





Abstract
Here we show that multiple DNA sequences, similar to the mitochondrial cytochrome oxidase I (COI) gene, occur within single individuals in at least 10 species of the snapping shrimp genus Alpheus. Cloning of amplified products revealed the presence of copies that differed in length and (more frequently) in base substitutions. Although multiple copies were amplified in individual shrimp from total genomic DNA (gDNA), only one sequence was amplified from cDNA. These results are best explained by the presence of nonfunctional duplications of a portion of the mtDNA, probably located in the nuclear genome, since transfer into the nuclear gene would render the COI gene nonfunctional due to differences in the nuclear and mitochondrial genetic codes. Analysis of codon variation suggests that there have been 21 independent transfer events in the 10 species examined. Within a single animal, differences between the sequences of these pseudogenes ranged from 0.2% to 20.6%, and those between the real mtDNA and pseudogene sequences ranged from 0.2% to 18.8% (uncorrected). The large number of integration events and the large range of divergences between pseudogenes and mtDNA sequences suggest that genetic material has been repeatedly transferred from the mtDNA to the nuclear genome of snapping shrimp. Unrecognized pseudogenes in phylogenetic or population studies may result in spurious results, although previous estimates of rates of molecular evolution based on Alpheus sister taxa separated by the Isthmus of Panama appear to remain valid. Especially worrisome for researchers are those pseudogenes that are not obviously recognizable as such. An effective solution may be to amplify transcribed copies of protein-coding mitochondrial genes from cDNA rather than using genomic DNA.
Introduction
Many studies have focused on DNA sequence variation using mtDNA because animal mtDNA in particular displays several highly desirable features (Avise2000) : extensive polymorphism, a faster rate of evolution than is found in single-copy nuclear DNA, rapid genetic sorting, and asexual and maternal inheritance (normally without intermolecular recombination, but see Awadalla, Walker, and Maynard-Smith[1999] ). Nevertheless, recent studies show that analyses of mtDNA data can be confounded by heteroplasmy, “leakage” from the paternal lineage, and nuclear duplications (reviewed in Zhang and Hewitt 1996 ). These problems, together with variable rates of evolution and nonneutrality, have profound implications for both phylogenetic and population genetic studies. However, verification of the true nature of mtDNA variation is rarely attempted because it is difficult and time-consuming.
The present study was undertaken after a phylogenetic study of Alpheus snapping shrimp revealed consistent problems with obtaining good sequence for some species. In some cases, sequences were initially clear but then rapidly deteriorated; these sequences always deteriorated at the same places, for both forward and reverse sequences (later found to result from attempts to sequence a cocktail of sequences that differed in length). In other cases, clear double peaks were observed in both forward and reverse sequences. In contrast, good sequences were consistently obtained from products amplified from cDNA, which coincidentally was being used to amplify several nuclear genes.
These patterns suggested that multiple sequences were being amplified, a possibility of considerable concern given the previous use of Alpheus taxa separated by the Isthmus of Panama to estimate rates of molecular evolution (Knowlton et al. 1993 ; Knowlton and Weigt 1998 ). In order to examine this possibility, we amplified and cloned COI both from total genomic DNA (gDNA) and from cDNA (where tissue preservation techniques allowed) and examined the level of sequence variation revealed. We found multiple copies of COI in all of the species examined, the details of which we present below.
Materials and Methods
For this study, we focused on 10 species of Alpheus for which good sequences for COI were difficult to obtain from gDNA. A single individual was examined from each species. Shrimp were collected from the rocky intertidal shoreline (in crevices or under stones), from within coral rubble close to the low tide level, or from mud flats in or near mangroves. Sample locations are listed in table 1 . These shrimp represented a small subset of the samples from a phylogenetic study using COI and two nuclear genes (GPI and EF-1α) (Williams et al.2001) . Shrimp from Panama (usually males or nonovigerous females) were frozen in liquid nitrogen and stored at −80°C; the single individual from the Cape Verde Islands was preserved in a salt-saturated dimethyl sulfoxide (DMSO) solution (Seutin, White, and Boag 1991 ).
RNA and DNA Extraction
Total genomic RNA and DNA were sequentially extracted from whole shrimp (chelae excluded for large animals) in a multistep guanidinium thiocyanate/acid phenol : chloroform extraction (Totally RNA extraction kit, Ambion) as per manufacturer's instructions. RNA was incubated at 37°C for 30 min with 1 μl (1 unit) of DNAase to remove any contaminating gDNA, and then DNAase was inactivated by heat denaturation at 80°C for 30 min.
RNA could not be extracted from the individual collected from Cape Verde, since it was preserved in a salt-saturated DMSO buffer. Total gDNA was obtained from this animal using a PureGene DNA isolation kit following the manufacturer's protocols except that the sample was digested overnight at 65°C with 6 μl of Proteinase K (20 mg/ml). Total genomic DNA enriched with mtDNA was also extracted from a small piece of muscle tissue from the Cape Verde specimen using the lysis method of Tamura and Aotsuka (1988) .
