





Abstract
Class II fumarase sequences were obtained by polymerase chain reaction from five trichomonad species. All residues known to be highly conserved in this enzyme were present. Nuclear run-on assays showed that one of the two genes identified in Tritrichomonas foetus was expressed, whereas no fumarase transcripts were detected in the related species Trichomonas vaginalis. These findings corroborate previous biochemical data. Fumarase genes were also expressed in Monocercomonas sp. and Tetratrichomonas gallinarum but not in Pentatrichomonas hominis, Trichomonas gallinae, Trichomonas tenax, and Trichomitus batrachorum under the culture conditions used. Molecular trees inferred by likelihood methods reveal that trichomonad sequences have no affinity to described class II fumarase genes from other eukaryotes. The absence of functional mitochondria in protists such as trichomonads suggests that they diverged from other eukaryotes prior to the alpha-proteobacterial symbiosis that led to mitochondria. Furthermore, they are basal to other eukaryotes in rRNA analyses. However, support for the early-branching status of trichomonads and other amitochondriate protists based on phylogenetic analyses of multiple data sets has been equivocal. Although the presence of hydrogenosomes suggests that trichomonads once had mitochondria, their class II iron-independent fumarase sequences differ markedly from those of other mitochondriate eukaryotes. All of the class II fumarase genes described from other eukaryotes are of apparent alpha-proteobacterial origin and hence a marker of mitochondrial evolution. In contrast, the class II fumarase from trichomonads emerges among other eubacterial homologs. This is intriguing evidence for an independent acquisition of these genes in trichomonads apart from the mitochondrial endosymbiosis event that gave rise to the form present in other eukaryotes. The ancestral trichomonad class II fumarase may represent a prokaryotic form that was replaced in other eukaryotes after the divergence of trichomonads with the movement of endosymbiont genes into the nucleus. Alternatively, it may have been acquired via a separate endosymbiotic event or lateral gene transfer.
Introduction
Trichomonads constitute a group of protists belonging to the phylum Parabasala (Cavalier-Smith 1998 ). They are aerotolerant, anaerobic organisms that lack mitochondria and peroxisomes but possess specialized organelles called hydrogenosomes, which are involved in metabolic processes that extend glycolysis (reviewed by Müller 1993 ; Kulda 1999 ). These protists are mostly parasitic or commensal flagellates inhabiting oxygen-poor environments. Among these microorganisms, Trichomonas vaginalis is the causative agent of human trichomoniasis (Graves and Gardner 1993 ), and Tritrichomonas foetus is recognized as a significant cause of bovine infertility (Yule, Skirrow, and Bondurant 1989 ). Phylogenies based on large-subunit ribosomal RNA (LSU rRNA) (Viscogliosi et al. 1993 ), small-subunit ribosomal RNA (SSU rRNA) (Sogin et al. 1996 ), elongation factors (EF-1α and 2) (Hashimoto et al. 1997 ; Roger et al. 1999 ), carbamate kinase (Minotto, Edwards, and Bagnara 2000 ), and enolase (Keeling and Palmer 2000 ) all propose that trichomonads could represent one of the oldest branches of the eukaryotic tree. It has been suggested that the absence of functional mitochondria in taxa occupying many of the deepest branches of the eukaryotic tree, such as parabasalids, is proof that these lineages are primitively amitochondriate (Cavalier-Smith 1989 ; for review, see Keeling 1998 ). Recently, genes have been identified in T. vaginalis and Giardia that phylogenetic analyses indicate are of mitochondrial origin (for review, see Embley and Hirt 1998 ). They include heat shock proteins Hsp10, cpn60, Hsp70 (Bui, Bradley, and Johnson 1996 ; Germot, Philippe, and Le Guyader 1996 ; Horner et al. 1996 ; Roger, Clark, and Doolittle 1996 ), adenylate kinase (Lange, Rozario, and Müller 1994 ), and valyl-tRNA synthetase (Hashimoto et al. 1998 ), although the latter has produced phylogenies that are in disagreement with accepted taxonomic relationships. In addition, it has been shown that the mechanism whereby proteins are targeted to the hydrogenosome is similar to that of mitochondria (Plümper, Bradley, and Johnson 1998 ), and a member of the mitochondrial carrier family has been identified in hydrogenosomes (Dyall et al.2000) . All these data strongly suggest that mitochondria and hydrogenosomes arose from a common ancestral endosymbiont (Bradley et al. 1997 ; reviewed in Embley, Horner, and Hirt 1997 ).
Within the trichomonads, evolutionary history is still uncertain. Phylogenetic relationships among the trichomonads were first proposed on the basis of marked morphological differences (Honigberg 1963 ; Brugerolle 1976 ), and later inferred from SSU rRNA (reviewed by Viscogliosi et al. 1999 ), LSU rRNA (Viscogliosi et al. 1993 ), iron-containing superoxide dismutase (FeSOD) (Viscogliosi et al. 1996 ), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Viscogliosi and Müller 1998 ). The molecular data have generated new hypotheses on the systematics of trichomonads. However, comparisons of the phylogenies based on these macromolecules reveal uncertainties in the positions of some species, indicating a need for sequences from new taxa or the use of additional phylogenetic markers.
