





Abstract
Low levels of genetic diversity and divergence at nuclear loci have previously been observed for cycloidea and fil1-like genes within and between several Antirrhinum species, and divergence at these loci is also low between species in genera at different levels of relatedness in the former family Scrophulariaceae (Digitalis and Verbascum). The low divergence values are surprising, because (based on the sequences of chloroplast loci) the Scrophulariaceae are thought to be polyphyletic, with two anciently diverged clades, and the species we compared belonged to the two different clades. Here, we extend our studies of sequence divergence to more nuclear genes: fil2, far, globosa, and Adh. Detailed studies revealed that in Antirrhinum these genes belong to gene families. Low levels of divergence between Antirrhinum and Verbascum were observed for four of the loci studied, fil2-1, fil2-2, far-L, and globosa, similar to our previous observations. We discuss hypotheses to explain these low synonymous divergence values. For Adh, no cases of very similar sequences were found, but, rather, our sequences from the three different genera (Antirrhinum, Digitalis, and Verbascum) were all very diverged. Repeated gene duplication and loss of elements in the Adh gene family is likely in these lineages, making it impossible to determine orthology of the Adh genes.
Introduction
In Antirrhinum, studies of DNA sequence diversity and divergence of genes of the cycloidea and fil1 gene families have revealed surprisingly little variation within species, as well as very low divergence between several Antirrhinum species and Digitalis purpurea (Vieira, Vieira, and Charlesworth 1999 ; Vieira and Charlesworth 2001 ). Low divergence was also observed for two fil1 genes for which orthologous Verbascum nigrum sequences were obtained (Vieira and Charlesworth 2001 ). The low divergence of members of these two gene families is unexpected, because the species analyzed are not thought to be very closely related. Phylogenetic studies using chloroplast gene sequences (rbcL, ndhF, and rps2) suggest polyphyly of the Scrophulariaceae, as well as an ancient split between two clades (Olmstead and Reeves 1995 ; Wolfe and dePamphilis 1998 ; Soltis and Soltis 2000 ; Olmstead et al. 2001 ). Scroph I, which includes the genus Verbascum, is still called Scrophulariaceae (Olmstead et al. 2001 ). Scroph II includes Antirrhinum and Digitalis and species formerly assigned to several other families, including Plantaginaceae, and has been renamed Veronicaceae (Olmstead et al. 2001 ). The relationship between these two clades is clear, despite the slow evolution of chloroplast gene sequences (see Wolfe, Sharp, and Li 1989 ; Clegg, Cummings, and Durbin 1997 ; Gaut 1998 ). A combined analysis of the nuclear 18S rDNA gene and two chloroplast genes does not resolve the phylogenetic relationships of these species (Soltis, Soltis, and Chase 1999 ), but this result is due to the slow rate of evolution of 18S rDNA sequences (which evolve slower than the chloroplast genes; Soltis et al. 1997 ), rather than to close relatedness.
In contrast to the low divergence of the genes we have sequenced in Antirrhinum and Verbascum, allozyme divergence has been found between populations within and between Antirrhinum species. In Antirrhinum lopesianum, Antirrhinum mollissimum, and Antirrhinum microphyllum, allozyme diversity He for the species ranged from 0.19 to 0.52, and Nei's genetic identity values between populations averaged less than 0.96, or D = 0.04 (Nei 1987), for A. mollissimum and only 0.91 (D = 0.09) for A. microphyllum; between the first species and the others, identities were 0.5 and 0.46 based on 14 loci (Mateu-Andres 1999 ). These D values are unusually high compared with those of other plant species (Crawford 1989 ). Even in two narrow endemic species, within-population diversity was almost 0.1 (Mateu-Andres and Segarra-Moragues 2000 ). Similar allozyme differentiation has been found within and between various other species of the former Scrophulariaceae (Elisens and Crawford 1988 ; Ritland 1989 ; Schoen and Brown 1991 ; Elisens 1992 ; Elisens and Nelson 1993 ). Allozymes may not be an unbiased sample of loci, since they may be chosen on the basis of having variants within species. The fact that differences are found nevertheless leads one to expect some silent or intron site differences, even between Antirrhinum species (although some species can hybridize; Mather 1947 ; Harrison and Darby 1955 ; Rothmaler 1956 ), and certainly between Antirrhinum and Verbascum.