COI PCR from gDNA
Diluted total gDNA (15–80 ng) was used in a polymerase chain reaction (PCR) to amplify a portion of COI. The primer combination COIF (CCA GCT GGA GGA GGA GAY CC; Kessing et al. 1989 ) and H7188 (CAT TTA GGC CTA AGA AGT GTT G; Knowlton et al. 1993 ) or COIF and COI(10) (TAA GCG TCT GGG TAG TCT GAR TAK CG; Baldwin et al. 1998 ) was used to amplify 640 or 677 bp of COI, respectively. Reactions were performed in 50-μl volumes containing 0.4 μM of each primer, 200 μM of each dNTP, 1.5 mM magnesium chloride, 1.5 U of AmpliTaq DNA polymerase, and 5 μl of AmpliTaq buffer (10×). Thermal cycling was performed as follows: initial denaturation for 2 min at 95°C, followed by 30 cycles of 60 s at 94°C, 60 s at 50°C, and 80 s at 72°C, with a final extension of 30 min at 72°C.
cDNA Synthesis and RT-PCR of COI
First-strand synthesis of cDNA was performed using Superscript II RNase H− reverse transcriptase (Gibco BRL) and a T18 primer following the manufacturer's instructions except for the addition of 20 U of recombinant RNasin ribonuclease inhibitor (Promega).
A 2–3-μl aliquot of cDNA was used in a PCR to amplify 757 bp of COI. Reactions were performed in 50-μl volumes containing 0.1 μM of COIF and COI(10) primer, 200 μM of each dNTP, 1.5 mM magnesium chloride, 2.5 U of AmpliTaq DNA polymerase, and 5 μl of AmpliTaq buffer (10×). Thermal cycle parameters were 3 min at 94°C, 40 cycles of 30 s at 94°C, 30 s at 50°C, and 80 s at 72°C, and a 30-min final extension at 72°C. Faint bands were excised from a 1.2% (w/v) GTG low-melt agarose gel (40 mM Tris-acetate [pH 8], 100 μM EDTA), and 2 μl was used to seed a new amplification reaction using the same PCR protocol.
Cloning
PCR amplicons were gel-purified prior to cloning on a 1.2% (w/v) GTG low-melt agarose gel (40 mM Tris-acetate [pH 8], 100 μM EDTA) by excising and treating the band with Gelase (Epicentre). The liquefied gel sample was further purified using columns from a QIAquick PCR purification kit (Qiagen). Purified products were cloned into JM109 cells using the pGEM-T Easy Vector System II (Promega) following manufacturer's instructions. Individual bacterial colonies were picked from the bacterial plate using wide-bore pipette tips and resuspended by vortexing in 10 μl of water. The presence of an insert of the appropriate size was confirmed by amplification of the insert using 2 μl of the bacteria/water mix and M13 forward and reverse primers in a 25-μl reaction (same concentrations as for RT-PCR) and gel electrophoresis. Cycle parameters were 8 min at 95°C, 40 cycles of 1 min at 94°C, 1 min at 56°C, and 80 s at 72°C, and a final 15-min extension. Those PCR reactions with the correctly sized insert were subjected to an enzyme clean-up, which required incubation of each PCR reaction with 8 U of exonuclease I and 1.5 U of shrimp alkaline phosphatase for 2 h at 37°C followed by 15 min at 80°C to inactivate the enzymes.
Sequencing
Automated sequencing was performed directly on purified PCR products using a dRhodamine Kit (Perkin Elmer). Protocols for cycle sequencing and subsequent purification followed manufacturer's instructions except that reactions were halved (total of 10 μl), and half of the dRhodamine mix was replaced with HalfTerm. The products of sequencing reactions were run on a 377 Applied Biosystems automated sequencer. Ten or 11 clones were sequenced from each individual PCR product obtained from RT-PCR, between 8 and 17 clones were sequenced from gDNA PCR products, and 15 clones were sequenced for the one specimen for which mtDNA-enriched DNA was used to amplify COI (table 1 ). Each clone was sequenced using both forward and reverse PCR primers.
Identification of Taq Error
Sequences obtained for each species from both cDNA and total gDNA, or, in the case of Alpheus holthuisi, mtDNA-enriched DNA and gDNA, were compared. All sequences from a single individual that differed by less than five unique substitutions (any substitution unique to one clone) were grouped, since previous studies based on RT-PCR clones have shown that singletons that differ by up to 5 bp are probably due to Taq error. In a phylogenetic study of more than 60 species of Alpheus where two nuclear genes were amplified from cDNA and cloned and between 7 and 20 clones were sequenced per individual, 24% of cloned sequences in one gene (elongation factor–1α) and 28% in another (glucose-6-phosphate isomerase) showed some evidence of Taq error, including base substitutions and/or indels of 1–5 bp (approximate rate of error 7.5%) (Williams et al.2001) . However, more than two sequences showing the same substitution(s) (even if they differed from another sequence by less than 5 bp) were reported, since this exceeds the level of Taq error observed in previous studies (the same base substitution at a single site has been observed to occur in two clones as a result of Taq error, albeit very infrequently; Williams et al.2001) . Sequence from one clone from Alpheus canalis “orange antennae” appeared to be a chimera between two more common sequences and has not been included.
Given the scale of previous studies, we are confident that these limits make it unlikely that we are reporting Taq error as real variation. In addition, the estimates of Taq error we used (based on Williams et al.2001) were probably exaggerated for gDNA clones for two reasons. First, estimates were based on clones resulting from multiple cycles of amplification. Second, the initial RT-PCR involved the use of reverse transcriptase, which has a higher error rate of replication error than DNA polymerases. Error rates for gDNA clones, although lower than those obtained for cDNA clones, would not reflect error rates obtained by direct sequencing from PCR products, which would presumably be much lower again.
