
| NIJ-12073e | TCGGATTCTCCTTCTGCAAAGTC | 15120–15142 |
| ND1-1750F | TACCATAATCTAACCTCAACTCTAAACA | 15410–15437 |
| aND1-385F | ACTCTCCCTCAAACTATCAAACAAATC | 15823–15849 |
dBonnie Bowen, unpublished data.
| Inversidens | Lampsilis | Katharina | Littorina | Cepaea | Albinaria | Roboastra | Pupa | Loligo | |
|---|---|---|---|---|---|---|---|---|---|
| Inversidens | — | 10 | 13 | 26 | 13 | 30 | 16 | 26 | 20 |
| Lampsilis | 56 | — | 16 | 20 | 16 | 30 | 20 | 20 | 26 |
| Katharina | 30 | 36 | — | 26 | 23 | 23 | 16 | 13 | 16 |
| Littorina | 26 | 43 | 50 | — | 16 | 23 | 16 | 16 | 26 |
| Cepaea | 16 | 36 | 20 | 20 | — | 16 | 20 | 16 | 13 |
| Albinaria | 23 | 26 | 26 | 26 | 33 | — | 13 | 26 | 13 |
| Roboastra | 40 | 36 | 23 | 23 | 30 | 36 | — | 23 | 23 |
| Pupa | 33 | 36 | 26 | 30 | 30 | 30 | 26 | — | 16 |
| Loligo | 30 | 33 | 50 | 50 | 20 | 33 | 23 | 30 | — |
| Inversidens | Lampsilis | Katharina | Littorina | Cepaea | Albinaria | Roboastra | Pupa | Loligo | |
|---|---|---|---|---|---|---|---|---|---|
| Inversidens | — | 10 | 13 | 26 | 13 | 30 | 16 | 26 | 20 |
| Lampsilis | 56 | — | 16 | 20 | 16 | 30 | 20 | 20 | 26 |
| Katharina | 30 | 36 | — | 26 | 23 | 23 | 16 | 13 | 16 |
| Littorina | 26 | 43 | 50 | — | 16 | 23 | 16 | 16 | 26 |
| Cepaea | 16 | 36 | 20 | 20 | — | 16 | 20 | 16 | 13 |
| Albinaria | 23 | 26 | 26 | 26 | 33 | — | 13 | 26 | 13 |
| Roboastra | 40 | 36 | 23 | 23 | 30 | 36 | — | 23 | 23 |
| Pupa | 33 | 36 | 26 | 30 | 30 | 30 | 26 | — | 16 |
| Loligo | 30 | 33 | 50 | 50 | 20 | 33 | 23 | 30 | — |
aPercentage of identical amino acid positions.
bPercentage of amino acid positions that belong to the same chemical group.
| Inversidens | Lampsilis | Katharina | Littorina | Cepaea | Albinaria | Roboastra | Pupa | Loligo | |
|---|---|---|---|---|---|---|---|---|---|
| Inversidens | — | 10 | 13 | 26 | 13 | 30 | 16 | 26 | 20 |
| Lampsilis | 56 | — | 16 | 20 | 16 | 30 | 20 | 20 | 26 |
| Katharina | 30 | 36 | — | 26 | 23 | 23 | 16 | 13 | 16 |
| Littorina | 26 | 43 | 50 | — | 16 | 23 | 16 | 16 | 26 |
| Cepaea | 16 | 36 | 20 | 20 | — | 16 | 20 | 16 | 13 |
| Albinaria | 23 | 26 | 26 | 26 | 33 | — | 13 | 26 | 13 |
| Roboastra | 40 | 36 | 23 | 23 | 30 | 36 | — | 23 | 23 |
| Pupa | 33 | 36 | 26 | 30 | 30 | 30 | 26 | — | 16 |
| Loligo | 30 | 33 | 50 | 50 | 20 | 33 | 23 | 30 | — |
| Inversidens | Lampsilis | Katharina | Littorina | Cepaea | Albinaria | Roboastra | Pupa | Loligo | |
|---|---|---|---|---|---|---|---|---|---|
| Inversidens | — | 10 | 13 | 26 | 13 | 30 | 16 | 26 | 20 |
| Lampsilis | 56 | — | 16 | 20 | 16 | 30 | 20 | 20 | 26 |
| Katharina | 30 | 36 | — | 26 | 23 | 23 | 16 | 13 | 16 |
| Littorina | 26 | 43 | 50 | — | 16 | 23 | 16 | 16 | 26 |
| Cepaea | 16 | 36 | 20 | 20 | — | 16 | 20 | 16 | 13 |
| Albinaria | 23 | 26 | 26 | 26 | 33 | — | 13 | 26 | 13 |
| Roboastra | 40 | 36 | 23 | 23 | 30 | 36 | — | 23 | 23 |
| Pupa | 33 | 36 | 26 | 30 | 30 | 30 | 26 | — | 16 |
| Loligo | 30 | 33 | 50 | 50 | 20 | 33 | 23 | 30 | — |
aPercentage of identical amino acid positions.
bPercentage of amino acid positions that belong to the same chemical group.
