





Abstract
The relatively recent origin of sex chromosomes in the plant genus Silene provides an opportunity to study the early stages of sex chromosome evolution and, potentially, to test between the different population genetic processes likely to operate in nonrecombining chromosomes such as Y chromosomes. We previously reported much lower nucleotide polymorphism in a Y-linked gene (SlY1) of the plant Silene latifolia than in the homologous X-linked gene (SlX1). Here, we report a more extensive study of nucleotide diversity in these sex-linked genes, including a larger S. latifolia sample and a sample from the closely related species Silene dioica, and we also study the diversity of an autosomal gene, CCLS37.1. We demonstrate that nucleotide diversity in the Y-linked genes of both S. latifolia and S. dioica is very low compared with that of the X-linked gene. However, the autosomal gene also has low DNA polymorphism, which may be due to a selective sweep. We use a single individual of the related hermaphrodite species Silene conica, as an outgroup to show that the low SlY1 diversity is not due to a lower mutation rate than that for the X-linked gene. We also investigate several other possibilities for the low SlY1 diversity, including differential gene flow between the two species for Y-linked, X-linked, and autosomal genes. The frequency spectrum of nucleotide polymorphism on the Y chromosome deviates significantly from that expected under a selective-sweep model. However, we detect population subdivision in both S. latifolia and S. dioica, so it is not simple to test for selective sweeps. We also discuss the possibility that Y-linked diversity is reduced due to highly variable male reproductive success, and we conclude that this explanation is unlikely.
Introduction
Heteromorphic sex chromosomes have evolved independently many times (Bull 1983 ), and the properties of sex chromosomes are very similar, suggesting that similar evolutionary processes have operated in the evolution of sex chromosomes in different groups of organisms. The ancient sex chromosome systems of mammals and Drosophila species are characterized by the absence of recombination and exhibit genetic degeneration (loss of functional copies of most Y-chromosome loci; see Lucchesi 1978 ). The chief reason for Y-chromosome genetic degeneration is thought to be low efficacy of selection in nonrecombining regions. This could be manifested as slow adaptive evolution of Y-linked genes compared with genes on the X chromosome (Orr and Kim 1998 ) and as reduced effectiveness of purifying selection, causing accumulation of deleterious mutations and gradual degeneration of the Y chromosome. It is thought that accumulation of deleterious mutations occurs due to three population genetic processes. One process is hitchhiking (fixation of deleterious mutations due to linkage to favorable mutations spreading in the population; Rice 1987 ). A second possibility is background selection (selection against deleterious mutations at linked genes, resulting in reduced effective population size and stochastic accumulation of mildly deleterious mutations; Charlesworth, Morgan, and Charlesworth 1993 ; Charlesworth, Charlesworth, and Morgan 1995 ). These two processes should considerably reduce the effective population size of a nonrecombining region, facilitating the operation of the third process, Muller's ratchet (stochastic loss of chromosomes with the fewest mutations; Charlesworth 1978 ), which is slow unless the size of the population is small (Charlesworth and Charlesworth 1997 ; Gordo and Charlesworth 2000 ). Any or all of these processes may have led to Y chromosomes gradually accumulating deleterious mutations, such that Y-linked genes have become less and less functional. A further consequence of these processes is reduction of the effective population size of Y-linked genes, which should therefore have reduced neutral variability. In chromosome regions with no recombination, such as Y chromosomes, these effects on diversity should be detectable in any loci, not just at loci that are involved in the hypothesized selective processes.
To test the theories for Y-chromosome degeneration, it will therefore be helpful to study neutral diversity of genes in the nonrecombining regions of Y chromosomes to see whether diversity at Y-linked loci is lower than expected from the ploidy differences between Y chromosomes and other chromosomes. Effective population sizes of Y-linked genes are expected to be lower than values for autosomal and X-linked loci, even without the effects of selection just outlined. Ne for Y-linked genes is, in theory, one fourth of that for autosomal genes and one third of that for X-linked genes (Caballero 1995 ). Under neutrality, the nucleotide polymorphism maintained in a population is proportional to the product of the neutral mutation rate and the effective population size of the population (Kimura 1983 ). Thus, Y-linked genes should have approximately one third of the nucleotide variation observed for X-linked genes and one fourth of that for autosomal loci, assuming equal neutral mutation rates for all genes. These differences should lead to lower Y-chromosomal diversity even in sex chromosome systems that are not actively degenerating.
Some data on the diversity of Y-linked loci are available for the neo-Y chromosomes of Drosophila americana (McAllister and Charlesworth 1999 ) and Drosophila miranda (Yi and Charlesworth2000) . The first of these species has a recent X-autosome fusion, and the second has a somewhat older translocation onto the Y chromosome. In both, nucleotide polymorphism of Y-linked genes was decreased relative to the homologous genes on the X chromosome and autosomes, taking into account the ploidy differences. Here, we extend our previous study of plant sex-linked genes, which also demonstrated very low variability at a Y-linked locus (Filatov et al.2000) .
The genus Silene contains about 700 species (Mabberley 1997 ), most of them hermaphroditic or gynodioecious. Two groups of dioecious species apparently evolved independently (Desfeux et al. 1996 ). One group includes the white-flowered Silene latifolia and its close relative, Silene dioica (pink flowered). These two species are closely related and form viable and fertile hybrids in nature (Baker 1948 ; Goulson and Jerrim 1997 ). They are estimated to have diverged from a nondioecious ancestor about 20 MYA based on divergence of ITS sequences (Desfeux et al. 1996 ). Silene latifolia is an agricultural weed and commonly grows in open sunny fields, particularly along the edges of paths and roads, while S. dioica generally grows in more shaded sites, such as woodlands and shady hedgerows, and has a rather more northerly distribution in Europe; S. dioica is absent from North America, while S. latifolia is an introduced weedy species there. The two species have similar chromosomal sex-determination systems (XX female and XY male; see Westergaard 1958 ). In S. latifolia, a small proportion of dihaploid plants, with a Y chromosome but no X chromosome, are viable (Vagera, Paulikova, and Dolezel 1994 ), although haploids with Y alone are not. In contrast, haploids carrying an X chromosome are viable (Ye et al. 1990 ). This suggests that the S. latifolia Y chromosome is at least partly degenerated, although it is not heterochromatic (Vyskot et al. 1993 ).