Genetic Distance Between Sequences
Distances between mtDNA sequences and pseudogene copies were estimated as uncorrected and corrected using Kimura's (1980) two-parameter (K2P) model. The K2P model was chosen to calculate genetic distances because it had been used in previous Alpheus studies (e.g., Knowlton et al. 1993 ; Knowlton and Weigt 1998 [although gamma corrected in Knowlton and Weigt 1998] ). Gaps were treated as missing data and affected sites were ignored in pairwise analyses.
Phylogenetic Analysis
We examined the phylogenetic relationship among all pseudogene and mtDNA sequences for the 10 individuals analyzed. We also included previously obtained sequences of transisthmian geminates of the taxa used in this study (or the most closely related transisthmian pair, where known, for taxa that did not have a transisthmian geminate) in order to estimate the timing of the origin of the pseudogenes. GenBank accession numbers for these additional species are listed in the legend of figure 1 . We used maximum-parsimony (MP) analysis in the computer program PAUP* 4.0b3a (Swofford 1999 ) so that we could include indel information to identify relationships among sequences. Gaps were treated as fifth characters and all characters were unweighted. We employed a heuristic search, with starting trees obtained via stepwise addition, tree bisection-reconnection (TBR) branch swapping, and closest taxa. Bootstrap estimates were also obtained using both neighbor-joining (NJ) with K2P and pairwise deletion of missing sites, and MP with fast stepwise addition (each 10,000 replicates).
Two caveats about the phylogeny presented here should be stressed. The first is that several of the assumptions of phylogenetic hypotheses are violated, since nuclear and mtDNA copies may be evolving at different rates, and mtDNA copies are presumably under some selective constraint, whereas nuclear copies are probably not (Bensasson, Zhang, and Hewitt2000) . Additionally, previous phylogenetic studies show that COI data begin to show evidence of saturation between some of the more differentiated geminate pairs (Williams et al.2001) . Therefore, deeper nodes are not well resolved in our phylogeny.
Determination of Separate Nuclear Integration Events
We used an approach developed by Bensasson, Zhang, and Hewitt (2000) to determine the number of independent integrations of pseudogenes from the mitochondrion to the nuclear genome. This approach makes use of the fact that functional sequences in the mitochondria show a bias in the number of absolute differences observed at each codon position (more at the third and first positions than at the second). Once a mitochondrial gene is moved to the nuclear genome, it is no longer functional and therefore can evolve without selective constraint. Thus, if a pseudogene sequence shows a bias in the amount of variation at each codon position, it is likely to reflect differences that accumulated before that sequence moved to the nuclear genome and consequently is evidence of a separate integration. This method can provide only a minimum estimate, since integrations that arose from similar mtDNA haplotypes would not be distinguished (Bensasson, Zhang, and Hewitt2000) .
The absolute number of differences between pseudogene sequences (including two sequences from A. holthuisi; see Results) were calculated for each codon position using PAUP*. Bases in two sequences (from A. holthuisi and Alpheus rostratus; see Results) that were inserted relative to cDNA data were removed in order to maintain the same reading frame observed in the true mitochondrial gene. Sites with missing data were ignored in pairwise analyses. Significant deviation from equal numbers of differences (1:1:1) for each codon site was tested using a chi-square test (df = 2, P < 0.05), and results were Bonferroni-corrected. Pseudogenes which did not show significant deviation from equal numbers of differences among codon sites were grouped together as a single nuclear integration event (Bensasson, Zhang, and Hewitt2000) .
Results
Pseudogene and new mtDNA sequences (for Alpheus bahamensis and A. holthuisi) have been deposited in GenBank (accession numbers for new mtDNA gene sequences: AY010896 and AY010897; pseudogene sequences: AF321325–AF321360). mtDNA sequences for the other eight species are identical to those reported previously (for the same individuals) in Williams et al. (2001) (GenBank accession numbers AF309878, AF309883, AF309896, AF309899, AF309915, AF309929, AF309924, and AF309932).
In all species, sequencing of cDNA clones revealed only one sequence (within the range of Taq error observed in earlier studies; see Materials and Methods). We believe that these sequences amplified from cDNA are representative of the functional mtDNA COI gene because they have been transcribed. On the other hand, sequencing of COI obtained from total gDNA revealed the presence of multiple sequences (table 1 ). The cDNA sequence was sometimes (for six of nine animals) but not always observed in the gDNA sequences and was often rare when it did occur. The presence of multiple copies in gDNA that are not found in the cDNA suggests that these are nonfunctional copies, most probably located in the nuclear genome (see Discussion). Hereinafter, the cDNA sequence is referred to as the mtDNA sequence.
For A. holthuisi, amplifications from DNA following mtDNA enrichment resulted in one common sequence (10/15 clones; table 1 ), whereas amplifications from gDNA obtained by “normal” procedures resulted in three sequences, one of which had an insertion of 7 bp that resulted in a frameshift. The other sequence, which was rare in the mtDNA amplification (3/15 clones) but common in gDNA clones (10/15), was not frameshifted and did not have stop codons. It differed from the putative mtDNA by 49 base substitutions and at two amino acid positions (Ala-Ser, Gln-Arg). The two amino acid changes are radical changes (Taylor 1986 ) at highly conserved positions which have the same amino acid in 52 species of Alpheus (105 individuals) and one outgroup species, Alpheopsis trigonus. Thus, the most frequent sequence amplified from DNA following mtDNA enrichment is probably from the mtDNA gene.