Our aim in this study was to test whether the low divergence we found for several loci was general or an unusual feature of the fil1 and cyc gene families. We therefore sequenced additional genes that have been identified in Antirrhinum and compared sequences from one Antirrhinum species with sequences from Verbascum and, for some loci, Digitalis. The Antirrhinum genes studied here include Adh genes and three genes involved in flower development, fil2, far, and globosa. fil2 is a flower-specific gene that encodes a protein of the extracellular matrix with an LRR signal peptide at the N-terminus (Steinmayr et al. 1994 ). A BlastX search reveals that fil2 has more than 62% amino acid identity to polygalacturonase genes from several plants (Actinida, AF263465; Prunus, Af020785, Z49063; Citrus, AB0162206, AB015356, AB016204; Vitis, AF305093). Farinelli (far) and globosa are floral homeotic genes that control petal and stamen development. far belongs to class C, and globosa to class B, of the MADS-box genes (Tröbner et al. 1992 ; Davies et al. 1999 ).
As we previously found for the fil1 and cyc genes, we show that in Antirrhinum all of these genes, including the alcohol dehydrogenase (Adh) genes, belong to gene families. Like the fil1 genes, four genes belonging to the fil2, far, and globosa gene families yielded identical sequences from Verbascum, but the results from the Adh genes were quite different. Divergence of the Adh sequences was high, even between Antirrhinum and Digitalis, and was at least roughly consistent with the rbcL and ndhF results, although gene duplications and losses make it impossible to establish orthology between Adh gene family members in the different taxa studied.
As explained above, the low divergence between Antirrhinum and Verbascum sequences appears to conflict with the hypothesis of an ancient split between two clades of Scrophulariaceae. We therefore also searched GenBank for pairs of gene sequences that were similar in Antirrhinum and other species of Scrophulariaceae in order to find further potential orthologs whose divergence between species of Scrophulariaceae could be compared. As will be seen, all of the few pairs of sequences that we found were nonorthologous; i.e., the “same” gene was not found in two different species. This suggests that gene families, and perhaps the birth and death of genes, may be common in these plant species, making it impossible at present to estimate sequence divergence between different species in this family.
Materials and Methods
Plant Material and PCR Amplification
Leaves of Antirrhinum majus subsp. cirrhigerum (Ficalho) Franco were collected in the field in the Aveiro in the north of Portugal (Vieira, Vieira, and Charlesworth 1999 ; Vieira and Charlesworth 2001 ). Verbascum nigrum, Verbascum thapsus, and Digitalis purpurea leaves were collected from wild individuals growing on the Edinburgh University campus. Genomic DNA was prepared from leaves of individual plants using the method of Ingram et al. (1997) .
PCR primers (table 1 ) were designed based on the GenBank sequences of the A. majus genes fil2, farinelli (far), and globosa (accession numbers X76995, AJ239057, and X68831, respectively). The regions of each of the genes analyzed are shown in figures 1–3 . fil2 is known to be a gene family in a number of species, including tomato, melon, maize, and willow (Hadfield and Bennett 1998 ; Futamura et al. 2000 ). Homologs of far and globosa have also been described as belonging to gene families in several angiosperm species (Yu et al. 1999 ; Theissen et al. 2000 ), and there is evidence for both ancient and recent duplications of globosa-like genes (Kramer, Dorit, and Irish 1998 ). However, among Antirrhinum sequences in GenBank, only far shows nucleotide similarity to any other known Antirrhinum gene (in this case, plena; Davies et al. 1999 ).