Searches for genes involved in sex determination have yielded several S. latifolia genes (Matsunaga et al. 1996 ; Barbacar et al. 1997 ; Delichère et al. 1999 ), some of them sex-linked (Guttman and Charlesworth 1998 ). The genes studied here are the X- and Y-linked genes SlX1 and SlY1 (Delichère et al. 1999 ; Filatov et al. 2000 ). These genes were isolated in screens for male organ-specific genes, although SlX1 is expressed in both sexes (Delichère et al. 1999 ). We previously found that nucleotide sequence diversity in the SlY1 gene of S. latifolia is only about one twentieth that of the X-linked ortholog, SlX1 (Filatov et al.2000) . To test whether Y-linked diversity in S. latifolia and S. dioica is reduced, versus the alternative that X-linked variability is unusually high, diversity data for autosomal genes are also needed. Here, we add diversity data on the autosomal gene CCLS37.1 (Barbacar et al. 1997 ; Laporte and Charlesworth 2001 ) In addition, we used outgroup sequences from Silene conica, a hermaphroditic species closely related to S. latifolia and S. dioica (Desfeux et al. 1996 ) to compare divergence in the X-linked, Y-linked, and autosomal genes and to test whether the Y-chromosomal mutation rate is low. We also present more detailed studies to test whether the pattern of nucleotide diversity in the SlY1 gene can be explained by a selective-sweep model, which our initial study seemed to rule out (Filatov et al.2000) . Finally, we examine possible effects of the population structure within the species (which could obscure evidence of selective sweeps) and gene flow between them (which could increase diversity at some loci).
Materials and Methods
Species and Populations
We studied nucleotide diversity in two closely related dioecious Silene species: S. latifolia and S. dioica. White-flowered plants were taken to be S. latifolia, and ones with deep pink flowers were taken to be S. dioica, following Baker (1948) and Goulson and Jerrim (1997) . The S. latifolia sample consisted of three males from laboratory strains and 24 from 10 natural populations, 12 of them used in our previous study (Filatov et al.2000) , while the S. dioica sample consisted of 18 males from four natural populations (table 1 ). In one Scottish population (North Berwick [NB]), S. latifolia, S. dioica, and hybrid plants with pale pink flowers were growing together. From this population, two S. latifolia, three S. dioica and two hybrid males (numbers 270 and 271) were collected (hybrids were oversampled relative to their abundance in the population). In addition, one individual initially classified as S. dioica was reclassified as a hybrid based on sequence data (see Results). A single S. conica plant was used to provide an outgroup.
Genes
Exon-intron structures for the regions studied in the three genes were inferred by comparing genomic DNA sequences with cDNA. In the region sequenced, the intron-exon structures of the sex-linked genes SlX1 and SlY1 (Delichère et al. 1999 ; GenBank accession numbers SLA18517 and SLA18519) were identical, with 15 exons and introns (Filatov et al.2000) . The sequences analyzed here spanned intron 10 to exon 15. The exon-intron structure of the autosomal CCLS37.1 gene (Barbacar et al. 1997 ; accession number SLZ93048) is known only for the portion we sequenced from genomic DNA. This contains one intron about 1 kb long, between nucleotides 308 and 309 of the GenBank CCLS37.1 cDNA sequence. All three genes were sequenced for most plants (table 1 ). However, CCLS37.1 was not studied in the Denmark S. latifolia population. For S. conica, we sequenced the CCLS37.1 homolog and a gene corresponding to SlX1 and SlY1 (denoted Sc).
Molecular Methods
Genomic DNA was isolated from leaves of individual Silene plants by a CTAB plant miniprep method (described in Filatov and Charlesworth 1999 ). For PCR amplification, Taq polymerase (Promega) or Expand long template PCR (Boehringer Mannheim; for amplification of a 5-kb region of the S. dioica SlY1 gene) was used. The PCR products were run on 1% agarose gels with standard 1 × TBE buffer (pH 8) and extracted from gels using the QIAquick gel extraction kit (Qiagen). The PCR products of Sc1 from S. conica and CCLS37.1 from all three species were cloned into the pCR4-TOPO vector using the TA-TOPO cloning kit (Invitrogen). Sequencing of clones and direct sequencing of PCR products were performed on an ABI Prism 377 automatic sequencer (Perkin Elmer) using ABI Prism BigDye terminator cycle sequencing kit (PE Applied Biosystems).
For the S. latifolia SlX1 and SlY1 genes and S. dioica SlX1, we used the same primers and studied the 2.3-kb region previously described (Filatov et al.2000) . The Y-specific primer (“−8”; see Filatov et al.2000) did not anneal to the S. dioica SlY1 gene, so a new primer specific to SlY1 was designed based on a sequence within intron 1 (primer “+18”: 5′-CCTCTTAACAAGATTCACTACGTCTC-3′). This was used with primer “−10” (see Filatov et al.2000) for long-range PCR amplification of a region of about 5 kb spanning introns 1 to 13 of the S. dioica SlY1 gene. This PCR product was used as a template for reamplification (with primers “+11” and “−10” as in Filatov et al.2000) of a 1.7-kb region spanning introns 10–13. Thus, the SlY1 region studied in S. dioica was about 0.5 kb shorter than that in S. latifolia. SlX1 and SlY1 PCR products of both dioecious species were sequenced directly. For S. conica, primers “+11” and “−10” were used to amplify a part of the homologous Sc1 gene for cloning and sequencing. The silent-site divergence of this gene from the SlX1 and SlY1 genes was about 7%, and the Sc1 sequence aligned with SlX1 and SlY1 even in the introns. However, most (about 0.5 kb) of intron 12 was deleted from the S. conica Sc1 gene, which reduced the number of nucleotides analyzed to about 1 kb.