Across the 10 species analyzed, pseudogene sequences always differed from the mtDNA sequence within a single individual by base substitutions. Indels occurred in nearly one third of all sequences (10/36 pseudogene sequences, 6/10 species), and only one of these indels did not result in a frameshift (a single amino acid deleted in an A. canalis “orange antennae” pseudogene). Only one sequence without an indel (in A. bahamensis) coded for a stop codon (using the mtDNA genetic code).
The average indel size was 3.8, with a range of 1–11. Most common were 1-bp deletions (4/10 sequences with indels). Deletions were much more common than insertions, which occurred only twice: there was a 1-bp insertion in A. rostratus pseudogene 3 (after bp 505 in the 564 aligned sites), and there was a 7-bp insertion in one pseudogene sequence from A. holthuisi (after bp 104; GenBank accession number AF321347). One sequence had two indels: one of the A. canalis “orange antennae” pseudogene sequences from family 2 (AF321335) had one indel of 1 bp and another of 2 bp (missing bp 151, 504, and 505). The other A. canalis “orange antennae” sequence from pseudogene family 2 (AF321336) also had a 3-bp deletion (resulting in the loss of an amino acid, and therefore no frameshift); however, it was not at the same place as the other sequence in the same pseudogene family. The largest deletion (of 11 bp) was in Alpheus bouvieri “Caribbean” pseudogene 2 (missing bp 476–486). One “hot spot” for indels, usually consisting of a string of five C's in the functional gene, was found to affect four sequences, with 1–2 bp being added or deleted. Two pseudogene sequences from Alpheus estuarensis pseudogene family 2 (GenBank accession numbers AF321343 and AF321340) were missing 1 bp (bp 505), one member of A. canalis “orange antennae” pseudogene family 2 (AF321335) was missing 2 bp (bp 504 and 505, as well as base 151), and one C was inserted in A. rostratus pseudogene 3 (after bp 505).
Nuclear Integrations
Pairwise examination of the absolute number of differences among 37 different pseudogenes (630 comparisons) suggest that there have been 21 unique horizontal transfers of COI copies from the mtDNA to the nuclear genome. Our data suggest that four individuals had at least three independent transfer events, three had two independent events, and three had one (table 1 and fig. 1 ). Three individuals have nuclear families (i.e., sequences derived from one transfer event) of five sequences, and three individuals have nuclear families of two (fig. 1 ).
Indel data are generally consistent with these groupings. The same 7-bp deletion is found in two sequences within A. bahamensis pseudogene family 3, the same 1-bp deletion is found in two sequences within A. estuarensis pseudogene family 2, and two completely different deletions are found in two different pseudogene integrations in A. bouvieri “Caribbean.” However, in A. canalis “orange antennae,” two sequences in pseudogene family 2 had different indels.
Phylogenetic Analysis
Of the 572 total characters examined (equivalent to the 564 sites used in previous studies, plus insertions), 348 were variable sites, and of these, 255 were phylogenetically informative. Phylogenetic analyses produced six equally most-parsimonious trees (1,796 steps, consistency index = 0.313, retention index = 0.654, homoplasy index = 0.687). We present here the strict consensus of these trees (fig. 1 ).
Pseudogenes often cluster most closely with the mtDNA sequence from the individual from which they were both isolated. For example, pseudogenes of Alpheus floridanus sp. A “Caribbean,” Alpheus paracrinitus “spot,” and A. paracrinitus “no spot” all cluster with the mtDNA sequence from the Caribbean individuals from which they were isolated and not with the Pacific sister species. There are, however, some exceptions; pseudogene 1 from A. bouvieri “Caribbean” clusters with the mitochondrial sequence of A. bouvieri “Pacific.” Some older pseudogenes appear to have been transferred to the nuclear genome before the geminate sister species separated by the Isthmus of Panama diverged, because they lie outside the geminate species pair to which the individual from which they were derived belongs; examples include A. bouvieri “Caribbean” pseudogene sequences 2 and 3, A. canalis “orange antennae” pseudogene family 2, and the A. rostratus pseudogenes.
Discussion
There are four possible reasons for the observation of multiple copies of an mtDNA gene: (1) contamination by other organisms (e.g., parasitic or ingested organisms) or by exogenous DNA in the laboratory, (2) heteroplasmy, (3) paternal leakage, and (4) duplication of the mtDNA gene. We do not believe that our results were affected by contamination, since phylogenetic analysis shows that all sequences amplified were of Alpheus and all sequences from a single animal clustered with other sequences derived from the same individual (or, in the case of some older pseudogenes, with closely related species). Additionally, both the DNA and the RNA used to produce cDNA were sequentially extracted from the same tissue, and there was no evidence of contamination with cDNA.