We did not characterize the fil2, far, and globosa gene families in detail in either Antirrhinum or Verbascum. However, BlastX searches in the Arabidopsis Information Resource (TAIR) database with A. majus query sequences indicated large families in the Arabidopsis thaliana genome. We took a value of more than 50 amino acid identities in a region more than 100 amino acids long as our criterion for identifying homologous genes. Antirrhinum majus globosa has 34 A. thaliana homologs, including pistillata, Apetala3, and many agamous-like genes. The results are similar for the A. majus far gene (more than 50 homologs, many of them overlapping those found for globosa). Finally, there were more than 100 fil2 homologs, including polygalacturonases, disease resistance genes, and kinase-like receptors. In A. thaliana, all three of these types of genes are known to be large gene families (Rounsley, Ditta, and Yanofsky 1995 ; Torki et al. 1999 ). It is unclear why no evidence for gene families was found in Antirrhinum when these genes were first described (Tröbner et al. 1992 ; Steinmayr et al. 1994 ).
Since these genes are clearly members of gene families in Antirrhinum also (see below), additional primers were designed based on the new sequences obtained (see Results). To amplify single members of these gene families, seminested PCR (Cubas, Vincent, and Coen 1999 ) was carried out on the product of the initial PCR reactions using further internal primers.
Antirrhinum Adh sequences were not available in GenBank. Primers (adA and adB; see table 1 ) were therefore designed for conserved regions of 20 bp identified based on the alignment of Adh1, Adh2, and Adh3 genes of other dicotyledon species (Solanum tuberosum, M25154, M25153, M25152; Leavenworthia stylosa, AF037564, AF037558, AF037560; Leavenworthia crassa, AF037563). These primers amplify a small region (392 bp) corresponding to part of exon 4 in A. thaliana (D63464).
Standard amplification conditions were 35 cycles of denaturation at 94°C for 30 s, primer annealing at 48°C for 30 s, and primer extension at 72°C for 2 min. Because we were working with very similar sequences, it was important to be extremely careful to avoid contamination. Standard negative controls (PCR cocktail without genomic DNA) were routinely included to ensure that similar sequences from species between which divergence was expected could not be attributed to contamination of the DNA samples or to the equipment used; these controls never yielded PCR products. Also, the PCR amplification was repeated with at least six A. majus individuals, and two of them were always sequenced. Since only one V. nigrum individual was used, DNA from several leaves was extracted independently and treated as different samples. The PCR of these samples always gave the same products.
All PCR amplification products were checked for homogeneity by digestion with several four-cutter restriction enzymes. If the number and/or size of the bands obtained after digestion was not compatible with that of the reference sequence (from which the primers were designed), the product was classified as heterogeneous. In such cases, we cloned the product and screened several colonies until several of each of the sequence types had been identified, and then we determined their DNA sequences. Cloning was performed using the TA cloning kit (Invitrogen). If more than one band was systematically obtained, we always cloned and sequenced all of them. Because differences can arise from nucleotide misincorporation during amplification, we determined the DNA sequences of plasmids from at least three different colonies (from the same PCR reaction) and obtained a consensus sequence. DNA sequencing was performed with an Applied Biosystems model 377 DNA sequencing system with the ABI PRISM BigDye cycle-sequencing kit (Perkin Elmer), using specific primers or the primers for the M13 forward and M13 reverse priming sites of the pCR 2.1 vector.
Sequence Analyses
The DNA sequences were deposited in GenBank (accession numbers AF307068–AF307071 for fil2-1, AF307072–AF307075 for fil2-2, AF307063–AF307064 for farL, AF307065–AF307067 for farS, AF307076–AF307078 for globosa1, and AF307054–307062 for Adh). The nucleotide sequences to be compared were aligned using ClustalX, version 1.64b (Thompson et al. 1997 ), and minor manual adjustments were made using SeqPup, version 0.6f. Intron/exon boundaries within the genes were deduced by comparison with the Antirrhinum GenBank cDNA sequences corresponding to the genes from which the primers were designed. The numbers of synonymous and nonsynonymous differences between pairs of sequences were calculated using DnaSP software, version 3.0 (Rozas and Rozas 1999 ). Divergence estimates were corrected for multiple hits using Jukes-Cantor correction (Jukes and Cantor 1969 ), and neighbor-joining trees were generated for the Adh genes with MEGA, version 1.01 (Kumar, Tamura, and Nei 1994 ).