Since fumarase (fumarate hydratase; EC 4.2.1.2) plays a key role in the Krebs cycle as well as other pathways, it might be informative about relationships that span a wide range of evolutionary distances. This enzyme catalyzes the reversible hydration of fumarate into l-malate. Fumarase is present in mitochondria matrix as well as cytosol of Metazoa, Plantae, and Fungi. To provide for this compartmentalization, a single gene harboring two unique start sites codes for both the mitochondrial and the cytosolic forms of fumarase in rat liver (Suzuki et al. 1989 ) and yeast (Wu and Tzagoloff 1987 ). Two distinct classes of fumarases have been so far identified. Class I fumarases (including similar fumA and fumB enzymes) are homodimeric, thermolabile, iron-sulfur-containing enzymes of approximately 120 kDa. Class I fumarases occur in bacteria, including Escherichia coli, and are also present in mitochondria of the eukaryote Euglena gracilis (Woods, Schwartzbach, and Guest 1988 ). Class II fumarases (or fumC enzymes) are homotetrameric, thermostable, iron-independent enzymes with a molecular mass of 200 kDa. They occur in several bacteria, such as E. coli (Guest et al. 1985 ) and the archaeon Sulfolobus solfataricus (Colombo et al. 1994 ), and resemble the eukaryotic class II fumarases from fungi (Wu and Tzagoloff 1987 ; Friedberg et al. 1995 ), mammals (Suzuki et al. 1989 ), and higher plants (Nast and Müller-Röber 1996 ; Behal and Oliver 1997 ). No molecular data are yet available on fumarases extracted from protozoa. There is no obvious amino acid sequence homology between the class I and class II fumarases. However, both classes of fumarase belong to a superfamily of metabolic enzymes that are functionally related, with fumarate as a product (Williams et al. 1992 ; Weaver et al. 1995 ; Turner et al. 1997 ). Other members of this superfamily include δ crystallin, aspartase, adenylosuccinate lyase, arginosuccinate lyase, and 3-carboxy-cis,cis-muconate lactonizing enzyme. In trichomonads, the available data about fumarases are limited. Tritrichomonas foetus possesses a succinate production pathway extending from malate through fumarate and catalyzed by fumarase and fumarate reductase, respectively (Ryley 1955 ; Kulda 1999 ). The fumarase is located in the cytosol, and the reaction catalyzed by it proceeds in the opposite direction of that in mitochondria (Lindmark et al. 1989 ). This succinate production pathway is absent in the metabolism of the related species T. vaginalis under the conditions tested, suggesting an absence of fumarase activity (Müller 1990 ; Kulda 1999 ). It is possible, however, that variations in fumarase activity in closely related species might reflect suboptimal experimental conditions.
In this study, we report the cloning and sequencing of class II fumarase partial gene sequences from various trichomonads and the presence of a nonexpressed fumarase gene in the parasite T. vaginalis. We compared trichomonad class II fumarases with class II fumarases from prokaryotes and eukaryotes in order to propose an origin of the protein in this group of amitochondriate protists. Phylogenetic analysis of available fumarase gene sequences reveals an origin of trichomonad fumarases that is distinctly different from that of other eukaryotes. We also used class II fumarases to further examine the phylogenetic relationships among trichomonads.
Materials and Methods
Cloning and Sequencing of Trichomonad Fumarase Genes
The organisms studied are listed in table 1 . All isolates used were grown axenically at 37°C or 27°C in trypticase/yeast extract/maltose (TYM) medium (Diamond 1957 ) without agar and supplemented with 10% horse serum (GibcoBRL, Life Technologies, Cergy Pontoise, France). Genomic DNA was isolated as described (Riley and Krieger 1992 ). Initially, a pair of degenerate PCR primers 1F-5R were designed based on two highly conserved stretches of the class II fumarase. These 17mer primers corresponded to the internal stretches 95-QTGSGT-100 (5′-CARACHGGHTCHGGHAC-3′) and 319-SIMPGK-324 (5′-TTDCCWGGCATDATDGA-3′) (numbering of residues follows E. coli fumarase II amino acid sequence). PCRs were carried out in 100 μl according to standard conditions of the EurobioTaq II DNA polymerase (Eurobio, Les Ulis, France). After the denaturation step at 94°C for 5 min, 40 cycles of amplification were performed with a GeneAmp PCR System 9700 apparatus (PE Biosystems, Courtaboeuf, France) as follows: 1 min at 94°C, 1 min at 45°C, and 3 min at 72°C. The final extension step was continued for an additional 15 min. The products were separated by agarose gel electrophoresis, and the band of the expected size (about 700 bp) was electroeluted and inserted by the TA overhang method into the pGEM-T Easy Vector System (Promega, Charbonnieres, France) and amplified in JM 109 competent cells. Five clones containing inserts were selected from each trichomonad species. A second pair of degenerate primers, 3F-6R, was used for PCR using T. foetus DNA as a template. These 17mer degenerate primers were deduced from relatively well conserved domains of class II fumarase and corresponded to the amino-terminal stretch 24-AQTQRS-29 (5′-GCHCARACHCARCGYTC-3′) and to the carboxyl-terminal stretch 407-MLVTAL-412 (5′-AGDGCDGTRACRAGCAT-3′). PCR reaction, purification, and cloning of the expected band (about 1,200 bp) were done under the same conditions as those described above. One positive clone was obtained. The alkaline lysis method was used for the preparation of plasmid DNA. The resulting plasmids were sequenced on both strands with the dideoxy termination method with primer walking using T7 DNA sequencing kits (Amersham Pharmacia Biotech, Orsay, France). The nucleotide sequences obtained in this study have been submitted to the GenBank database under the following accession numbers: T. foetus, AF308585 and AF308586; Monocercomonas sp., AF308581; Trichomitus batrachorum, AF308583; T. vaginalis, AF308584; and Tetratrichomonas gallinarum, AF308582.
Southern Blotting and Run-On Assays
Southern blots were performed with standard methods. Genomic DNA from T. foetus was digested with several restriction enzymes (Gibco BRL, Life Technologies), size-fractionated in 0.8% agarose gels, and blotted to a nitrocellulose membrane (Hybond N+, Amersham Pharmacia Biotech). Blots were hybridized to the gel-purified T. foetus 1fumC and T. foetus 2fumC32P-labeled probes corresponding to PCR products obtained with the 1F-5R primer pair.