Although mtDNA homoplasmy can be assumed for somatic cells in most organisms (Avise2000) , heteroplasmy has been widely reported (e.g., Drosophila [Solignac, Monnerot, and Mounolou 1983] , lizards [Densmore, Wright, and Brown 1985] , and crickets [Harrison, Rand, and Wheeler 1985] ). It may arise at low frequencies in somatic cells more or less continuously (Comas, Paabo, and Bertranpetit 1995 ) and could potentially explain some of the sequences which differed only slightly from the true mtDNA gene. Heteroplasmy cannot, however, explain why these sequences were not then observed in the cDNA, nor can it explain the presence of other, highly differentiated sequences which are obviously not functional. Paternal leakage is also an unlikely explanation, since it is usually a low-level and transient phenomenon (e.g., Meusel and Moritz 1993 ; Anderson et al. 1995 ; Shitara et al. 1998 ) and, again, cannot explain the absence of these sequences from cDNA or the presence of obviously nonfunctional sequences. On the other hand, there is a growing body of evidence to suggest that there is a high rate of transfer of mitochondrial genes to the nuclear genome (Adams et al. 2000; Berg and Kurland2000) , and pseudogene copies of mitochondrial genes have been observed in a wide range of organisms (e.g., Kamimura et al. 1989 ; Lopez et al. 1994 ; Sunnucks and Hales 1996 ; Schnieder-Broussard and Neigel 1997; Bensasson, Zhang, and Hewitt 2000 ).
Two lines of evidence suggest that multiple copies of COI in 10 Alpheus species is best explained by duplications of a portion of the mtDNA and subsequent horizontal transfer events to the nuclear genome. First, only one sequence was ever obtained from cDNA clones. Since only transcribed genes (or processed pseudogenes from contaminating gDNA) may be amplified from cDNA, it seems likely that these sequences correspond to the functional mitochondrial gene. Second, multiple sequences were obtained from gDNA, which, since they were not found in the cDNA, suggests that they are not transcribed. This is best explained if the copies are found in the nuclear genome, because in some cases the gDNA copies vary only slightly from the true copy, and not in such a way that should affect its functionality if they were maintained in the mitochondrion.
Transfer of even a functioning copy of COI into the nuclear genome would render it nonfunctional because differences in the nuclear and mitochondrial genetic codes would result in three stop codons (within the portion sequenced in this study). Moreover, transfer of any mtDNA gene to the nuclear DNA is unlikely to result in a functional gene, not only because of differences in the genetic codes, but also because (1) some reading frames lack a termination codon (in other cases, the termination codon is AGA or AGG, which codes for an arginine in the nucleus), and (2) there is only a single promoter for transcription of either strand in mtDNA, and not individual promoters for each gene as in the nucleus (Lewin 1994 , p. 743). On the other hand, it is possible to amplify transcribed mtDNA genes by using the method outlined in our study because mitochondrial mRNA has a short polyA sequence at its 3′ end (Lewin 1994 , p. 743). This is important because RT-PCR uses an oligo (T) primer which binds to this region, thereby amplifying all mRNA transcripts which are generally precursors to functioning proteins.
This is one of the first reported examples of pseudogenes in crustaceans, although there is a growing body of unpublished data. Schnieder-Broussard and Neigel (1997) showed the presence of a nuclear duplication of the 16S gene in stone crabs which differed at only 3% of their nucleotide bases and in length by 2 bp, Schubert et al. (personal communication) identified a COI pseudogene in the fiddler crab Ocypode quadrata that had an altered reading frame, and Bucklin et al. (1999) identified a putative COI pseudogene in a copepod that differed at 36% of the nucleotides examined. The Alpheus pseudogenes identified in this study span both extremes. For example, while many of the pseudogenes were obviously not functional due to frameshifts, others were of identical length to the functional copy and did not include stop codons (using the mitochondrial genetic code). Thus, in many cases, it would be extremely difficult to identify the true sequences without resorting to amplification from cDNA. Additionally, in some cases the true mitochondrial sequence was not obtained at all from gDNA amplifications. This could be explained if some of the primer sequences were a better match to pseudogene copies than the original mtDNA gene. Pseudogenes have often been shown to coamplify with, or in some cases instead of, the desired mtDNA target (e.g., Collura and Stewart 1995 ).
To calibrate the mutation rate for mtDNA, we used the uncorrected divergence of 5.2% observed between the most recently diverged, mangrove-associated species pair, A. estuarensis/A. colombinesis, and assume that this represents a separation of 3.1 Myr when the closure of the Isthmus of Panama was complete (Duque-Caro 1990 ). This gives μ1 = 1.7 × 10−8 substitutions per site per year for cytoplasmic mitochondrial DNA. We have no Alpheus-based calibration for divergence between nuclear pseudogenes, and therefore we used the estimate of Li, Gojobori, and Nei (1981) of μ2 = 4.7 × 10−9 substitutions per site per year for nuclear pseudogene divergences. These two estimates suggest that divergence times range from 82,000 years (for A. paracrinitus “spot” pseudogene 1/mtDNA) to 8.7 Myr (for A. rostratus pseudogene 3/mtDNA).
This method probably seriously underestimates the timing of the oldest integration events for several reasons. First, we know that the mtDNA COI saturates very rapidly at distances above 50 differences (Williams et al.2001) , and there are 106 differences between A. rostratus mtDNA and pseudogene 3. Second, the estimate for pseudogene divergence rates (Li, Gojobori, and Nei 1981 ) is based on mammals, and there is some evidence to suggest that mutation rates of both mitochondrial and nuclear genes are generally higher in homeotherms than in poikilotherms (Martin 1999 ). In support of this suggestion, the A. rostratus pseudogene 3 integration appears to have preceded the divergence of the A. paracrinitus “spot”/A. rostratus lineage from the A. paracrinitus “no spot” lineage (fig. 1 , but see caveat in Materials and Methods section), which occurred approximately 18 MYA (according to calculations in Knowlton and Weigt [1998] ).