To find homologous genes for comparison between Antirrhinum and other species in the Scrophulariaceae, we used BlastX searches, which use only protein-coding regions (Altschul et al. 1997 ), using as queries GenBank sequences from species of Scrophulariaceae other than Antirrhinum. Of the 62 non-Antirrhinum genes available, only six had amino acid homology with Antirrhinum sequences using the criterion described above. These, with their GenBank accession numbers were as follows: ACS1, AF083814 (homology with a gene from Striga hermonthica, AF090351); PHYA, U08142 (homology with a gene from Digitalis lanata, AJ002525); GADPH, X59517 (homology with a gene from Craterostigma plantagineum, X78307); MADS-box transcription factor, Y10750 (homology with a gene from Paulownia kawakamii, AF060880); Chs, X03710 (homology with a gene from Digitalis lanata, AJ002526); TFNS5, AB028151 (homology with a gene from Torenia hybrida, AB028152). One sequence, SUT1 from Asarina barclaiana (AF191024), was similar to a non-Antirrhinum gene (Alonsoa meridionalis, AF191025). For comparative purposes, we also included a sequence of each gene from a species in the related families Geraniaceae (Pelargonium hortorum, U17231), Solanaceae (Sophora affinis, U78835; Petunia, X60346; Solanum tuberosum, AF008651; Nicotiana tabacum, X82276), and Fabaceae (Perilla frutescens, AB002582; Glycine max, D83968).
Results
Evidence that fil2, far, globosa, and Adh Are Members of Gene Families
fil2
Primers Fil2A and Fil2B (table 1 ) amplified a PCR product of the predicted size (845 bp) from two A. majus plants. Digesting this band with several restriction enzymes revealed two DNA sequences. We therefore cloned this PCR product and sequenced several clones corresponding to both types of sequence, from both individuals. Each individual had both sequence types, which we denote by fil2-1 and fil2-2, and each of these was identical in the two individuals. Between the fil2-1 and fil2-2 sequences, there were 17 nucleotide differences (9 nonsynonymous and 8 synonymous) and one 4-bp indel in the putative intron (fig. 1 ). Twelve A. majus plants were tested for the presence of the fil2-1 and fil2-2 sequences, and both sequences amplified from all individuals, strongly suggesting that they represent two different genes. Blast searches with these two types of sequences revealed high nucleotide sequence similarity to the Antirrhinum fil2 gene, but both fil2-1 and fil2-2 had several differences from the GenBank sequence. For fil2-1, there were 12 nonsynonymous and 10 synonymous differences in the coding sequences compared, along with five nucleotide differences plus eight indels in the putative intron; for fil2-2, there were five nonsynonymous and four synonymous differences in the coding sequence, along with five nucleotide differences plus seven indels in the intron (fig. 1 ). fil2-2 was similar to fil2-1 at the 5′ end, while at the 3′ end it was similar to the A. majus GenBank fil2 sequence (fig. 1 ). This is not the result of PCR recombination, since identical results were obtained in four independent PCR reactions using three different sets of primers (Fil2A and Fil2B for A. majus, and Fil2A with fil2-1R or fil2-2R for V. nigrum and V. thapsus).