Synthesis of nascent mRNAs was investigated by means of a modified run-on assay according to Ullu and Tschudi (1990) . Briefly, trichomonads were washed and resuspended in transcription buffer (150 mM sucrose, 20 mM glutamate potassium, 3 mM MgCl2, 1 mM dithiothreitol, 10 mg/ml leupeptin, 20 mM HEPES-Tris [pH 7.9]) and permeabilized in the presence of 1 mg/ml lysolecithin. This suspension was mixed with the transcription mixture (4 mM ATP, 2 mM GTP, 2 mM CTP, 50 mM creatine phosphate, 1.2 mg/ml creatine phosphokinase, 32P-UTP, 5 mM glutamate potassium, 1.5 mM MgCl2, 5 mM dithiothreitol, 1 mM HEPES) and incubated for 20 min at 35°C. RNAs were isolated by trizol extraction according to the protocol of the manufacturer (GibcoBRL, Life Technologies). 32P-labeled transcripts were hybridized to DNA-specific probes immobilized on nitrocellulose. Blots were analyzed using storage phosphor autoradiography (PhosphorImager SI, Molecular Dynamics Inc., Cambridge, Mass.). The β-tubulin and malic enzyme probes (positive controls) were prepared by PCR amplification using T. vaginalis DNA as template. The β-tubulin-specific primers corresponded to the internal stretches 170-IVPSPK-175 (5′-CATCGTCCCATCTCCAAAGG-3′) and 255-AVNLVP-260 (5′-AATGGAACAAGGTTGACAGC-3′). The malic-enzyme-specific primers corresponded to the amino-terminal stretch 42-EERDR-46 (5′-AGGAAGAACGTGACCGCC-3′) and to the carboxyl-terminal stretch 504-DHDMGN-509 (5′-GTTGCCCATATCGTGGTC-3′). Numbering of residues follows T. vaginalis β-tubulin (Katiyar and Edlind 1994 ) and malic enzyme (Hrdý and Müller 1995a ) amino acid sequences. PCR products (275 and 1,405 bp, respectively) were inserted into a pGEM-T cloning vector (Promega). Nucleotide sequences of both PCR products were verified by sequencing using the ABI Prism 310 Genetic Analyzer (PE Biosystems).
Phylogenetic Analysis
Deduced amino acid sequences of the clones obtained from trichomonad species were aligned with all class II fumarase sequences retrieved from the GenBank database by visual editing using the ED program of the MUST program package, version 1.0 (Philippe 1993 ). Accession numbers of the sequences included in the data set are listed in the legend to figure 4. Phylogenetic analyses of the amino acid sequences were performed using the protein maximum-likelihood program PROTML, version 2.2 (Adachi and Hasegawa 1996 ) with the heuristic quick-add OTU search and local rearrangement (R-search) methods with the Jones, Taylor, and Thornton (JTT-F) amino acid replacement model. Regions of ambiguous alignment were removed, yielding 211 sites for the data set, including six partial trichomonad class II fumarase sequences, and 377 sites for the data set, including one almost complete trichomonad sequence. Local bootstrap proportions (1,000 replicates) for protein maximum likelihood (ML) were determined with the resampling estimated log likelihood (RELL) method (Hasegawa and Kishino 1994 ). Phylogenetic analysis was also performed using both the protein ML distance and the ML quartet puzzling methods (ML-QP) implemented in the PUZZLE program, version 5.0 (Strimmer and von Haeseler 1996 ), with an eight-category discrete gamma model of among-sites rate variation (JTT+Γ). The gamma shape parameter α was estimated by ML optimization on a neighbor-joining topology to be 0.56. Phylogenetic trees were inferred from the ML distances using the Fitch-Margoliash algorithm with global rearrangements (FITCH, version 3.5; Felsenstein 1995 ). ML distance bootstrap values for bipartitions were calculated by analysis of 100 resampled data sets generated with the SEQBOOT program and analyzed with the PUZZLEBOOT script (http://www.tree-puzzle.de/) in conjunction with PUZZLE, FITCH, and CONSENSE. Full protein ML analyses using the quartet puzzling algorithm in PUZZLE utilized 1,000 puzzling steps.
Likelihood-mapping (LM; Strimmer and von Haeseler 1997 ) was performed using PUZZLE, version 5.0, to (1) determine support for the monophyly of trichomonad class II fumarases and (2) confirm the separation of trichomonad class II fumarase genes from other eukaryotic and alpha proteobacterial homologs. This method is based on an analysis of the maximum likelihoods for the three resolved tree topologies that can be computed for four groups of sequences, a so-called quartet. The tree-likeness for the four clusters can be visualized in a single graph. Sequences in our data set excluding those for the archaea were assigned to one of four groups. In both LM analyses, 10,000 quartets were performed by random choice.
To further test support for the monophyly of trichomonad class II fumarase genes, MacClade, version 4.0 (Maddison and Maddison 1992 ), was used to force a branch rearrangement that placed the branch leading to T. gallinarum and T. vaginalis at the base of the clade that includes the four other trichomonads. This arbitrarily constructed tree was imported into PUZZLE along with the tree in figure 4 . Both trees were compared using a Kishino-Hasegawa (K-H) test (Kishino and Hasegawa 1989 ) using the JTT model of substitution and gamma-distributed rates.
Results and Discussion
Nucleotide Sequences
The primers 1F and 5R produced 637–655-bp PCR products representing around 50% of the open reading frame (ORF) coding for class II fumarase sequences in five trichomonad species (fig. 1 ). Five clones were sequenced in each species, and they exhibited the same nucleotide sequences except for T. foetus, in which two fumarase gene copies were found and designated Tf1fumC and Tf2fumC, respectively. Interestingly, one fumarase gene copy has been found in T. vaginalis in which no fumarase activity has been detected (Y. Hrdý, personal communication). The T. foetus result agrees with data obtained from Southern blots (fig. 2 ). Tritrichomonas foetus genomic DNA cleaved with the restriction enzymes HindIII, PvuII, and NdeI was hybridized with either Tf1fumC or Tf2fumC PCR products. The PCR products did not contain restriction sites for any of those enzymes. Under our conditions of stringency, the Tf1 and Tf2fumC probes annealed to different restriction fragments, and therefore represent different genes. We did not observe cross-hybridization between the Tf1fumC and Tf2fumC probes, which are 75.7% identical at the nucleotide level. These data indicate that there are at least two genes coding for class II fumarase in T. foetus. The trichomonad fumarase genes showed species-dependent differences in codon usage (data not shown). Trichomonas vaginalis and its closest relative, T. gallinarum, have the most restricted set of codons, as noted for FeSOD (unpublished observations) and GAPDH (Viscogliosi and Müller 1998 ) genes. This variation does not correlate with either the overall G+C content of the genes or the G+C content of the third positions of the codons, as shown for T. vaginalis (McInerney 1997 ).