In any event, all estimates, in combination with the high number of integration events, suggest that transfer of COI to the nuclear genome has occurred repeatedly over millions of years. In the only other crustacean for which pseudogene data are available for more than one species, putative nuclear sequences from two different species were closely related, suggesting a single nuclear integration event (Schnieder-Broussard and Neigel 1997). Moreover, in Alpheus, the presence of more than one sequence per integration event suggests that there have been duplication events in the nuclear genome as well (although some sequences may represent different alleles of a single locus). Pseudogene sequences which are thought to have arisen from duplications within the nucleus after a single transfer event from the mitochondrion (a “family” of pseudogenes) were identified in six Alpheus individuals. Using the most recently arisen sequence in the “family” (which presumably is the most closely related to the mtDNA sequence because it was the most closely related to the original transferred sequence) to estimate time of divergence showed that in every case families of pseudogenes were older than other pseudogenes from the same individual, which were found only as single sequences. Comparing the divergence times of all pseudogene sequences for all species using the minimum divergence time for each pseudogene family shows that four out of the six family groups diverged between 3.8 and 5.1 MYA and include those families with the largest numbers of sequences (two, five, five, and six sequences per family). The other two families diverged 2.8 and 6.4 MYA (both with two sequences per family). Neither the newest (less than 2.8 Myr old) nor the oldest (6.6 and 8.7 Myr old) pseudogenes were found in families. This suggests that, as might be expected, the newest pseudogenes have not evolved multiple nuclear copies, but the lack of families in the oldest pseudogenes may reflect either the general removal of nongenic DNA within this time frame or that sequences have diverged so much at priming sites that they are unlikely to be amplified.
Multiple independent integration events have been widely assumed but rarely observed (e.g., Fukuda et al. 1985 ; Sorenson and Fleischer 1996 ; Sunnucks and Hales 1996 ; Bensasson, Zhang, and Hewitt 2000 ). In each of these examples, sampling effort (especially the number of individuals and the number of clones sequenced per individual) and sampling design (e.g., the use of primers designed specifically to amplify pseudogene sequences in closely related species) differed; therefore, they are not directly comparable. However, seven COI-II pseudogenes were identified in three species of Sitobion aphids, each thought to have arisen independently (Sunnucks and Hales 1996 ), which reflects a level of introgression similar to that seen in Alpheus. Data for duck pseudogenes suggested that although there were multiple integrations within a single genus, there was only one pseudogene per species, one of which was shared between two closely related species (Sorenson and Fleischer 1996 ). Bensasson, Zhang, and Hewitt (2000) found 12 independent transfer events in one grasshopper species and evidence for other transfer events in other species. The highest number of introgressions to date has been inferred for humans by Fukuda et al. (1985), who used Southern blot data to suggest that 10–130 copies of four different regions of mtDNA occur in the nuclear genome. Perhaps most impressively, Adams et al. (2000) have shown that in many plants the relative rate of transfer of a (still functional) ribosomal-protein gene from the mitochondrion to the nuclear genome actually exceeds the rate of nucleotide substitution at a single silent site.
The number of pseudogenes found varies greatly depending on the taxa examined; for instance, very few pseudogenes are known for Drosophila, despite the large amount of molecular data amassed for this genus (Jeffs and Ashburner 1991 ). It has been noted that pseudogene abundance appears to correlate with genome size (Bensasson, Zhang, and Hewitt2000) , which may reflect the rate at which nongenic DNA is removed from the genome (Petrov et al. 1998 ; Bensasson et al. 2001 ). This would suggest that the Alpheus genome may be larger than that of Drosophila, and while no known data for Alpheus exist to verify this, the maximum genome size for Crustacea is greater than that of other animals except for some bony fish and amphibians (Li 1997 , p. 382). It has also been noted that insertions in pseudogene sequences occur more rarely than deletions and that they occur most commonly after short runs of a single base (Bensasson et al.2001) . We observed the same pattern not only in this study, but also by previous observations of Taq error in Alpheus (unpublished data), suggesting that PCR errors may reflect potential DNA polymerase problems. It has been thought that indels are likely to occur most frequently in older pseudogenes, and this is consistent with our finding that five pseudogene sequences with indels (one of which has two separate sites of deletions) may have diverged from their mtDNA sequence over 6 MYA. The youngest pseudogene sequence with an indel (a 1-bp deletion) is estimated to have diverged 3.9 MYA. The two sequences with insertions include the oldest pseudogene sequence found in this study (A. rostratus pseudogene 3) at 8.7 MYA and one that diverged from its mtDNA about 4 MYA (based on the “younger” of two sequences in the A. holthuisi pseudogene family).