For CCLS37.1, we used the primers 37.1F (forward) (5′-AGGGATTCAATGGTGGTCGTGG-3′) and 37.1Rb (reverse) (5′-GTGCAAATGAAATTCAGAGGAC-3′). These amplify a single band of about 1.2 kb from both S. latifolia and S. dioica and a band of about 1 kb from S. conica. The PCR products of CCLS37.1 from all species were cloned and sequenced using 37.1F, 37.1Rb, and three internal primers, 37.1Fb (5′-CTTTGCCACATCATCCATAG-3′), 37.1F6 (5′-TGCCTTCGTTTTTGTCTCTTGA-3′), 37.1R7 (5′-GGTACTGAAATACCAGGCCAAGAG-3′). Both strands were sequenced for most of the region studied, but for 11 of the 21 sequences, the first 400 bp were sequenced twice in the same direction. Since cloning of PCR products (unlike direct sequencing) may result in incorporation of Taq polymerase errors, which will inflate the number of singletons in the sample, we checked all the singleton polymorphic sites by additional direct sequencing of PCR products.
Sequence Alignment and Analysis
Sequences were aligned using ClustalX, version 1.64 (Thomson et al. 1997 ), followed by manual adjustment using ProSeq, version 2.71 (Filatov2001) . Estimates of nucleotide diversity, population subdivision, and gene flow statistics, as well as permutation-based tests of significance (Hudson, Boos, and Kaplan 1992 ), were performed using ProSeq, version 2.71. The neighbor-joining tree (fig. 2 ) was created using MEGA, version 1.01 (Kumar, Tamura, and Nei 1993 ).
Within each species, nucleotide diversities were compared between genes using HKA tests (Hudson, Kreitman, and Aguadé 1987 ), taking ploidy differences into account using DNAsp, version 3.5 (Rozas and Rozas 1999 ). Interspecific divergence values were estimated using a single S. conica individual. Because the S. conica sequence contains deletions in introns relative to those of S. latifolia and S. dioica from the SlX1 and SlY1 sequences, the total number of sites in this analysis was 1,012 nt. To estimate the recombination statistic, CHud (Hudson 1987 ), and for the FS neutrality tests (Fu 1997 ), we also used DNAsp, version 3.5, and P values for the FS neutrality test were estimated by coalescent simulations without recombination.
The possibility of different evolutionary rates of the X and Y chromosomes was tested by a likelihood ratio test using the local-molecular-clock method (Yoder and Yang2000) . To compare mutation rates in the SlX1 and SlY1 genes, we rooted the branches of the SlX1 and SlY1 sequences using the homologous S. conica sequence, Sc1. A maximum-likelihood tree for all the SlX1 and SlY1 sequences of either S. latifolia or S. dioica, plus the S. conica sequence, was constructed by the PHYLIP dnaml program (Felsenstein 1993 ). A model with three different evolutionary rates (for SlX1, SlY1, and Sc1) in the ancestry of the sequences was then tested against one with just two evolutionary rates, one for the non-sex-linked homolog Sc1 and another common to both the SlX1 and the SlY1 genes. This was done using the baseml program in the PAML package (Yang2000) to calculate likelihoods for the two models. As the log likelihood ratio of these values is χ2-distributed (Muse and Weir 1992 ), the significance of the differences between the two models can be evaluated. Since a separate evolutionary rate for Sc1 was allowed in both models, the rate on the S. conica branch does not affect the results of the analysis.
Tests for Gene Flow Between S. latifolia and S. dioica
Two approaches were used to test whether our data from S. latifolia and S. dioica differed significantly from the predictions of a model with no gene flow. First, we used coalescent simulations (Hudson 1990 ) to model a split into two populations of the same size without subsequent gene flow, but with recombination within populations (using ProSeq, version 2.71; Filatov2001) . The simulations were conditioned on the actual number of segregating sites in the pooled sample of the two species and were run with a recombination rate equal to the average estimate for the two species. Until the time of the split (Ta) is reached (going back in time), coalescence and recombination events occur within each of the two populations. At time Ta, the two populations are united, and the process continues until the most recent common ancestor is reached. We obtained bounds for the values of several descriptive statistics by comparing the values estimated from the data with those obtained from simulations with different divergence times (Ta) between the two species (scaled in terms of Ne values). The statistics calculated for each run were the net divergence Da (Nei 1987 ), the population subdivision statistics Fst (Hudson, Boos, and Kaplan 1992 ), and the numbers of polymorphic sites that were fixed and shared between the two populations. After 1,000 runs, the 5% and 95% percentiles for the distributions of all the statistics were calculated and used as confidence intervals for the values of the statistics given the speciation time Ta.
Second, we used Wakeley and Hey's (1997) model of population divergence in a two-island model without gene flow, taking into account differences in effective population sizes between the two modern and the ancestral species. The approximate age of the split between the extant populations and the scaled mutation rates (𝛉 = 4Neμ) of all three populations were estimated using ProSeq, version 2.71 (Filatov2001) . The program uses the numbers of shared polymorphic sites, fixed sites, and polymorphic sites exclusive to each of the two populations sampled to obtain numerical solutions of equations 12–16 in Wakeley and Hey (1997) by Newton-Raphson iteration (Press et al. 1992 ).
Results
Polymorphism and Population Structure Within Species in the SlY1, SlX1, and CCLS37.1 Genes
Nucleotide diversity in the three S. latifolia and S. dioica genes is summarized in table 2 . The lengths of coding and noncoding regions differ between the genes (for instance, our CCLS37.1 sequences contain only 18% nonintron sites, compared with 25% for SlX1). One cannot, therefore, directly compare their diversity. At least in Drosophila, silent sites in exons typically have higher diversity than intron sites (Moriyama and Powell 1995 ). We therefore estimated nucleotide diversity separately in noncoding and coding sites of the three genes. In both species, X-linked and autosomal sequences are less variable at replacement sites than at silent or intron sites. For SlX1, diversity values (π) range from 1% to 2%, with no consistent difference in diversity between synonymous and intron sites (table 2 ). For all types of sites, the SlY1 gene of both species had lower diversity than SlX1 or CCLS37.1. Among the nine S. dioica SlY1 1.5-kb sequences, we found only one nucleotide variant (in intron 11 of sequences from the Corrèze population; fig. 1 ). In the 22 S. latifolia SlY1 sequences for which the longer sequences (2 kb) were analyzed, we found five nucleotide variants (four in introns, and a Pro/Leu amino acid replacement; fig. 1 ).