Based on two fixed differences between fil2-1 and fil2-2 at positions 493 and 495 (fig. 1 ), specific primers were designed to amplify each of these genes separately (table 1 ). Using these primers together with Fil2A, PCR amplification products were also obtained from genomic DNA of V. nigrum and V. thapsus, and their sequences were determined. Both sequences were present in Verbascum, supporting the conclusion that at least two separate fil2 genes exist. These primers were not expected to amplify the GenBank sequence, since the relevant sequences differed from the GenBank sequence. Thus, further sequences might also be present in these species. The V. nigrum and V. thapsus sequences were identical for both fil2-1 and fil2-2. In a comparison of the A. majus and Verbascum sequences, only one difference was found (in the intron) out of 477 bp that could be compared for the fil2-1 gene (table 2 and fig. 1 ; Ks = 0.0032 based on 318 intron and synonymous sites), and none were found in the fil2-2 gene.
far
Two bands of different sizes, 905 bp (L) and 731 bp (S), were amplified from A. majus using primers FarF and FarR (table 1 ). Both bands were obtained in all six individuals tested. For two individuals, both L and S were cloned and sequenced in order to establish whether these bands were both specific amplification products. The corresponding sequences from these two individuals were identical. Both the L and the S sequences were similar to the GenBank far sequence (96% and 88% nucleotide identity, respectively). The GenBank A. majus far sequence was most similar to the L sequence, but there were several differences (five synonymous nucleotide differences, 18 nucleotide differences in the putative introns, and five intronic indels), and it is possible that they were not allelic (fig. 2 ).
Between the putative coding regions of the L and S sequences, there were 15 silent-site and seven replacement-site differences (fig. 2 ). There were also length differences in the putative introns. Intron 1 of the L sequences was 171 bp, versus 121 bp in the S sequences; the respective intron 3 lengths were 80 versus 97 bp, and those of intron 4 were 254 versus 106 bp. Because of these length differences in the putative introns, the L and S sequences could not be reliably aligned.
To compare orthologs between V. nigrum and A. majus, a new forward primer based on the S sequence was designed in order to amplify this sequence specifically (again, this will not amplify the GenBank sequence). No attempt was made to amplify or further study the V. nigrum far-L. The S-band-specific forward primer spans the end of exon 2 and the beginning of intron 3 (table 1 and fig. 1 ) and yields a fragment of 662 bp in both species. The V. nigrum and A. majus S-band sequences were identical in this region (table 2 ). This finding, together with the number of differences between the two A. majus band size types, strongly suggests that the L and S types of sequences represent two different genes (which we denote by far-S and far-L).
globosa
Primers GloF1 and GloR (table 1 ) amplified a PCR product in A. majus. From all six individuals studied from this species, the size of this amplification product was 994 bp, rather than the expected 864 bp based on the Antirrhinum GenBank sequence. We cloned and sequenced the product from two individuals; both were identical. Our sequence was similar in both the intron and the coding region with the GenBank globosa sequence (>95% nucleotide identity for both regions). The size difference was mainly due to a duplication of a 129-bp region of the intron of the GenBank sequence (fig. 3 ). Apart from this, out of the 826 sites compared, there were 17 nucleotide differences between our sequence and the one in GenBank (two synonymous differences in putative coding regions and 15 in the intron; fig. 3 ), plus five indels in the intron.
To test for the presence of a similar sequence in V. nigrum, we designed a new primer (Glogap; see table 1 and fig. 1 ) specific for the region of the gene that is duplicated relative to the GenBank sequence (see above). Since this region was present twice in the target sequence, two bands with different sizes (512 bp and 641 bp) were expected. Both were obtained and sequenced in both V. nigrum and A. majus using primers Glogap and GloR. As expected, there were no differences between the 512-bp band and the corresponding region of the 622-bp band. The 622-bp sequences were identical in V. nigrum and A. majus (table 2 ; there were no differences in the exon regions, but too few sites were compared to permit divergence estimates). Once again, gene duplication was indicated for the globosa gene. If there were only a single copy of this gene, it is very unlikely that a sequence from a distantly related species would be identical to our A. majus sequence but different from that in GenBank.