Amino Acid Sequences
The inferred trichomonad amino acid sequence (212–218) residues were aligned with class II fumarases from E. coli and Saccharomyces cerevisiae (fig. 1 ). The alignment required the insertion of seven gaps, most of them in the sequences of T. vaginalis and T. gallinarum. The single ORFs of the trichomonad clones could be easily aligned with homologous sequences, suggesting the lack of introns, as also noted for all other trichomonad genes sequenced so far. After elimination of gaps, the trichomonad fumarase sequences exhibited a higher degree of similarity to bacteria (55.5%–62.6%), including E. coli, than to eukaryotes (48.8%–57.4%), including S. cerevisiae (full alignment not shown). They also showed 54.0%–99.5% identity to each other (table 2 ). The most divergent sequences within the trichomonads were those of T. vaginalis and T. gallinarum. We noted that the similarity values between these two taxa and the remaining trichomonad species were very low (54.0%–56.4%) in comparison with those observed for other enzymes. For instance, we calculated 67.0% (194 positions compared) and 72.0%–75.5% (322 positions) identity between T. vaginalis and T. foetus FeSOD and GAPDHs, respectively, whereas the values were 55.5%–56.4% (211 positions) for class II fumarases. In addition, the evolutionary distance between T. vaginalis/T. gallinarum and E. coli was as great as the distance between both species and the remaining trichomonads.
As stated before, fumarases belong to a superfamily of enzymes which use fumarate as a substrate. Although very little sequence similarity is observed across the superfamily, three regions of highly conserved amino acid sequences have been identified and implicated in the common catalytic mechanism (Williams et al. 1992 ; Weaver et al. 1995 ; Weaver and Banaszak 1996 ). The three domains are 129–146, 181–200, and 312–331, based on the numbering from E. coli class II fumarase. Sequence comparisons reveal that almost all of the well-conserved residues composing the two first domains are present in the trichomonad fumarase sequences. The third domain corresponds most closely to the amino acid residues complementary to the antisense primer 5R. On the basis of crystallographic (Weaver et al. 1995, 1998 ; Weaver and Banaszak 1996 ) and site-directed mutagenesis (Bourgeron et al. 1994 ; Weaver, Mees, and Banaszak 1997 ) studies, the residues Ser-139, Ser-140, Asn-141, Thr-187, His-188, and Glu-315 have been shown to be involved in the catalytic activity of class II fumarases. All these residues are invariant in trichomonad sequences.
A second pair of degenerate primers, 3F-6R, corresponding to well-conserved domains among class II fumarases, was used in order to increase the length of the trichomonad sequences (over 80% of most of the ORFs of class II fumarase genes) and to better assess the positions of these protists in further phylogenetic reconstructions. A PCR product of the expected size (1,132 bp after exclusion of the primers) was obtained for T. foetus and named TfExfumC. This sequence codes for the class II fumarase and is composed of 377 amino acid residues. The sequence of the TfExfumC clone is identical to that of the Tf1fumC gene obtained with the 3F-5R primer pair in their overlapping region. Unfortunately, no PCR product or clones encoding class II fumarases were obtained in other trichomonad species, suggesting that the 3F and 6R priming sites are not well conserved among trichomonads. The amino acid sequence of the long Tf1fumC clone was aligned with all known class II fumarase sequences (fig. 1 and data not shown). The alignment revealed a high overall similarity (40.6%–61.7% for 360 common positions), and the long Tf1fumC sequence exhibited a higher degree of similarity to bacterial enzymes from E. coli (61.7%) and Myxococcus xanthus (60.6%) than to eukaryotic homologs (55.6%–59.7%). The long Tf1fumC sequence shared a two-amino-acid deletion (positions 38 and 39 in the S. cerevisiae sequence) with bacterial class II fumarases (with the exception of Thermus aquaticus and Thermus thermophilus) that is absent in eukaryotic class II fumarases. We also found additional residues involved in the catalytic activity of the enzyme (Saribas, Schindler, and Viola 1994 ; Weaver et al. 1995 ; Weaver and Banaszak 1996 ), such as Thr-100, Lys-324, and Asn-326 (numbering as in the E. coli sequence). As previously stated, three domains are conserved among enzymes belonging to the lyase class, for which fumarate is a substrate. The high degree of conservation of the last domain (312–331) has led to a consensus sequence that defines this family of proteins. It contains the motif GSSXMPXKXN, also found in the long Tf1fumC sequence, and represents the only significant region of similarity between the class I and class II fumarases (Saribas, Schindler, and Viola 1994 ; Weaver et al. 1998 ).
Expression of Trichomonad Fumarase Genes
DNA-specific probes immobilized on nitrocellulose membranes were used for hybridization with total nascent radioactively labeled RNAs from T. foetus and T. vaginalis (fig. 3A ). β-tubulin and malic enzyme T. vaginalis probes were used as positive controls, whereas pGEM-T Easy Vector was used as a negative one. We did not observe cross-reactions between Tf1fumC and Tf2fumC probes in Southern blots, indicating the strong specificity of each probe. We did not obtain a signal for Tf2fumC probe with T. foetus RNAs under our conditions of growth, indicating nonexpression of this gene. In addition, T. vaginalis RNAs did not hybridize with the TvfumC probe, strongly suggesting nonexpression of the TvfumC gene. This corroborated the absence of fumarase activity described in T. vaginalis (I. Hrdý, personal communication) and might help to explain the low similarity values observed between the fumarase sequence of this taxon and those of most of the remaining trichomonads. Indeed, the nonexpressed TvfumC gene could be under less evolutionary constraint or could have been recruited to a new function. Since the fumarase sequences of the related species T. vaginalis and T. gallinarum exhibit a very high level of similarity, this suggests that the absence of fumarase activity might be a common feature of the Trichomonadinae. In order to test this hypothesis, run-on assays were performed with RNAs from the Trichomonadinae T. gallinarum, Pentatrichomonas hominis, Trichomonas gallinae, and Trichomonas tenax, from a close relative of T. foetus in our study, Monocercomonas sp., and from T. batrachorum (fig. 3B ). The Tf2fumC probe was not used, since it previously gave no signal with both T. foetus and T. vaginalis RNAs. As expected, RNAs from Monocercomonas sp. hybridized with both Tf1fumC and TvfumC probes. Interestingly, T. gallinarum RNAs hybridized strongly with the TvfumC probe and weakly with the Tf1fumC probe, whereas no signal was obtained with RNAs from P. hominis, both species of Trichomonas genera, T. gallinae and T. tenax, and T. batrachorum. The positive signal obtained with T. gallinarum RNAs clearly contradicts our preliminary hypothesis that the absence of fumarase activity, and globally, of succinate production, is a common feature of the Trichomonadinae.