In addition, indel polymorphisms were more abundant in the X-linked and autosomal genes than in the Y-linked gene. Treating regions with overlapping indels as single-indel sites, there are at least 28 insertion/deletion (indel) polymorphisms in the 24 S. latifolia SlX1 sequences and 17 in the 11 SlX1 S. dioica sequences. Among the CCLS37.1 sequences, there are seven indels in the 21 S. latifolia sequences and four in the 15 sequences from S. dioica. In the sample of S. latifolia SlY1 sequences, however, there are only two indels, and there is one in the S. dioica SlY1 sequences. Indel regions were excluded from most of the further analyses, but they were included in analyses of shared and fixed variants in S. latifolia and S. dioica (see below). The S. latifolia SlY1 sequences fall into four haplotypes, which differ by several nucleotide substitutions and indels (fig. 1 ).
All the genes studied show evidence of population subdivision in both S. latifolia and S. dioica, and all Fst estimates except that for the S. latifolia CCLS37.1 gene are significantly greater than zero (P < 0.05). The S. latifolia SlY1 haplotypes are associated with the geographic locations from which the samples originated, although the association is not complete, and several populations have two haplotypes, even with our small samples (fig. 1 ). The Fst estimates for the SlY1, SlX1, and CCLS37.1 genes in S. latifolia were 0.76, 0.46, and 0.36, respectively. For the SlX1 and CCLS37.1 genes of S. dioica, Fst estimates were 0.79 and 0.18, respectively (Fst was not calculated for the S. dioica SlY1 because only one polymorphic site was found in the sample).
HKA Tests and Tests for Mutation Rate Differences in the SlX1 and SlY1 Genes
To compare nucleotide diversity in the SlY1, SlX1, and CCLS37.1 genes, we used the HKA test (Hudson, Kreitman, and Aguadé 1987 ). This test assumes that sequences follow a neutral coalescent process in which polymorphism is proportional to divergence, which may not be true for a subdivided population such as that from which our samples come (see previous section). However, Wakeley (1999) has shown that the coalescent approach may still be useful in subdivided populations. The genealogy of such samples can be considered as having two phases: a very short recent “scattering phase” and a much longer “collecting phase,” which starts (going backward in time) when each lineage ancestral to the sample is in a separate deme. Wakeley (1999) demonstrated that the genealogy of ancestral lineages during the collecting phase is a coalescent. Because the collecting phase lasts much longer than the scattering phase, the scattering phase can be ignored, provided that a large enough number of populations are sampled. Since our S. latifolia sample included 10 natural populations, the genealogy may thus be well approximated by a coalescent process, and the HKA test may be used. This may not, however, be legitimate for the S. dioica sample from only four populations.
The HKA test results were similar for both species (table 3 ). The SlY1 gene has significantly (P < 0.05) less diversity than SlX1, taking ploidy into account. However, for both species, SlX1 has significantly (P < 0.05) higher nucleotide polymorphism than CCLS37.1, while the HKA tests for the CCLS37.1/SlY1 comparison are nonsignificant.
The HKA test takes into account possible mutation rate differences between genes, so the reduced SlY1 diversity is not likely to be due to a lower neutral mutation rate of this gene. Moreover, using likelihood ratio tests, we did not detect significant evolutionary rate differences in the X- and Y-linked genes: a model with two rates (one for Sc1 and another for the SlX1 and SlY1 genes) accounts for the data, as does one with three rates (for SlX1, SlY1, and Sc1); χ2 = 2.48 for S. latifolia and 1.33 for S. dioica (df = 1, P > 0.05 for both). The evolutionary rates in the SlX1 and SlY1 genes are thus not significantly different. Differences in evolutionary rates cannot therefore explain the low SlY1 polymorphism.
We can also eliminate the possibility that the lower diversity of CCLS37.1 compared with SlX1 could simply be due to different amounts of coding and noncoding sequence from the two genes. Our data show no sign of lower intron diversity (table 2 ). Moreover, the HKA test remains significant for SlX1 and CCLS37.1 introns only (table 3 ).
Possible Causes of Low Diversity in the CCLS37.1 Gene
The much lower diversity in the autosomal gene than in the X-linked gene is surprising. As noted above, the effective population size for autosomal genes is expected to be four thirds that of X-linked ones, and thus neutral diversity of autosomal genes should be somewhat higher than that for X-linked loci. We tested several other possibilities for this difference, including differences in recombination rates, and effects of either balancing selection (which could inflate SlX1 diversity) or selective sweeps (which could have reduced diversity in CCLS37.1).
Estimates of Recombination Rates for the X-Linked and Autosomal Genes
One possible cause of the low CCLS37.1 diversity is the well-known effect of reduced diversity in regions of low recombination (e.g., Begun and Aquadro 1992 ; Stephan and Langley 1998 ). To examine this possibility, we estimated recombination rates using Hudson's (1987) measure of recombination rate per nucleotide, CHud, and calculated Kelly's (1997)ZnS statistic, a summary of linkage disequilibrium for all sites. In both species, SlX1 appears to experience less recombination than CCLS37.1, the opposite of the difference in DNA diversity. For S. latifolia, CHud = 0.012 for SlX1 and 0.122 for CCLS37.1, and ZnS = 0.131 for SlX1 and 0.08 for CCLS37.1. For S. dioica, both statistics suggest lower recombination than in S. latifolia (CHud = 0.004 for SlX1 and 0.013 for CCLS37.1; ZnS = 0.291 for SlX1 and 0.098 for CCLS37.1). These estimates assume that the populations are at mutation-drift equilibrium, an assumption which may be violated for the loci and populations studied here. However, subject to this caveat, there is no evidence in either species that lower recombination causes lower diversity in CCLS37.1 than in SlX1. Since this approach often appears to underestimate recombination frequencies (Andolfatto and Przeworski2000) , this conclusion is conservative.