Adh
Primers adA and adB (table 1 ) were designed for sequences in the coding region that are conserved in the Adh-1, Adh-2, and Adh-3 genes of two distantly related genera (see Materials and Methods). PCR products of the expected size (392 bp) were obtained from species of all three genera studied, A. majus, D. purpurea, and V. thapsus. As the primers are based on a conserved region of several paralogous Adh genes, the PCR product is expected to be heterogeneous, and it should include all Adh genes of this family in our species (generally two or three for diploid angiosperms; Small and Wendel 2000 ). On average, 30 clones from each species (from several different PCR reactions) were therefore digested with various restriction enzymes (AciI, AluI, RsaI, and DdeI) to determine the different types of sequences amplified. There were three types of clones in A. majus, one in D. purpurea, and two in V. nigrum. For all species, at least four clones of each type were sequenced.
Five different sequences were obtained from a single A. majus individual. A Blast search revealed that all sequences shared the highest amino acid similarity (>62% amino acid identity) with an Adh-like sequence from S. tuberosum (accession number X92179). Since A. majus is a diploid species, the presence of five different sequences implies the presence of at least three genes. We denote them by Adhant1, Adhant2, and Adhant3; these numbers do not imply orthology with other Adh plant genes with the same numbers. The mean pairwise difference per synonymous site between these genes (Ks) ranges from 0.192 (between Adhant1 and Adhant2) to 1.41 (Adhant2 vs. Adhant3). Despite the small portion of Adh coding region sequenced, even the lowest divergence value (0.192, based on 83 synonymous sites) differed significantly (P < 0.001 by a 2 × 2 χ2 test) from even the shortest coding sequence region from the other genes (fil2-1, 0/53 sites; see table 2 ). The two Adhant1 sequences from the single individual studied were similar but not identical (Ks = 0.0247) and could be allelic. The same applies to the two Adhant2 sequences (Ks = 0.0244). These differences were much larger than the average synonymous site diversity for the cyc and fil1 genes of Antirrhinum species (Vieira, Vieira, and Charlesworth 1999 ; Vieira and Charlesworth 2001 ).
In V. thapsus, the restriction enzyme digestions revealed only two types of clones. Sequences were obtained for each type from a single individual. The Ks value for these sequences was 0.453. They thus appear to represent paralogs from an ancient duplication. Ten D. purpurea clones were sequenced. Two different sequences (digitalis1 and digitalis2; fig. 4 ) were found, differing at four nucleotide positions, two of them nonsynonymous; the Ks estimate based on just these two sequences was 0.0243, and they could be allelic. The differences in copy number made it uncertain which (if any) of these genes were orthologous.
Divergence between the sequences from the different species was considerable. Synonymous-site divergence between A. majus and Verbascum (with Jukes-Cantor correction) appeared to be saturated, with values of 1 or greater for all comparisons. Even Ka values ranged from >6% to 19.5%. Figure 4 presents an unrooted gene tree showing the relationships among our Adh gene sequences from species of Scrophulariaceae, plus sequences from Solanaceae taken from GenBank. Given the nature of our adA and adB primers, together with the fact that they evidently amplify very divergent sequences, it is unlikely that the Antirrhinum genome contains further Adh genes more closely related to the Verbascum sequences than the ones amplified. Furthermore, a primer (v1) designed based on the verbascum1 sequence, yielded no amplification product from A. majus with the reverse primer adB over a range of different annealing temperatures down to 45°C, whereas a PCR product of the expected size was always obtained in Verbascum. Thus, for this locus, A. majus has no detectable gene with a sequence similar to verbascum1, in contrast to all the other nuclear genes studied. Adh copy numbers have thus probably changed between these species.