Several putative pseudogenes recognized in our study must reflect very recent transfer events, since they differ from the mtDNA sequence by only a few substitutions (table 1 ) and appear to have occurred after divergence associated with the Isthmus of Panama (fig. 1 ; also based on divergence calculations). For example, in the single pseudogene observed in A. paracrinitus “no spot Caribbean,” there were 3 bp that differed from the mtDNA sequence; one substitution occurred at each codon site, two of which led to amino acid changes. In Alpheus malleator “Caribbean,” one pseudogene sequence (number 1) differed from mtDNA by only 7 bp, and in A. paracrinitus “spot,” the difference was just 1 bp. Although it is tempting to ascribe such similar sequences to Taq or sequencing error, these sequences were never found in cDNA. Moreover, slightly divergent pseudogenes were found in more than one Alpheus species; in each case, multiple peaks could be observed in both forward and reverse chromatograms of directly sequenced PCR products, and the presence of multiple sequences was confirmed by cloning. Ours is not the first study to find such low levels of divergence between mtDNA and some pseudogenes; pseudogenes differed from the mtDNA sequence by 1.4% in one grasshopper species (Bensasson, Zhang, and Hewitt2000) and by 0.6%–2.9% in one aphid species (Sunnucks and Hales 1996 ), and there are reports of human pseudogenes which are 100% homologous with the mtDNA gene (Fukuda et al. 1985 ).
The data presented here, although not from a random sample (we targeted species that were difficult to sequence directly), provide only a minimum estimate of the extent to which pseudogenes are present in the Alpheus genome. Although our analyses suggest that a large number of transfer events have occurred, it is possible that even so, we have underestimated the number of integrations, since pseudogenes arising from two (or more) integrations from similar mtDNA haplotypes would not be recognized (Bensasson, Zhang, and Hewitt2000) . Moreover, our use of an estimate of Taq error based on RT-PCR clones in this study for defining Taq error in gDNA clones errs on the side of being conservative: some of the variation observed, but not reported, probably also reflects additional pseudogenes. Estimates for the number of pseudogene copies and integration events may also increase (1) within individuals with the screening of more clones, (2) within species with the sequencing of a larger number of individuals, and (3) almost assuredly within the genus with the examination of additional species. For example, it is probable that some of the pseudogenes found in this study arose before the rise of the Isthmus of Panama (e.g., A. rostratus pseudogenes 2 and 3, based on both the phylogeny and the divergence calculations) and therefore before the species in question diverged from its closest sister species. Thus, it is very likely that where pseudogenes were found in one species, related sequences will also be found in its geminate pair in some cases. That they were not always found in this study reflects only our sampling procedure; pseudogenes were identified when the primers used were a poor match to the functional mtDNA gene and were not explicitly sought (e.g., by designing primers that matched pseudogenes). More generally, inconsistent results in sequencing and examination of chromatograms from previously obtained sequences suggest that pseudogenes are abundant within the genus.
Some probable Alpheus pseudogene sequences have been published in several papers covering a range of topics (Knowlton et al. 1993 ; Knowlton and Weigt 1997, 1998 ; Williams, Knowlton, and Weigt 1999 ). Fortunately, preliminary analyses suggest that the general conclusions of these papers remain valid. For instance, the staggered pattern of divergence across the Isthmus of Panama (Knowlton et al. 1993 ; Knowlton and Weigt 1998 ) persists when corrected sequences are substituted, as does the distinctiveness of previously identified cryptic species (Knowlton and Weigt 1997 ). On the other hand, a single-base-pair substitution which was thought to be indicative of a genetic break between Indian Ocean populations and Pacific Ocean populations of Alpheus lottini (Williams, Knowlton, and Weigt 1999 ) is apparently not real.
Pseudogenes probably occur far more often than is realized. They can cause great problems in phylogenetic or population studies and have even been the source of error in the identification of human diseases (e.g., Hirano et al. 1997 ; Wallace et al. 1997 ) and in the search for “ancient” DNA from fossils (Collura and Stewart 1995 ). Nuclear duplications almost certainly evolve at a different rate than the real mtDNA gene (e.g., Lopez et al. 1997 ) and may show a different model of amino acid substitution (Sunnucks and Hales 1996 ), with the rate of replacement to silent substitutions and the ratio of transversions to transitions being much higher. Translocated mitochondrial genes evolve much more slowly than their functional counterparts in the mitochondrion and therefore more closely reflect the ancestral, pretranslocated form of the gene (Arctander 1995 ). Thus, nuclear pseudogenes can appear to be more closely related to the most recent ancestral species than the real gene, which in turn could lead to underestimates of the time of differentiation between two species (e.g., Sunnucks and Hales 1996 ). One likely example in our study involves A. bouvieri pseudogene 1, which is closer to the mitochondrial gene of the Pacific member of the transisthmian pair than it is to the mitochondrial gene of the Caribbean individual from which it was isolated.
Some pseudogenes have been shown to differ from the real gene by >36% (Bucklin et al. 1999 ), and in this study by >20%, suggesting that inadvertent use of pseudogenes may result in nontrivial errors. It thus seems imperative to be sure that mitochondrial sequences are indeed just that. It may be advisable for all studies to do preliminary work with cDNA (multiple sequences may still be amplified from mtDNA-enriched DNA as shown in this study, and even from mtDNA purified on a cesium chloride gradient [Collura and Stewart 1995] ) before increasing sample sizes with more easily and rapidly obtainable gDNA.
Stephen Palumbi, Reviewing Editor
Present address: Mollusca Research Group, Department of Zoology, Natural History Museum, London, England.
Keywords: Alpheus cytochrome oxidase I pseudogene numt
Address for correspondence and reprints: S. T. Williams, Mollusca Research Group, Department of Zoology, Natural History Museum, Cromwell Road, London SW7 5BD, United Kingdom. [email protected].