Codon Usage in 13 Mitochondrial Protein-Coding Genes of Lampsilis ornata.
| AA | Codon | N | % |
|---|---|---|---|
| Phe (F) | TTT | 267 | 7.18 |
| TTC | 37 | 0.10 | |
| Leu (L1) | TTA | 188 | 5.06 |
| TTG | 183 | 4.92 | |
| Ser (S2) | TCT | 110 | 2.96 |
| TCC | 26 | 0.70 | |
| TCA | 51 | 1.37 | |
| TCG | 13 | 0.35 | |
| Tyr (Y) | TAT | 94 | 2.53 |
| TAC | 33 | 0.89 | |
| TERM | TAA | 4 | 0.11 |
| TAG | 8 | 0.22 | |
| Cys (C) | TGT | 52 | 1.40 |
| TGC | 6 | 0.16 | |
| Trp (W) | TGA | 46 | 1.24 |
| TGG | 67 | 1.80 | |
| Leu (L2) | CTT | 81 | 2.18 |
| CTC | 26 | 0.70 | |
| CTA | 83 | 2.23 | |
| CTG | 15 | 0.40 | |
| Pro (P) | CCT | 60 | 1.61 |
| CCC | 22 | 0.59 | |
| CCA | 45 | 1.21 | |
| CCG | 16 | 0.43 | |
| His (H) | CAT | 58 | 1.56 |
| CAC | 19 | 0.51 | |
| Gln (Q) | CAA | 35 | 0.94 |
| CAG | 26 | 0.70 | |
| Arg (R) | CGT | 10 | 0.27 |
| CGC | 4 | 0.11 | |
| CGA | 32 | 0.86 | |
| CGG | 13 | 0.35 | |
| Ile (I) | ATT | 188 | 5.06 |
| ATC | 55 | 1.48 | |
| Met (M) | ATA | 103 | 2.77 |
| ATG | 70 | 1.88 | |
| Thr (T) | ACT | 77 | 2.07 |
| ACC | 40 | 1.08 | |
| ACA | 43 | 1.16 | |
| ACG | 13 | 0.35 | |
| Asn (N) | AAT | 58 | 1.56 |
| AAC | 32 | 0.86 | |
| Lys (K) | AAA | 49 | 1.32 |
| AAG | 40 | 1.08 | |
| Ser (S1) | AGT | 55 | 1.48 |
| AGC | 16 | 0.43 | |
| AGA | 35 | 0.94 | |
| AGG | 49 | 1.32 | |
| Val (V) | GTT | 190 | 5.11 |
| GTC | 22 | 0.59 | |
| GTA | 68 | 1.83 | |
| GTG | 95 | 2.56 | |
| Ala (A) | GCT | 99 | 2.66 |
| GCC | 39 | 1.05 | |
| GCA | 40 | 1.08 | |
| GCG | 20 | 0.54 | |
| Asp (D) | GAT | 52 | 1.40 |
| GAC | 14 | 0.38 | |
| Glu (E) | GAA | 29 | 0.78 |
| GAG | 57 | 1.53 | |
| Gly (G) | GGT | 127 | 3.42 |
| GGC | 26 | 0.70 | |
| GGA | 60 | 1.61 | |
| GGG | 126 | 3.39 |
| AA | Codon | N | % |
|---|---|---|---|
| Phe (F) | TTT | 267 | 7.18 |
| TTC | 37 | 0.10 | |
| Leu (L1) | TTA | 188 | 5.06 |
| TTG | 183 | 4.92 | |
| Ser (S2) | TCT | 110 | 2.96 |
| TCC | 26 | 0.70 | |
| TCA | 51 | 1.37 | |
| TCG | 13 | 0.35 | |
| Tyr (Y) | TAT | 94 | 2.53 |
| TAC | 33 | 0.89 | |
| TERM | TAA | 4 | 0.11 |
| TAG | 8 | 0.22 | |
| Cys (C) | TGT | 52 | 1.40 |
| TGC | 6 | 0.16 | |
| Trp (W) | TGA | 46 | 1.24 |
| TGG | 67 | 1.80 | |
| Leu (L2) | CTT | 81 | 2.18 |
| CTC | 26 | 0.70 | |
| CTA | 83 | 2.23 | |
| CTG | 15 | 0.40 | |
| Pro (P) | CCT | 60 | 1.61 |
| CCC | 22 | 0.59 | |
| CCA | 45 | 1.21 | |
| CCG | 16 | 0.43 | |
| His (H) | CAT | 58 | 1.56 |