Tests for Selection
If diversity in CCLS37.1 has been reduced by a recent selective sweep (Kaplan, Hudson, and Langley 1989 ), the frequency spectrum of polymorphic sites should exhibit excess rare variants (Langley 1990 ; Braverman et al. 1995 ). If, however, SlX1 diversity is inflated due to balancing selection at a linked site, which can maintain diversity for a very long time (e.g., Strobeck 1983 ; Nordborg, Charlesworth, and Charlesworth 1996 ; Charlesworth, Nordborg, and Charlesworth 1997 ; Takahata and Satta 1998 ), the site frequency spectrum should have a bias toward frequent variants. Population subdivision affects the frequency spectrum similarly to balancing selection but should affect all the genes similarly.
The SlX1 spectrum does not deviate significantly from the distribution expected for neutral variants. Neither Tajima's (1989)D nor Fu and Li's (1993) D* statistic differs significantly from zero for either species studied (table 4 ). Thus, it is unlikely that SlX1 has unusually high diversity caused by balancing selection. These tests are also nonsignificant for CCLS37.1. However, Fu's (1997)FS statistic, which is very sensitive to the frequency spectrum bias toward rare polymorphisms, detects a significant deviation from neutrality for the CCLS37.1 gene in both species, despite the population subdivision, which would tend to bias the frequency spectrum in the opposite direction, masking the effect of a selective sweep (Strobeck 1983 ; Nordborg, Charlesworth, and Charlesworth 1996 ). This suggests a recent selective sweep in CCLS37.1. The frequency spectrum bias toward rare variants is significant for this gene. We therefore tentatively conclude that the CCLS37.1 has atypically low diversity for an autosomal locus and has perhaps experienced a selective sweep. In what follows, we therefore assume that the lower diversity at SlY1 than SlX1 requires further explanation, rather than seeking to explain why SlX1 diversity is high.
Gene Flow Between S. latifolia and S. dioica
A possible reason for the difference in diversity between the SlY1 and SlX1 genes is a difference in gene flow of these loci between S. latifolia and S. dioica. Hybridization clearly occurs between the two species. Both plants with an intermediate flower phenotype (pale pink; plants 270 and 271 from the North Berwick population in Scotland) have CCLS37.1 and SlY1 sequences (SlY1 sequenced only for plant 271) that cluster with the corresponding S. dioica sequences, while their SlX1 sequences cluster with those of S. latifolia SlX1 (fig. 2 ). In addition, one S. dioica plant (plant 484) out of four from the Corrèze population (France) had a CCLS37.1 sequence that clustered with those from S. latifolia, although its SlX1 and SlY1 sequences fell among those of S. dioica (fig. 2 ). On the basis of these sequence data, this plant was reclassified as a hybrid.
Differences in selective pressures against introgressed alleles for X-linked, Y-linked and non-sex-linked genes could, in theory (Barton and Bengtsson 1986 ), result in different rates of gene flow for the SlX1, SlY1, and CCLS37.1 genes. Does gene flow occur, does it occur at different rates for the loci studied, and could it account for the diversity differences observed? Table 5 compares nucleotide site and indel differences between the two species. In SlY1, no polymorphisms of either kind are shared between the species (excluding all three hybrids). Most of the variable sites are fixed differences, and Fst between the species is high. Gene flow between S. latifolia and S. dioica must therefore be low for this locus. For SlX1, there are few fixed differences and several shared polymorphic sites, such that net silent-site divergence and Fst are lower than for SlY1 (table 5 ). The autosomal gene, CCLS37.1, is even less diverged than SlX1, with only two fixed indels and no shared sites (out of 48 variable sites). The Fst estimates for CCLS37.1 are, however, similar to those for SlX1. Finally, even after removing from the data the 11 SlX1 sites with polymorphisms that are shared between the two species and could be caused by gene flow, the diversity differences between SlX1 and SlY1 remain significant for both species (HKA test; P < 0.05; see table 3 ).
Polymorphic sites shared between S. latifolia and S. dioica genes, such as those in SlX1, however, may not indicate gene flow, but could have persisted since the time of common ancestry, especially if the time since speciation is short. To test whether a split without further gene flow was compatible with our data from these loci or whether subsequent introgression was required to explain the results, we ran coalescent simulations assuming no gene flow (see Materials and Methods) and estimated divergence times between the two species (Ta). The Ta values, scaled in terms of Ne values, are consistent for the SlX1 and CCLS37.1 genes and suggest speciation between 2Ne and 4Ne generations ago. If there has been gene flow, this is an underestimate.
A model of population divergence without gene flow (Wakeley and Hey 1997 ) is also compatible with our data. Estimated numbers of generations since the split between the two populations, based on the SlX1 and CCLS37.1 data, are shown in table 6 . The parameter values estimated for S. latifolia and S. dioica by this model are similar to those estimated above within each species individually. With the same mutation rate (μ) in the ancestral and the modern populations, the estimated ancestral population size is close to the modern S. latifolia population size. Assuming no gene flow between the two species, the speciation event is estimated to have occurred about 2Ne generations ago (table 6 ), consistent with the time estimated from the coalescent simulations.
Discussion
Reduced Diversity on the Y Chromosome
Genetic degeneration of nonrecombining Y chromosomes by the processes mentioned above should be accompanied by considerable loss of nucleotide diversity (reviewed by Charlesworth and Charlesworth2000) . In the early stages of Y-chromosome evolution, when recombination has recently ceased, many functional genes will still be present, and there should thus be a high rate of both advantageous and deleterious mutations. Since the genes will be linked in a nonrecombining block, the reduction of diversity in the region should be severe. The S. latifolia Y chromosome has probably not yet become fully genetically degenerate, as active genes not thought to be involved in sex determination or male function, such as SlY1, are present. We previously reported very low SlY1 polymorphism (Filatov et al.2000) , and this is verified with the present larger sample. Although it is likely that diversity is reduced for the Y-linked SlY1 locus, rather than being inflated for the X-linked one, the low CCLS37.1 diversity makes this uncertain. There is, however, no evidence for balancing selection acting at or near the X-linked SlX1 locus, and we found some evidence for a selective sweep at CCLS37.1.