Discussion
The Antirrhinum fil2, far, globosa, and Adh Genes Are Members of Gene Families
All four kinds of genes studied here (fil2, far, globosa, and Adh genes) are clearly members of gene families in Antirrhinum. Our fil2, far, and globosa genes are probably not allelic with the A. majus sequences in GenBank (see Results). It is surprising that our primers did not amplify sequences similar to those in GenBank, since they were designed based on these sequences. A possible explanation is that the total copy numbers for these gene families, which are unknown in both Antirrhinum and Verbascum, may be very large. There is evidence that this is the case for A. thaliana (see Materials and Methods). In addition, studying genes that belong to gene families, even small ones, can produce artifactual sequences in several ways, including PCR recombination or combination of partial sequence data from different clones (based on the assumption that a gene is single-copy). It is helpful to check that the entire sequence can be amplified from genomic DNA, but this is not always done. Because of their high sensitivity, PCR approaches are likely to reveal gene families even when Southern blotting suggests a single copy. Heterogeneity of the PCR product may not be immediately apparent (unless the region amplified includes length differences, a band of the expected size will be found) but can readily be detected by studying multiple clones, as described here.
Copy Number and Sequence Differences in the Adh Gene Family
The fact that the genes we have studied are members of gene families complicates molecular evolutionary analysis, as it is essential to determine orthology before comparing sequences. This is not a problem for the fil2, far, and globosa genes, as we found identical sequences in the different species. However, it is clear from the gene tree that duplications and losses have obscured orthology among the Adh genes of Antirrhinum, Verbascum, and Digitalis (fig. 4 ). This is consistent with our finding of different copy numbers in the three species studied.
The Adh sequences from the different species are highly diverged, suggesting that some of the gene duplication events are ancient. Sequences from the Solanaceae and Scrophulariaceae form separate clusters, so several gene duplications must have occurred after these two families split (fig. 4 ). A minimum of three duplications are required in the lineages of Scrophulariaceae analyzed here, one in each lineage (fig. 4 ). In the Solanaceae, at least five duplications must have occurred to explain the data shown in the tree (fig. 4 ). Phylogenetic analyses have revealed repeated gains and losses of Adh genes in other genera, even among closely related species (Gaut et al. 1996, 1999 ; Morton, Gaut, and Clegg 1996 ; Clegg, Cummings, and Durbin 1997 ; Small and Wendel 2000 ).
The uncertain orthology makes it unclear whether the divergence between the Adh genes of Antirrhinum, Verbascum, and Digitalis is truly more extensive than that for the other genes studied, as pairwise comparisons make it appear. For example, the Adhant3 sequence could be orthologous to those from Verbascum and Digitalis, with considerable divergence (see fig. 4 ). The Adh gene Ks values would then be consistent with the high synonymous-site divergence in chloroplast gene sequences between Antirrhinum and Verbascum (about 0.1 based on GenBank sequences of ndhF and trnL), assuming a fivefold faster nuclear gene substitution rate (Gaut 1998 ). A formal alternative is that none of our Adh sequences are orthologous. This alternative, however, is less parsimonious when examined closely, as follows.
Antirrhinum has two gene types (the Adhant3 type just mentioned and the two sequences Adhant1 and Adhant2, which are quite similar to one another). Since there are clearly three different types of sequences in the taxa studied, paralogy would, by definition, require two duplications before the taxa diverged, with each species having retained only one of the genes. The common ancestor of Antirrhinum and Digitalis must have had all three types of genes, since Antirrhinum has two of them and Digitalis has the third, so the losses must be recent (after the split from Antirrhinum). Digitalis must have lost all but the third type, and Antirrhinum and Verbascum must have lost the third type, during this period. The only alternative is that although the very divergent (on this hypothesis, paralogous) Digitalis sequence amplifies with our primers, more similar orthologs in the other species do not. This seems unlikely but, if true, implies at least some coding sequence difference, unlike our results for the other genes. We must also account for the absence of an ortholog of Adhant1 and Adhant2 in Verbascum (whose two sequences, presumably due to more recent duplication in this lineage, differ greatly from these). Therefore, either there must be a hypothetical ortholog that fails to amplify (i.e., has diverged), or this hypothesis requires yet another duplication in the Verbascum lineage (after its split from Antirrhinum/Digitalis; otherwise further gene losses are required), with additional loss of the Adhant3 ortholog in Verbascum. Moreover, this implies that the Verbascum sequence diverged from Adhant3 later than the divergence times for the other (orthologous) genes studied, so the much greater sequence divergence remains puzzling. Compared with the hypothesis that Adhant3 is orthologous to the Verbascum and Digitalis sequences, we thus require at least two more duplications, and three more gene losses (or failure to amplify). It is therefore arguable that some sequence divergence almost certainly happened in the Adh sequences and that these genes therefore behave differently from the other genes studied.