| CAC | 19 | 0.51 | |
| Gln (Q) | CAA | 35 | 0.94 |
| CAG | 26 | 0.70 | |
| Arg (R) | CGT | 10 | 0.27 |
| CGC | 4 | 0.11 | |
| CGA | 32 | 0.86 | |
| CGG | 13 | 0.35 | |
| Ile (I) | ATT | 188 | 5.06 |
| ATC | 55 | 1.48 | |
| Met (M) | ATA | 103 | 2.77 |
| ATG | 70 | 1.88 | |
| Thr (T) | ACT | 77 | 2.07 |
| ACC | 40 | 1.08 | |
| ACA | 43 | 1.16 | |
| ACG | 13 | 0.35 | |
| Asn (N) | AAT | 58 | 1.56 |
| AAC | 32 | 0.86 | |
| Lys (K) | AAA | 49 | 1.32 |
| AAG | 40 | 1.08 | |
| Ser (S1) | AGT | 55 | 1.48 |
| AGC | 16 | 0.43 | |
| AGA | 35 | 0.94 | |
| AGG | 49 | 1.32 | |
| Val (V) | GTT | 190 | 5.11 |
| GTC | 22 | 0.59 | |
| GTA | 68 | 1.83 | |
| GTG | 95 | 2.56 | |
| Ala (A) | GCT | 99 | 2.66 |
| GCC | 39 | 1.05 | |
| GCA | 40 | 1.08 | |
| GCG | 20 | 0.54 | |
| Asp (D) | GAT | 52 | 1.40 |
| GAC | 14 | 0.38 | |
| Glu (E) | GAA | 29 | 0.78 |
| GAG | 57 | 1.53 | |
| Gly (G) | GGT | 127 | 3.42 |
| GGC | 26 | 0.70 | |
| GGA | 60 | 1.61 | |
| GGG | 126 | 3.39 |
Note.—AA = amino acid; N = total number of particular codon in all proteins; % = percentage of total codon usage; TERM = termination codons. The total number of codons is 3,717. Abbreviated stop codons were excluded.
Codon Usage in 13 Mitochondrial Protein-Coding Genes of Lampsilis ornata.
| AA | Codon | N | % |
|---|---|---|---|
| Phe (F) | TTT | 267 | 7.18 |
| TTC | 37 | 0.10 | |
| Leu (L1) | TTA | 188 | 5.06 |
| TTG | 183 | 4.92 | |
| Ser (S2) | TCT | 110 | 2.96 |
| TCC | 26 | 0.70 | |
| TCA | 51 | 1.37 | |
| TCG | 13 | 0.35 | |
| Tyr (Y) | TAT | 94 | 2.53 |
| TAC | 33 | 0.89 | |
| TERM | TAA | 4 | 0.11 |
| TAG | 8 | 0.22 | |
| Cys (C) | TGT | 52 | 1.40 |
| TGC | 6 | 0.16 | |
| Trp (W) | TGA | 46 | 1.24 |
| TGG | 67 | 1.80 | |
| Leu (L2) | CTT | 81 | 2.18 |
| CTC | 26 | 0.70 | |
| CTA | 83 | 2.23 | |
| CTG | 15 | 0.40 | |
| Pro (P) | CCT | 60 | 1.61 |
| CCC | 22 | 0.59 | |
| CCA | 45 | 1.21 | |
| CCG | 16 | 0.43 | |
| His (H) | CAT | 58 | 1.56 |
| CAC | 19 | 0.51 | |
| Gln (Q) | CAA | 35 | 0.94 |
| CAG | 26 | 0.70 | |
| Arg (R) | CGT | 10 | 0.27 |
| CGC | 4 | 0.11 | |
| CGA | 32 | 0.86 | |
| CGG | 13 | 0.35 | |
| Ile (I) | ATT | 188 | 5.06 |
| ATC | 55 | 1.48 | |
| Met (M) | ATA | 103 | 2.77 |
| ATG | 70 | 1.88 | |
| Thr (T) | ACT | 77 | 2.07 |
| ACC | 40 | 1.08 | |
| ACA | 43 | 1.16 | |
| ACG | 13 | 0.35 | |
| Asn (N) | AAT | 58 | 1.56 |
| AAC | 32 | 0.86 | |
| Lys (K) | AAA | 49 | 1.32 |