Salient Points in the Molecular Phylogeny Inferred from Class II Fumarase Amino Acid Sequences
Class II fumarase genes were used to explore the evolutionary history of this gene in the context of a larger data set encompassing all known class II fumarase gene sequences. The trichomonad data set included six trichomonad amino acid sequences (211 positions) derived from the PCR products obtained with the 1F-5R primer pair. Phylogenetic analyses under ML were performed using the archaea Sulfolobus solfataricus and Aeropyrum pernix as outgroups in view of their basal position in rRNA phylogenies (Van de Peer et al. 1994 ; De Rijk et al. 1995 ; Sako et al. 1996 ). A first analysis was performed using 211 amino acid positions and including the six trichomonad sequences (fig. 4 ). This analysis was repeated using a data set that included 377 positions and included the long Tf1fumC sequence (fig. 5 ). What is clear from these analyses is that the origin of the trichomonad fumarase gene is different from that of described fumarase genes from other eukaryotes.
In both analyses, a large clade was formed by the eukaryotic fumarases from animals, plants, and fungi. Eukaryotic fumarases and their α-proteobacterial homologs grouped together in these trees with high support (94% and 95%, respectively). This result was repeated under ML distance criteria using a gamma model of rate heterogeneity using PUZZLE, version 5.0, and the Fitch-Margoliash method, as well as ML-QP methods implemented in PUZZLE, version 5.0 (Strimmer and von Haeseler 1996 ) (data not shown). This is consistent with a mitochondrial origin and localization of eukaryotic fumarases, with the exception of trichomonads. Indeed, the class II fumarases from trichomonads occupy distal positions in relation to all other eukaryotes. The first trichomonad group, including T. foetus, emerges within a heterogenous bacterial group including proteobacteria and low-G+C Gram-positive bacteria (figs. 4 and 5 ) whereas the second one, including T. vaginalis, did not exhibit a special relationship to one or another bacterial subgroup (fig. 4 ). Although there is little or no bootstrap support for these specific relationships, it is likely that trichomonad fumarases are not part of a well-resolved assemblage that includes mitochondria-bearing eukaryotes. Despite the fact that mitochondria may have been present in the ancestors of trichomonads, the emergence of trichomonad fumarases suggests that these protists have acquired class II fumarase genes through evolutionary events different from the endosymbiotic source of other eukaryotes. Such observations were confirmed by LM analysis (see below).
As stated above, a clear separation is observed between a first trichomonad group, including Monocercomonas sp., the two sequences of T. foetus, and T. batrachorum (Tritrichomonas group), and a second one, composed of the T. vaginalis and T. gallinarum sequences (Trichomonas group). This dichotomy was independent of the position of the root of the tree (data not shown). The unexpected polyphyly of the trichomonad fumarases could be in agreement with the low similarity values observed between the fumarase sequences of these two groups. However, in all other molecular trees, trichomonads form an independent, well-supported monophyletic group (Viscogliosi et al. 1993, 1996, 1999 ; Viscogliosi and Müller 1998 ), and the polyphyly of trichomonads was not supported by bootstrap values in our ML analysis. Moreover, neither the LM analysis nor the K-H test rejected the monophyly of trichomonads (see below).
Finally, among the bacteria, the order of emergence of the different clades was not well supported by bootstrap values (figs. 4 and 5 ), as also noted in rRNA-based reconstructions (Van de Peer et al. 1994 ; De Rijk et al. 1995 ). Different well-established eubacterial groups can be distinguished: the α-proteobacteria, including Bradyrhizobium japonicum and Rickettsia prowazekii; the ϵ-proteobacteria, comprising the sequences of Campylobacter jejuni and Helicobacter pylori and the radio-resistant micrococci; and the related group composed of two Thermus species. In contrast, the well-defined clade of the γ-proteobacteria including E. coli, Haemophilus influenzae, and Pseudomonas aeruginosa did not form a monophyletic group in this study. We attempted to optimize the resolution of the bacterial sequences. PROTML, version 2.2, was used to find the best tree in an exhaustive search that fit the criteria imposed by a user-defined constraint tree. In this tree, trichomonads were held together as a monophyletic group, and the topology of the eukaryotes and alpha proteobacteria above the 94% support node in figure 4 was fixed. Several groups were defined among the remaining sequences: the two Thermus sequences, the two ϵ-proteobacteria plus Bacillus subtilis, the γ-proteobacteria plus Myxococcus xanthus, and the two archaeal sequences. The best tree found under that search out of 1,000 trees was imported into PUZZLE, version 5.0, for calculation of branch lengths and was then compared with the tree in figure 4 using a K-H test (data not shown). There is not a significant difference at the 0.05 percent level following Kishino and Hasegawa (1989) between the two topologies (log L = −7,697.87 for our original tree vs. log L = −7,720.91 for our constrained tree). Thus, the order of emergence among the bacterial groups below the 94% node was unresolved and should therefore not be overinterpreted.
Investigation of Relationships Among Class II Fumarases Using the LM Analysis and the K-H Test
For LM analysis, sequences in our data set excluding those for the archaea were assigned to one of four groups, a–d. In our first analysis, we considered the monophyly of trichomonads, and the four groups were as follows: a = all eukaryotic and alpha proteobacterial sequences above the 94% support node in figure 4 ; b = T. foetus 1, T. foetus 2, Monocercomonas sp., and T. batrachorum (Tritrichomonas group); c = T. gallinarum and T. vaginalis (Trichomonas group); and d = all other prokaryotic sequences excluding the archaea. We noted that the tree-likeness of the class II fumarase data was relatively high (72.8 + 8.1 + 1.5 = 82.4%) and that 72.8% of all quartets between the four corresponding clusters supported the clustering of the Tritrichomonas and Trichomonas groups (fig. 6A ). Based on LM, the two trichomonad groups are more closely related to each other than they are to the eukaryotic and alpha proteobacterial or the miscellaneous other prokaryotic sequences. Thus, we cannot reject the hypothesis of monophyly of trichomonads. In parallel, we constrained the sequences from T. vaginalis and T. gallinarum with those of the Tritrichomonas group. The K-H test within PUZZLE showed that the two trees were not significantly different (log L = −7,697.87 for our original tree, and log L = −7,695.38 for the constrained tree) under a 5% significance test. This also indicates that monophyly of trichomonads cannot be rejected.