Possible Factors Reducing Nucleotide Diversity on the Y Chromosome
Our results enable us to eliminate several possible explanations for the low SlY1 diversity in S. latifolia and S. dioica. A lower mutation rate on the Y chromosome is one possibility. There is currently little information about mutation rates of genes on different chromosomes, other than for mammals (McVean and Hurst 1997 ; Smith and Hurst 1999 ) and Drosophila (Baur and Aquadro 1997 ). No data are available from plants. However, the assumption of equal mutation rates in the three genes can be tested by comparing their sequences with those of an outgroup species, since neutral divergence depends only on the mutation rate (Kimura 1983 ). Our results give no evidence of different mutation rates of the homologous X- and Y-chromosome genes. This conclusion is supported by HKA tests showing significantly reduced diversity in SlY1 compared with SlX1, taking into account their relative rates of divergence from the S. conica sequence.
Another possibility is a high variance in male mating success (including sexual selection) or a female-biased sex ratio. These situations will reduce the effective population size of Y-linked genes compared with that for X-chromosomal and autosomal loci (Caballero 1995 ; Charlesworth 1996 ) and can reduce (or even reverse) the difference in effective population sizes between X chromosomes and autosomes. X-chromosomal diversity can then reach values similar to, or slightly higher than, those of autosomal loci (Caballero 1995 ). It is not known whether plants are likely to have a high variance in male mating success, but this possible cause of the low SlY1 diversity should be testable by comparing nucleotide diversity in X-linked and autosomal genes. Our results do not support this explanation because nucleotide diversity is considerably higher in the X-linked gene, SlX1, than in the autosomal CCLS37.1.
Unlike the situation for animal male gametes, a high proportion of genes are expressed in at least one pollen grain nucleus (Tanksley, Zamir, and Rick 1981 ; Stinson et al. 1987 ; Vielle-Calzada et al. 2000 ). If X-linked genes expressed in pollen are important for mating success, pollen grains carrying Y chromosomes will be somewhat defective compared with X-bearing pollen. Indeed, a female-biased sex ratio is often observed in natural populations of S. latifolia and S. dioica (Correns 1928 ; Lloyd 1974 ) and some, but not all, S. latifolia families (Taylor 1994a, 1994b ). However, gene expression in the haploid pollen also allows the possibility of selection against deleterious mutations in pollen-expressed Y-linked genes (Haldane 1927 ). On the one hand, this may lead to background selection, reducing silent diversity in sequences on plant Y chromosomes, but on the other hand, it may slow down genetic degeneration, at least at loci under purifying selection during the pollination process.
Many other factors, including a gene's local recombination frequency, and selection within the locus, or at very closely linked loci, affect its diversity. The excess of singleton polymorphisms in CCLS37.1 indeed suggests that diversity in this gene is unusually low and has probably been reduced by a recent selective sweep. This gene may therefore not be a suitable reference locus for asking whether Y-linked genes have lower diversity than their X-linked homologs. Studies of diversity levels of further autosomal and X-linked loci are required to resolve this question.
Gene Flow as a Possible Cause of Elevated Diversity of X-Linked Genes
Yet another possibility is that hybridization between S. latifolia and S. dioica may contribute to the high SlX1 diversity in both species. Although hybridization certainly occurs, the S. latifolia or S. dioica sequences form separate clusters, and all sequences of the X-linked and autosomal genes cluster either with S. latifolia or S. dioica sequences. This might suggest that only very recent hybrids occur, and not older gene introgressions (which should have generated recombinant alleles), perhaps indicating some form of selection against hybrids. Consistent with this, the divergence time between the two Silene species for SlX1 and CCLS37.1, estimated by two methods that assume no gene flow since the time of speciation, is similar to that estimated directly from the SlY1 nucleotide divergence of the S. latifolia and S. dioica alleles. These analyses do not, however, conclusively rule out gene flow. The evidence of hybridization in our sequence data must underestimate its frequency; if data were available from more loci, more plants would probably be classified as hybrids. However, we found no evidence of markedly different rates of gene flow between S. latifolia and S. dioica for the different loci, although these tests did detect such differences between Drosophila pseudoobscura and its close relatives (Wang, Wakeley, and Hey 1997 ).
To be as conservative as possible in testing whether diversity is higher in SlX1 than in SlY1, we could assume that introgression is so frequent between the two species that diversity has reached equilibrium under gene flow. Since the Fst values between the two species for SlX1 are not much higher than those between different populations within each species, the two species may be viewed as a single subdivided population. Assuming conservative migration (Nagylaki 1998 ), the expected equilibrium within-species diversity (π) is independent of the migration rate and is given by 4NTμ, where NT is the total population size (Maruyama 1971, 1972 ; Slatkin 1987 ; Strobeck 1987 ). The SlX1 diversity would then be, at most, doubled as a result of gene flow (on the most conservative assumption, that migration occurs in both directions between the two species and that these two species have the same population size). We should thus halve the observed SlX1 diversity value; this gives 51 and 28 polymorphic sites in the S. latifolia and S. dioica SlX1 sequences, respectively, still considerably higher than the observed SlY1 diversity within either species. The HKA test remains significant (P < 0.05) for S. latifolia, but not for S. dioica. Thus, at least for S. latifolia, correction both for ploidy differences and for gene flow does not remove the diversity difference between SlX1 and SlY1.
The diversity difference between X- and Y-linked genes cannot, therefore, be explained by a low Y-chromosome mutation rate or by a high variance in male mating success, and probably not by different gene flow between the two species. This suggests that differences in effective population sizes of genes on the X and Y chromosomes are involved.
Testing Between Background Selection and Selective Sweeps
The different population genetic models to explain reduced genetic diversity in nonrecombining regions are, in principle, distinguishable because they lead to different predicted site frequency spectra of polymorphic variants. The selective-sweep model (Rice 1987 ) predicts a frequency spectrum biased toward rare variants (Langley 1990 ; Braverman et al. 1995 ). Under the background selection model, however, the site frequency spectrum should be close to that expected under neutrality, unless the deleterious mutations driving the process have very small selection coefficients (Charlesworth, Charlesworth, and Morgan 1995 ). No detailed study has yet been published of the effects of Muller's ratchet on diversity at neutral sites in a nonrecombining chromosome, but simulations show effects intermediate between the above two models, with a frequency spectrum less biased than that caused by selective sweeps but still potentially distinguishable from the neutral spectrum, at least for the population sizes studied so far (I. Gordo and B. Charlesworth, personal communication).