Table 1 Characteristics of Multiple COI-like Sequences in Alpheus
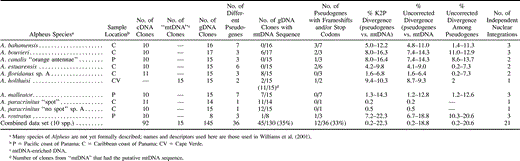
Table 1 Characteristics of Multiple COI-like Sequences in Alpheus
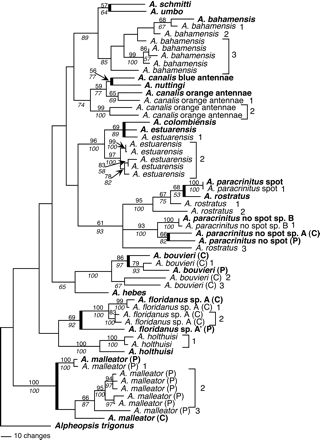
Fig. 1.—Strict consensus of six equally most parsimonious trees showing the phylogenetic relationship among selected COI sequences and pseudogene sequences found in this study. Mitochondrial sequences are indicated by bold text. Where important for species identification (see footnote to table 1 ), sample location is indicated: (P) Pacific coast of Panama, (C) Caribbean coast of Panama. Independent nuclear integrations are identified with numbers for each individual, and nuclear families (sequences arising from a single integration event) are grouped with square brackets. The nodes that link transisthmian sister species are indicated by a thick line. Numbers above each branch are maximum-parsimony bootstrap values using the fast heuristic approach, and numbers in italics below each branch are neighbor-joining bootstrap support values. Bootstrap values less than 50% are not shown. GenBank accession numbers for species not examined in this study (following the order in the figure) are as follows: AF309936, AF309941, AF309881, AF309921, AF309886, AF309929, AF309925, AF309927, AF309880, AF309908, AF309901, AF309913, AF309946). Other GenBank accession numbers are listed in Results
We thank Eyda Gomez and Javier Jara for assistance in the field and lab. Wes Toller and Ron Burton provided valuable discussion about laboratory techniques early in the project. David Reid, José Lopez, and Joe Neigel offered valuable criticisms of the manuscript. The Smithsonian Institution provided most of the support for this project. An NSF grant (97-11525) to Lee Weigt and N.K. provided the opportunity to obtain specimens from Cape Verde. We thank the governments of Panama (Autoridad Nacional del Ambiente and Recursos Marinos) and the Republic of Cape Verde for permission to collect. Special thanks go to Lee Weigt for providing access to chromatograms of previously published sequences.
References
Adams K. L., D. O. Daley, Y.-L. Qiu, J. Whelan, J. D. Palmer,
Anderson T. J. C., R. Komuniecki, P. R. Komuniecki, J. Jaenike,
Arctander P.,
Avise J. C.,
Awadalla P., A. Eyre-Walker, J. Maynard Smith,
Baldwin J. D., A. L. Bass, B. W. Bowen, W. H. Clark Jr,
Bensasson D., D. A. Petrov, D.-X. Zhang, D. L. Hartl, G. M. Hewitt,
Bensasson D., D.-X. Zhang, G. M. Hewitt,
Berg O. G., C. G. Kurland,
Bucklin A., M. Guarnieri, R. S. Hill, A. M. Bentley, S. Kaartvedt,
Collura R. V., C. Stewart,
Comas D., S. Paabo, J. Bertranpetit,
Densmore L. D., J. W. Wright, W. M. Brown,
Duque-Caro H.,
Fukuda M., S. Wakasugi, T. Tsuzuki, H. Nomiyama, K. Shimada,
Harrison R. G., D. M. Rand, W. C. Wheeler,
Hirano M., A. Shtilbans, R. Mayeux, M. M. Davidson, S. DiMauro, J. A. Knowles, E. A. Schon,
Jeffs P., M. Ashburner,
Kamimura N., S. Ishii, M. Liandong, J. W. Shay,
Kessing B., H. Croom, A. Martin, C. McIntosh, W. O. McMillan, S. Palumbi,
Kimura M.,
Knowlton N., L. A. Weigt,
———.
Knowlton N., L. A. Weigt, L. A. Solorzano, D. K. Mills, E. Bermingham,
Li W.-H., T. Gojobori, M. Nei,
Lopez J., M. Culver, J. C. Stephens, W. E. Johnson, S. J. O'Brien,
Lopez J. V., N. Yuhki, R. Masuda, W. Modi, S. J. O'Brien,
Martin A. P.,
Meusel M. S., R. F. A. Moritz,
Petrov D. A., Y.-C. Chao, E. C. Stephenson, D. L. Hartl,
Schneider-Broussard R., J. E. Neigel,
Seutin G., B. N. White, P. T. Boag,
Shitara H., J.-I. Hayashi, S. Takahama, H. Kaneda, H. Yonekawa,
Solignac M., M. Monnerot, J.-C. Mounolou,
Sorenson M. D., R. C. Fleischer,
Sunnucks P. D., F. Hales,
Swofford D. L.,
Tamura K., T. Aotsuka,
Wallace D. C., C. Stugard, D. Murdock, T. Schurr, M. D. Brown,
Williams S. T., N. Knowlton, L. A. Weigt,
Williams S. T., N. Knowlton, L. A. Weigt, J. A. Jara,

{kind=link}