In a second LM analysis, we tested the separation of trichomonad sequences from other eukaryotic and alpha proteobacterial homologs. The four groups were as follows: a = all eukaryotic and alpha proteobacterial sequences above the 94% support node in figure 4 ; b = Mycobacterium tuberculosis group (also including H. influenzae, Leptospira interrogans, Chlamydia trachomatis, Synechocystis sp., T. aquaticus, T. thermophilus, and P. aeruginosa; c = E. coli group (also including M. xanthus, C. jejuni, H. pylori, and B. subtilis); and d = all trichomonad sequences. As shown in figure 6B, 66.6 + 4.2 + 9.9 = 80.7% of the quartets belong to the regions that represent fully resolved trees. Most of them support the clustering of trichomonads with the E. coli group. In contrast, we find only very low support (4.2%) for the topology that pairs trichomonads with other eukaryotic and alpha proteobacterial representatives. This result reinforces our ML analyses and the observation that trichomonads do not belong in the same group with eukaryotes exhibiting mitochondrial class II fumarases.
Phylogenetic Evaluation of the Trichomonad Sequences
Class II fumarase genes were also used to explore phylogenetic relationships within the trichomonads. According to morphological data, an early separation of two lineages, one leading to the genus Tritrichomonas and the other leading to the genus Trichomonas, has been proposed (Honigberg 1963 ; Brugerolle 1976 ). These two lineages correspond to the two main subfamilies of Trichomonadidae: the Tritrichomonadinae (T. foetus) and the Trichomonadinae (T. vaginalis and T. gallinarum). This dichotomy is in agreement with our fumarase data, although fumarase genes do not hold trichomonads together as a monophyletic group. The ancestral trichomonad was assumed to be a Monocercomonas-like organism belonging to the family Monocercomonadidae. This species has a rudimentary cytoskeleton and was thought to have given rise to the other, more complex, Trichomonadidae such as T. batrachorum. In our tree, Monocercomonas sp. exhibited a derived position and is closely related to T. foetus with high bootstrap support (98% of the replicates). In SSU rRNA phylogenies (Viscogliosi et al. 1999 ) the clustering of T. foetus and Monocercomonas sp. was very tenuous. Nevertheless, the grouping of both taxa is found in phylogenies based on LSU rRNA (Viscogliosi et al. 1993 ), FeSOD (Viscogliosi et al. 1996 ), GAPDH (Viscogliosi and Müller 1998 ), and fumarase genes (this study), strongly suggesting that these two genera may be related. We also note that in this study, T. batrachorum has a basal position among the trichomonads in its group in 98% of the replicates, preceding the emergence of T. foetus and Monocercomonas sp. This taxon has been included in the Trichomonadinae on the basis of cytological studies (Brugerolle 1976 ), but it is clear that this genus is misclassified according to its position in molecular phylogenies. In SSU rRNA- (Edgcomb et al. 1998 ; Delgado-Viscogliosi et al. 2000 ), LSU rRNA- (Viscogliosi et al. 1993 ), and GAPDH-based phylogenies (Viscogliosi and Müller 1998 ), T. batrachorum exhibits an early emergence at the base of the Trichomonadinae-Tritrichomonadinae dichotomy, emphasizing its pivotal evolutionary position among the Trichomonadidae. This analysis is partially in agreement with these data, although the relationships between T. batrachorum and Trichomonadinae are difficult to interpret because of the unexpected phylogenetic position of T. vaginalis and T. gallinarum. However, the position of Monocercomonas sp. and T. batrachorum in our fumarase phylogeny strengthens the hypothesis that the relative morphological simplicity of Monocercomonadidae including Monocercomonas sp. might have arisen through the loss of cytoskeletal structures.
The Quest for the Origin of Class II Fumarases in Trichomonads
The small number of available class II fumarase sequences means that it would be premature to favor a single hypothesis concerning the origin of trichomonad fumarases. However, alternative scenarios can be considered on the basis of successive gains and losses of evolutionarily distinct fumarase genes. According to our data, there is no support for trichomonads having two separate origins for class II fumarase genes. Thus, we can discuss the origin of a likely ancestral class II fumarase gene common to all of the species analyzed in our study. It now seems clear that trichomonads have a class II fumarase origin separate from that of the mitochondrial enzyme found in other eukaryotes studied so far. It is possible that either trichomonads lost the “mitochondrial” fumarase gene and maintained an ancestral one in their genome, or an ancestral gene has been replaced by the mitochondrial one via an endosymbiotic event in eukaryotes, with the known exception of trichomonads. Moreover, it is known that T. vaginalis and other trichomonads internalize bacteria by phagocytosis (Rendon-Maldonado et al. 1998 ). The acquisition of genes from bacterial neighbors through phagocytosis has been proposed for microbial eukaryotes (for pertinent reference, see Doolittle 1998 ). Thus, lateral gene transfer from yet-unidentified bacteria could be hypothesized for the class II fumarase gene in trichomonads. Such transfers could be current events in T. vaginalis, since they have already been suggested for FeSOD (Smith, Feng, and Doolittle 1992 ), GAPDH (Viscogliosi and Müller 1998 ; Figge et al. 1999 ), pyruvate : ferredoxin oxidoreductase (Hrdý and Müller 1995b ), and, more recently, N-acetylneuraminate lyase (de Koning et al. 2000 ).
Mark Ragan, Reviewing Editor
Keywords: Parabasala trichomonads fumarase gene expression protein evolution molecular phylogeny
Address for correspondence and reprints: Eric Viscogliosi, Institut Pasteur, INSERM Unité 547, 1 Rue du Professeur Calmette, B.P. 245, 59019 Lille cedex, France. [email protected].