No bias in the frequency spectrum was detected for loci on the D. americana (McAllister and Charlesworth 1999 ) and D. miranda (Yi and Charlesworth2000) neo-Y chromosomes, which tends to support the background selection model for these sex chromosome systems in which functional genes are present on the Y chromosome. On the other hand, Zurovcova and Eanes (1999) report excess singleton polymorphisms in the D. melanogaster Y-linked dynein gene, supporting the selective-sweep model (perhaps caused by selection within this very large gene).
There are problems in testing site frequency spectra in subdivided populations such as those of Silene. Seed migration between populations separated by shorter geographic distances than those studied here is limited in these species based on studies using genetic markers (McCauley 1994 ; Giles, Lundqvist, and Goudet 1998 ; Ingvarsson and Giles 1999 ; Richards, Church, and McCauley 1999 ), and gene flow between populations appears to be mostly via pollen movement (Richards, Church, and McCauley 1999 ). Subdivision may therefore be less extensive for Y chromosomes than for autosomes and X chromosomes. On the other hand, the lower effective population size for Y-linked genes implies that genetic drift will affect these genes more than autosomal loci, causing them to have lower within-population variability and greater differentiation between populations. The frequency spectrum of variants in Y-linked loci might thus be particularly affected by subdivision. Further theoretical work is, however, needed to elucidate the net effects on sex-linked and autosomal loci of multiple sampling from demes in a subdivided population.
It is nevertheless clear that if deme sizes are small and gene flow between populations is low, mutations on the Y chromosome may quickly be fixed in local populations by drift and/or local selective sweeps, such that different variants will be present in different populations. Thus, if these species have a subdivided population structure, the site-frequency spectra will be affected. Sampling of multiple individuals per population generates samples in which all of the polymorphic sites on the Y chromosomes are non-singletons, such that a negative Tajima's D might become nonsignificant, and selective sweeps would be wrongly rejected. The only case in which we obtained a positive D statistic was that of the S. latifolia SlY1 (table 4 ), but it may nevertheless be more appropriate to sample a single sequence per population. We therefore tested whether such a sample changes the outcome of the Tajima's tests. Subsamples of S. latifolia SlY1 sequences, one from each natural population and one from the laboratory strain, were randomly generated. Figure 3 compares the average frequency spectrum in 1,000 such subsamples of 11 SlY1 sequences (fig. 3B) with the observed spectrum for the entire sample (fig. 3A). Tajima's D remains positive for the subsamples (mean value 0.418). Thus, the SlY1 frequency spectrum shows no bias toward rare polymorphic sites, as would be expected under the selective-sweep hypothesis.
This test is, however, based only on intuitive ideas about the expected behavior of subdivided populations. Ideally, we should argue based on results of models that include population subdivision. Assuming a high selection coefficient and a low migration rate between subpopulations (Nm < 1), a process of selective sweeps in a subdivided population is dominated by the restricted migration, and intrasubpopulation fixation can be ignored (Slatkin and Wiehe 1998 ). For a nonrecombining chromosome such as the Y chromosome, hitchhiking in Slatkin and Wiehe's (1998) model leads to fixation of one allele in the whole population, eliminating diversity in the entire linked region. This model is probably reasonable for Silene populations, since Fst is high for all loci. Moreover, our finding of four S. latifolia SlY1 haplotypes which differ by two or more substitutions, one of which is found in several different geographic locations (fig. 1 ), suggests that some gene flow of Y chromosomes between populations must occur. These data thus appear inconsistent with a simple advantageous hitchhiking model, and our conclusion that selective sweeps do not seem to be responsible for low diversity at the SlY1 locus is therefore probably valid if we take into account the subdivided population structure.
Wolfgang Stephan, Reviewing Editor
Present address: School of Biosciences, University of Birmingham, Birmingham, England.
Keywords: Y chromosomes sex linkage selective sweeps background selection gene flow
Address for correspondence and reprints: Dmitry A. Filatov, School of Biosciences, Diversity of Birmingham, Edgbaston, Birmingham B15 2TT, United Kingdom. [email protected] .
Table 1 Species and Populations Sampled in this Study
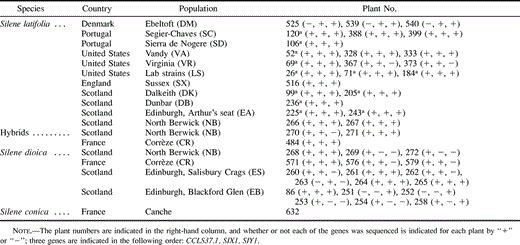
Table 1 Species and Populations Sampled in this Study
Table 2 DNA Polymorphism in Coding and Noncoding Regions of SlY1, SlX1, and CCLS37.1 Genes
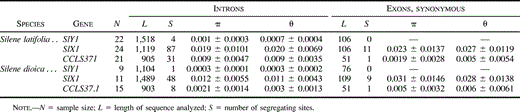
Table 2 DNA Polymorphism in Coding and Noncoding Regions of SlY1, SlX1, and CCLS37.1 Genes