| AAG | 40 | 1.08 | |
| Ser (S1) | AGT | 55 | 1.48 |
| AGC | 16 | 0.43 | |
| AGA | 35 | 0.94 | |
| AGG | 49 | 1.32 | |
| Val (V) | GTT | 190 | 5.11 |
| GTC | 22 | 0.59 | |
| GTA | 68 | 1.83 | |
| GTG | 95 | 2.56 | |
| Ala (A) | GCT | 99 | 2.66 |
| GCC | 39 | 1.05 | |
| GCA | 40 | 1.08 | |
| GCG | 20 | 0.54 | |
| Asp (D) | GAT | 52 | 1.40 |
| GAC | 14 | 0.38 | |
| Glu (E) | GAA | 29 | 0.78 |
| GAG | 57 | 1.53 | |
| Gly (G) | GGT | 127 | 3.42 |
| GGC | 26 | 0.70 | |
| GGA | 60 | 1.61 | |
| GGG | 126 | 3.39 |
| AA | Codon | N | % |
|---|---|---|---|
| Phe (F) | TTT | 267 | 7.18 |
| TTC | 37 | 0.10 | |
| Leu (L1) | TTA | 188 | 5.06 |
| TTG | 183 | 4.92 | |
| Ser (S2) | TCT | 110 | 2.96 |
| TCC | 26 | 0.70 | |
| TCA | 51 | 1.37 | |
| TCG | 13 | 0.35 | |
| Tyr (Y) | TAT | 94 | 2.53 |
| TAC | 33 | 0.89 | |
| TERM | TAA | 4 | 0.11 |
| TAG | 8 | 0.22 | |
| Cys (C) | TGT | 52 | 1.40 |
| TGC | 6 | 0.16 | |
| Trp (W) | TGA | 46 | 1.24 |
| TGG | 67 | 1.80 | |
| Leu (L2) | CTT | 81 | 2.18 |
| CTC | 26 | 0.70 | |
| CTA | 83 | 2.23 | |
| CTG | 15 | 0.40 | |
| Pro (P) | CCT | 60 | 1.61 |
| CCC | 22 | 0.59 | |
| CCA | 45 | 1.21 | |
| CCG | 16 | 0.43 | |
| His (H) | CAT | 58 | 1.56 |
| CAC | 19 | 0.51 | |
| Gln (Q) | CAA | 35 | 0.94 |
| CAG | 26 | 0.70 | |
| Arg (R) | CGT | 10 | 0.27 |
| CGC | 4 | 0.11 | |
| CGA | 32 | 0.86 | |
| CGG | 13 | 0.35 | |
| Ile (I) | ATT | 188 | 5.06 |
| ATC | 55 | 1.48 | |
| Met (M) | ATA | 103 | 2.77 |
| ATG | 70 | 1.88 | |
| Thr (T) | ACT | 77 | 2.07 |
| ACC | 40 | 1.08 | |
| ACA | 43 | 1.16 | |
| ACG | 13 | 0.35 | |
| Asn (N) | AAT | 58 | 1.56 |
| AAC | 32 | 0.86 | |
| Lys (K) | AAA | 49 | 1.32 |
| AAG | 40 | 1.08 | |
| Ser (S1) | AGT | 55 | 1.48 |
| AGC | 16 | 0.43 | |
| AGA | 35 | 0.94 | |
| AGG | 49 | 1.32 | |
| Val (V) | GTT | 190 | 5.11 |
| GTC | 22 | 0.59 | |
| GTA | 68 | 1.83 | |
| GTG | 95 | 2.56 | |
| Ala (A) | GCT | 99 | 2.66 |
| GCC | 39 | 1.05 | |
| GCA | 40 | 1.08 | |
| GCG | 20 | 0.54 | |
| Asp (D) | GAT | 52 | 1.40 |
| GAC | 14 | 0.38 | |
| Glu (E) | GAA | 29 | 0.78 |
| GAG | 57 | 1.53 | |
| Gly (G) | GGT | 127 | 3.42 |
| GGC | 26 | 0.70 | |
| GGA | 60 | 1.61 | |
| GGG | 126 | 3.39 |
Note.—AA = amino acid; N = total number of particular codon in all proteins; % = percentage of total codon usage; TERM = termination codons. The total number of codons is 3,717. Abbreviated stop codons were excluded.