Table 1 Organisms Used in this Study
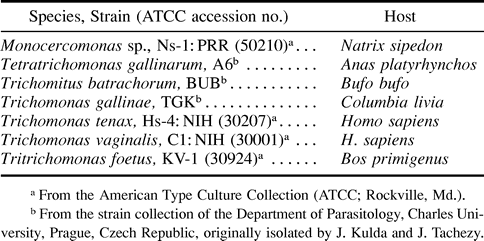
Table 1 Organisms Used in this Study
Table 2 Positional Amino Acid Identities (see fig. 1 for alignment) for Trichomonad Fumarase Genes
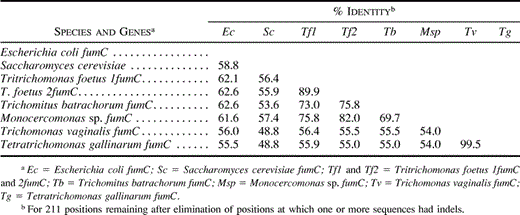
Table 2 Positional Amino Acid Identities (see fig. 1 for alignment) for Trichomonad Fumarase Genes

Fig. 1.—Alignment of the deduced amino acid sequences of class II fumarases of trichomonads with two related homologs, Escherichia coli fumC (Ec, P05042) and Saccharomyces cerevisiae fumC (Sc, P08417). Abbreviations are as follows: Tf1 and Tf2, Tritrichomonas foetus 1 and T. foetus 2fumC; Tb, Trichomitus batrachorum fumC; Msp, Monocercomonas sp. fumC; Tv, Trichomonas vaginalis fumC; Tg, Tetratrichomonas gallinarum fumC. Residues identical to those in the E. coli sequence are represented by dashes, and gaps are represented by asterisks. The forward arrow (>) indicates the omitted amino-terminal extension of the mitochondrial S. cerevisiae enzyme. Domains corresponding to the degenerate primers used in this study are underlined. Residues involved in the catalytic activity of the enzyme are represented by arrows. The three conserved regions among enzymes belonging to the lyase class have been boxed
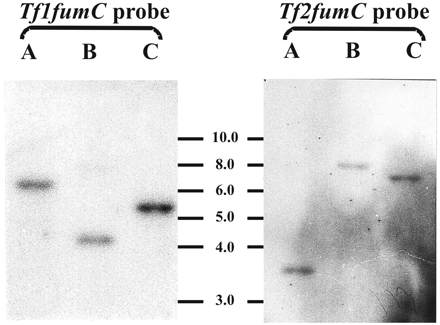
Fig. 2.—Southern blots of Tritrichomonas foetus gDNA digested with the restriction enzymes (A) HindIII, (B) PvuII, and (C) NdeI and hybridized with either Tf1fumC or Tf2fumC probes. Size markers are in kilobases
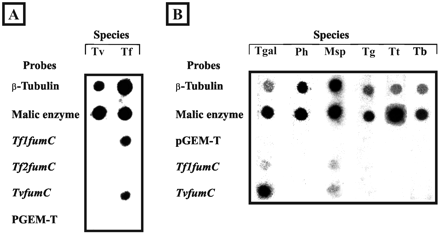
Fig. 3.—Nuclear run-on assays. A, Tritrichomonas foetus (Tf) and Trichomonas vaginalis (Tv) radiolabeled RNAs were hybridized to different probes immobilized on nitrocellulose membranes. B, Hybridization of RNAs from Tetratrichomonas gallinarum (Tgal), Pentatrichomonas hominis (Ph), Monocercomonas sp. (Msp), Trichomonas gallinae (Tg), Trichomonas tenax (Tt), and Trichomitus batrachorum (Tb) with different probes
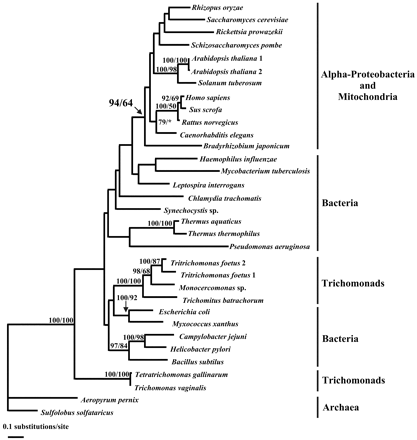
Fig. 4.—Phylogeny of class II fumarase proteins. The analysis was restricted to the sequences between domains corresponding to the pair of primers 1F-5R. After exclusion of all gaps from the alignment, 211 shared sites were analyzed with PROTML. The horizontal length of each branch is proportional to the estimated number of substitutions. The outgroup is represented by the sequences of Aeropyrum pernix and Sulfolobus solfataricus. Support values are given as percentages near individual nodes and are computed using both the resampling estimated log likelihood (RELL) and the maximum-likelihood (ML) distance methods. Only values above 50% are indicated. Database accession numbers of the species are as follows: Aeropyrum pernix, AP000062; Arabidopsis thaliana, AF020303 and U82202; Bacillus subtilis, AJ223978; Bradyrhizobium japonicum, P28894; Caenorhabditis elegans, AF025459; Campylobacter jejuni, Y16882; Chlamydia trachomatis, AE001358; Escherichia coli, P05042; Haemophilus influenzae, U32820; Helicobacter pylori, AE000634; Homo sapiens, P07954; Leptospira interrogans, AB010203; Mycobacterium tuberculosis, AL021897; Myxococcus xanthus, AF013216; Pseudomonas aeruginosa, JC4982; Rattus norvegicus, P14408; Rhizopus oryzae, JC4293; Rickettsia prowazekii, AJ235272; Saccharomyces cerevisiae, P08417; Schizosaccharomyces pombe, AL035260; Solanum tuberosum, X91615; Sulfolobus solfataricus, P39461; Sus scrofa, P10173; Synechocystis sp., D64000; Thermus aquaticus, D84646; Thermus thermophilus, AB010884
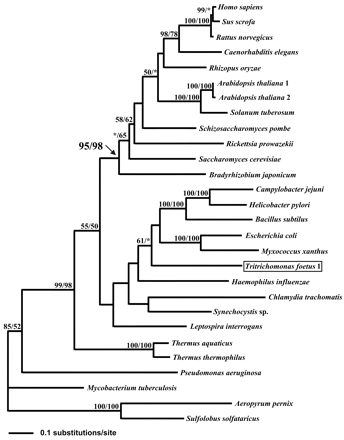
Fig. 5.—Phylogenetic reconstruction of class II fumarases using PROTML and including 377 positions corresponding to the sequences located between the pair of primers 3F-6R. The outgroup is represented by the sequences of Aeropyrum pernix and Sulfolobus solfataricus. The tree was constructed using PROTML. Support values shown at nodes are computed using both the resampling estimated log likelihood (RELL) and the maximum-likelihood (ML) distance methods
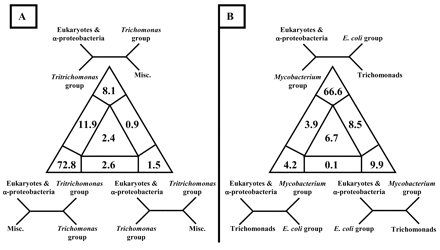
Fig. 6.—Four-cluster likelihood mapping of class II fumarase amino acid sequences testing (A) the monophyly of trichomonads and (B) the separation of trichomonad sequences from other eukaryotic and alpha proteobacterial homologs. In both analyses, sequences were split in four disjoint groups. The occupancies (in percentages) for the seven areas of attraction are indicated. The corners of the triangles are labeled with the corresponding fully resolved tree topologies. Misc. = miscellaneous group of bacteria excluding the archaea
We thank Monica Riley (Marine Biological Laboratory, Woods Hole, Mass.) for insightful discussions about the fumarase phylogeny, Miklos Müller (Rockefeller University, New York) for preliminary phylogenetic analysis, Ping Liang (Marine Biological Laboratory, Woods Hole) for help with phylogenetic analyses, and Ivan Hrdý (Charles University, Prague) for sharing his unpublished data with us. This work was funded by the Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale, and the Institut Pasteur de Lille (support provided to E.V.); by NASA Astrobiology Cooperative Agreement NCC2-1054 and NASA Exobiology grant NRA 97-OSS-11 to M.L.S.; and by grant GAUK 264/1999 to J.T.