Table 2 Extended
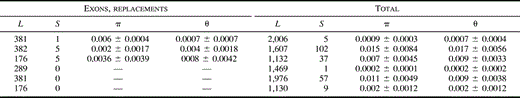
Table 2 Extended
Table 3 Results of Pairwise HKA Tests for the SlX1, SlY1, and CCLS37.1 Genes
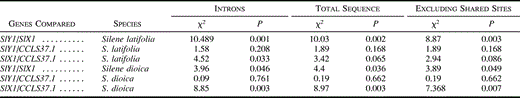
Table 3 Results of Pairwise HKA Tests for the SlX1, SlY1, and CCLS37.1 Genes
Table 4 Results of Neutrality Tests for the SlY1, SlX1, and CCLS37.1 Genes of Silene dioica and Silene latifolia
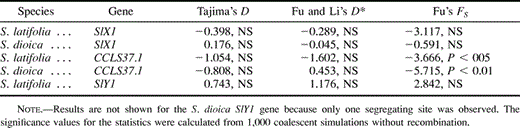
Table 4 Results of Neutrality Tests for the SlY1, SlX1, and CCLS37.1 Genes of Silene dioica and Silene latifolia
Table 5 Differences Between Sequences from Silene latifolia and Silene dioica

Table 5 Differences Between Sequences from Silene latifolia and Silene dioica
Table 6 Estimates of Population Parameters for Silene latifolia and Silene dioica

Table 6 Estimates of Population Parameters for Silene latifolia and Silene dioica

Fig. 1.—Polymorphic sites in the SlY1 region studied. Sequences are referred to by the species names, followed by the population abbreviation (see table 1 ) and the number of the individual plant. The types of sites are indicated by letters (i = intron; s = synonymous; r = replacement). Dots indicate nucleotides or indel sequences identical to the first sequence. The S. latifolia SlY1 sequences fall into four haplotypes, shown at the right-hand side of the figure. Indels: a = AC; b = CTG; c = AA; d = AT; e = 37 bp; f = CCC; g = CTG; h1 = 186 bp; h2 = 161 bp; h3 = 44 bp; h4 = 43 bp
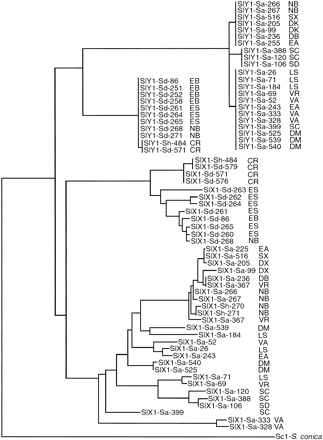
Fig. 2.—Neighbor-joining tree of Silene latifolia and Silene dioica SlX1 and SlY1 sequences, rooted by a single Silene conica sequence of a non-sex-linked gene, Sc1, homologous to SlX1 and SlY1 genes

Fig. 3.—Silene latifolia SlY1 site-frequency spectra. White bars show the expected relative neutral frequencies. The short horizontal lines indicate one standard deviation from the mean values, calculated from 1,000 coalescent simulations without recombination, conditioning on the actual number of segregating sites observed in the sample. A, The gray bars show the observed frequency spectrum for the pooled sample of 22 sequences. B, The gray bars show the average frequency spectrum for 1,000 random subsamples of 11 S. latifolia SlY1 sequences, one sequence from each population listed in table 1
We thank G. A. T. McVean, B. Charlesworth, and I. Gordo for discussions and advice on analyses, R. Goodwin for help with S. dioica sequences, Y. Piquot for providing S. conica material, and the University of Edinburgh greenhouse staff for plant care. D.A.F. was supported by a grant to D.C. from the Leverhulme Trust, V.L. by a grant from the European Science Foundation and a grant to D.C. from BBSRC, C.V. by the programme Magistère Universitaire, École Normale Supérieure, Paris, and D.C. by the Natural Environment Research Council of Great Britain.
References
Andolfatto P., M. Przeworski,
Baker H. G.,
Barbacar N., S. Hinnisdaels, I. Farbos, F. Moneger, A. Lardon, C. Delichère, A. Mouras, I. Negrutiu,
Barton N., B. O. Bengtsson,
Baur V. L., C. F. Aquadro,
Begun D. J., C. F. Aquadro,
Braverman J. M., R. R. Hudson, N. L. Kaplan, C. H. Langley, W. Stephan,
Caballero A.,
Charlesworth B.,
———.
Charlesworth B., D. Charlesworth,
Charlesworth B., M. T. Morgan, D. Charlesworth,
Charlesworth B., M. Nordborg, D. Charlesworth,
Charlesworth D., B. Charlesworth, M. T. Morgan,
Correns C.,
Delichère C., J. Veuskens, M. Hernould, N. Baarbacar, A. Mouras, I. Negrutiu, F. Monéger,
Desfeux C., S. Maurice, J. P. Henry, B. Lejeune, P. H. Gouyon,
Felsenstein J.,
Filatov D. A.,
Filatov D. A., D. Charlesworth,
Filatov D. A., F. Monéger, I. Negrutiu, D. Charlesworth,
Fu Y.-X.,
Giles B. E., E. Lundqvist, J. Goudet,
Gordo I., B. Charlesworth,
Goulson D., K. Jerrim,
Guttman D. S., D. Charlesworth,
Haldane J. B. S.,
Hudson R. R.,
Hudson R. R., D. D. Boos, N. L. Kaplan,
Hudson R. R., M. Kreitman, M. Aguad,
Ingvarsson P. K., B. E. Giles,
Kimura M.,
Kumar S., K. Tamura, M. Nei,
Langley C. H.,
Laporte V., D. Charlesworth,
Lucchesi J. C.,
McAllister B., B. Charlesworth,
McCauley D. E.,
McVean G. T., L. D. Hurst,
———.
Matsunaga S., S. Kawano, H. Takano, H. Uchida, A. Sakai, T. Kuroiwa,
Moriyama E. N., J. R. Powell,
Nordborg M., B. Charlesworth, D. Charlesworth,
Orr H. A., Y. Kim,
Press W. H., S. A. Teukolsky, W. T. Vetterling, B. P. Flannery,
Rice W. R.,
Richards C. M., S. Church, D. E. McCauley,
Rozas J., R. Rozas,
Slatkin M.,
Slatkin M., T. Wiehe,
Smith N. G. C., L. D. Hurst,
Stephan W., C. H. Langley,
Stinson J. R., A. J. Eisenberg, R. P. Willing, M. E. Pe, D. D. Hanson, J. P. Mascarenhas,
Strobeck C.,
———.
Tajima F.,
Takahata N., Y. Satta,
Tanksley S. D., D. Zamir, C. M. Rick,
Taylor D. R.,
———.
Thomson J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, D. G. Higgins,
Vagera J., D. Paulikova, J. Dolezel,
Vielle-Calzada J.-P., R. Baskar, U. Grossniklaus,
Vyskot B., A. Araya, J. Veuskens, I. Negrutiu, A. Mouras,
Wang R. L., J. Wakeley, J. Hey,
Westergaard M.,
Yang Z.,
Ye D., P. Installe, D. Ciupercescu, J. Veuskens, Y. Wu, G. Salesses, M. Jacobs, I. Negrutiu,
Yi S., B. Charlesworth,
Yoder A., Z. Yang,

{kind=link}
{kind=link}
{kind=link}