The authors would like to thank David Oppenheimer for providing guidance, bench space, and use of equipment; Mary Pollock and Sujatha Krishnakumar for providing discussion and assistance in the lab; the UA labs of Guy and Kim Caldwell, Margaret Johnson, and Janis O'Donnell for use of equipment; and Bonnie Bowen who generously provided sequence for cob primers. We thank Rafael Zardoya for protocol discussion; Rafael Zardoya and Gonzalo Giribet for providing their pre-published manuscripts; Kevin Helfenbein for helpful suggestions for identification of tRNAs; and Kevin Roe for support, insightful comments, and encouragement during the project. We also thank anonymous reviewers who provided suggestions and constructive comments to the manuscript. Financial support was provided by the Alabama Department of Conservation and Natural Resources and the National Science Foundation (multiuser equipment grant # DBI-0070351 to C.L.). This research is in partial fulfillment of the Ph.D. program in the Department of Biological Sciences at the University of Alabama for J.M.S.
Literature Cited
Avise, J. C.
Beagley, C. T., R. Okimoto, and D. R. Wolstenholme.
Black, W. C. IV, and R. L. Roehrdanz.
Boore, J. L., and W. M. Brown.
Boore, J. L., T. M. Collins, D. Stanton, L. L. Daehler, and W. M. Brown.
Boore, J. L., D. V. Lavrov, and W. M. Brown.
Boore, J. L., and J. L. Staton.
Burzynski, A., M. Zbawicka, D. O. F. Skibinski, and R. Wenne.
Folmer, O., W. R. Hoeh, M. B. Black, and R. L. Vrijenhoek.
Garey, J. R., and D. R. Wolstenholme.
Garrido-Ramos, M. A., D. T. Stewart, B. W. Sutherland, and E. Zouros.
Giribet, G., and W. Wheeler.
Grande, C., J. Templado, J. L. Cervera, and R. Zardoya.
Hatzoglou, E., G. C. Rodakis, and R. Lecanidou.
Helfenbein, K. G., W. M. Brown, and J. L. Boore.
Hoeh, W. R., D. T. Stewart, B. W. Sutherland, and E. Zouros.
Hoffman, R. J., J. L. Boore, and W. M. Brown.
Kumazawa, Y., H. Ota, M. Nishida, and T. Ozawa.
Kurabayashi, A., and R. Ueshima.
Ladoukakis, E. D., and E. Zouros.
Lavrov, D. V., J. L. Boore, and W. M. Brown.
Lewis, D. L., C. L. Farr, and L. S. Kaguni.
Liu, H.-P., J. B. Mitton, and S.-K. Wu.
Lowe, T. M., and S. R. Eddy.
Lydeard, C., W. E. Holznagel, M. Glaubrecht, and W. F. Ponder.
Lydeard, C., M. Mulvey, and G. M. Davis.
Maddison, D. R., and W. P. Maddison.
Moritz, C., and W. M. Brown.
Morrison, C. L., A. W. Harvey, S. Lavery, K. Tieu, Y. Huang, and C. W. Cunningham.
Ojala, D., J. Montoya, and G. Attardi.
Okimoto, R., H. M. Chamberlin, J. L. Macfarlane, and D. R. Wolstenholme.
Passamonti, M., and V. Scali.
Quesada, H., D. A. G. Skibinski, and D. O. F. Skibinski.
Rawlings, T. A., T. M. Collins, and R. Bieler.
Roe, K. J., and C. Lydeard.
Salvini-Plawen, L., and G. Steiner.
Serb, J. M., J. E. Buhay, and C. Lydeard.
Shao, R., J. H. Campbell, and S. C. Barker.
Skelton, P. W., and M. J. Benton.
Skibinski, D. O. F., C. Gallagher, and C. M. Beynon.
Staton, J. L., L. L. Daehler, and W. M. Brown.
Staton, J. L., L. L. Daehler, C. C. Moritz, and W. M. Brown.
Swofford, D. L.
Terrett, J. A., S. Miles, and R. H. Thomas.
Thompson, J. D., D. G. Higgins, and T. J. Gibson.
Tomita, K., S. Yokobori, T. Oshima, T. Ueda, and K. Watanabe.
White, L. R., B. A. Mcpheron, and J. R. Stauffer, Jr.
Wilding, C. S., P. J. Mill, and J. Grahame.
Yamazaki, N., R. Ueshima, and J. A. Terrett, et al. (12 co-authors).
Zardoya, R., A. Garrido-Pertierra, and J. M. Bautista.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}