References
Adachi J., M. Hasegawa,
Behal R. H., D. J. Oliver,
Bourgeron T., D. Chretien, J. Poggi-Bach, S. Doonan, D. Rabier, P. Letouze, A. Rotig, P. Landrieu, P. Rustin,
Bradley P. J., C. J. Lahti, E. Plumper, P. J. Johnson,
Brugerolle G.,
Bui E. T. N., P. J. Bradley, P. J. Johnson,
Colombo S., M. Grisa, P. Tortora, M. Vanoni,
de Koning A. P., F. S. L. Brinkman, S. J. M. Jones, P. J. Keeling,
Delgado-Viscogliosi P., E. Viscogliosi, D. Gerbod, J. Kulda, M. L. Sogin, V. P. Edgcomb,
De Rijk P., Y. Van de Peer, I. Van den Broeck, R. De Wachter,
Diamond L. S.,
Doolittle W. F.,
Dyall S. D., C. M. Koehler, M. G. Delgadillo-Correa, P. J. Bradley, E. Plümper, D. Leuenberger, C. W. Turck, P. J. Johnson,
Edgcomb V. P., E. Viscogliosi, A. G. B. Simpson, P. Delgado-Viscogliosi, A. J. Roger, M. L. Sogin,
Embley T. M., D. S. Horner, R. P. Hirt,
Felsenstein J.,
Figge R. M., M. Schubert, H. Brinkmann, R. Cerff,
Friedberg D., Y. Peleg, A. Monsonego, S. Maissi, E. Battat, J. S. Rokem, I. Goldberg,
Germot A., H. Philippe, H. Le Guyader,
Graves A., W. A. Gardner,
Guest J. R., J. S. Miles, R. E. Roberts, S. A. Woods,
Hasegawa M., H. Kishino,
Hashimoto T., Y. Nakamura, T. Kamaishi, M. Hasegawa,
Hashimoto T., L. B. Sánchez, T. Shirakura, M. Müller, M. Hasegawa,
Honigberg B. M.,
Horner D. S., R. P. Hirt, S. Kilvington, D. Lloyd, T. M. Embley,
Hrd I., M. Müller,
———.
Katiyar S. K., T. D. Edlind,
Kishino H., M. Hasegawa,
Lange S., C. Rozario, M. Müller,
Lindmark D. G., B. L. Eckenrode, L. A. Halberg, I. D. Dinbergs,
Maddison W. P., D. R. Maddison,
Minotto L., M. R. Edwards, A. S. Bagnara,
Müller M.,
Nast G., B. Müller-Röber,
Philippe H.,
Plümper E., P. J. Bradley, P. J. Johnson,
Rendon-Maldonado J. G., M. Espinosa-Cantellano, A. Gonzalez-Robles, A. Martinez-Palomo,
Riley D. E., J. N. Krieger,
Roger A. J., C. G. Clark, W. F. Doolittle,
Roger A. J., O. Sandblom, W. F. Doolittle, H. Philippe,
Ryley J. F.,
Sako Y., N. Nomura, A. Uchida, Y. Ishida, H. Morii, Y. Koga, T. Hoaki, T. Maruyama,
Saribas A. S., J. F. Schindler, R. E. Viola,
Smith M. W., D. F. Feng, R. F. Doolittle,
Sogin M. L., H. G. Morrison, G. Hinkle, J. D. Silberman,
Strimmer K., A. von Haeseler,
———.
Suzuki T., M. Sato, T. Yoshida, S. Tuboi,
Turner M. A., A. Simpson, R. R. McInnes, P. Lynne Howell,
Ullu C., C. Tschudi,
Van de Peer Y., J. M. Neefs, P. de Rijk, P. de Vos, R. de Wachter,
Viscogliosi E., I. Durieux, P. Delgado-Viscogliosi, D. Bayle, D. Dive,
Viscogliosi E., V. P. Edgcomb, D. Gerbod, C. Noël, P. Delgado-Viscogliosi,
Viscogliosi E., M. Müller,
Viscogliosi E., H. Philippe, A. Baroin, R. Perasso, G. Brugerolle,
Weaver T. M., L. Banaszak,
Weaver T. M., M. Lees, V. Zaitsev, et al. (12 co-authors)
Weaver T. M., D. G. Levitt, M. I. Donnely, P. P. Wilkens Stevens, L. Banaszak,
Weaver T. M., L. Mees, L. Banaszak,
Williams S. E., E. M. Woolridge, S. C. Ransom, J. A. Landro, P. C. Babbitt, J. W. Kozarich,
Woods S. A., S. D. Schwartzbach, J. R. Guest,
Wu M., A. Tzagoloff,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}