Turnover in Plant Gene Families
Our results add to other data showing that gene families are common in plants (e.g., Clegg, Cummings, and Durbin 1997 ; Kramer, Dorit, and Irish 1998 ; Meyers et al. 1999 ; Durbin, McCaig, and Clegg 2000 ; Oberholzer, Durbin, and Clegg 2000 ; Pan et al. 2000 ; Theissen et al. 2000 ; Zhang, Pond, and Gaut 2001 ). The frequency of duplications is difficult to estimate. Few studies describe the total copy numbers for gene families in a group of species. Based on the average number of independent lineages inferred within Poaceae, Asteraceae, Fabaceae, and Solanaceae, Clegg, Cummings, and Durbin (1997) suggest a faster rate of duplication for the Chs and rbcS gene families than for the Adh gene family. These authors also suggest that new gene copies arise infrequently within families. However, this may be the result of poor species representation in each family. For instance, for Adh, only one genus was included for each of the families Malvaceae, Vitaceae, Asteraceae, and Pinaceae. Our data suggest duplications of Adh within the Scroph II clade similar to those inferred in the genus Gossypium (Small and Wendel 2000 ) and in the Brassicaceae (Koch, Haubold, and Mitchell-Olds 2000 ).
Orthology Among Scrophulariaceae GenBank Sequences
To obtain additional evidence on sequence divergence between species plants related to our study species, we also searched GenBank for pairs of homologous nuclear genes in taxa from the former Scrophulariaceae. For any given gene, the divergence values for synonymous sites, Ks (and perhaps nonsynonymous divergence, Ka), for orthologous loci should, of course, reflect the relationships between the species being compared, and more distantly related species should have larger Ks and Ka values than more closely related species. Only seven gene pairs were found (see Materials and Methods). The species from which the comparison sequences are available are not in the same clade as Antirrhinum and Digitalis (Veronicaceae, according to Olmstead et al. 2001 ). No phylogenetic data are available for Asarina or Craterostigma. Striga and Paulownia belong to lineages separate from both Scroph I and Scroph II (they are assigned, respectively, to Orobanchaceae and Paulowniae by Olmstead et al. 2001 ). Four of the genes compared between the different species (ACS1, PHYA, GADPH, and the MADS-box transcription factor; fig. 5 ) clearly cannot be orthologs. Ks values for these genes between different species of the former Scrophulariaceae are as high as, or higher than, those between these species and members of other plant families. For the other three genes (Chs, TFNS5, and SUT1), substitution at silent sites is saturated between the species compared, and Ks values are as high as those for two of the first four genes. It is therefore very unlikely that any of these pairs are orthologous genes. Again, these results indicate that gene families are important in the genomes of these species; otherwise, orthologs would be found.
Low Divergence Values Between the Distantly Related A. majus and Verbascum
Table 2 summarizes the new results presented here, together with our previous work on the same taxa, but excluding those from the alcohol dehydrogenases, for which we were unable to determine orthology. In addition, we found identical sequences for five fil1-like genes in A. majus subsp. cirrhigerum and D. purpurea, as well as for one of the cyc-like genes in these two species (cyc4; one sequence from A. majus subsp. cirrhigerum and two from Misopates orontium were identical to the Digitalis cyc4 sequence; the cyc-like genes in which sequence differences were found may not be single loci; see Vieira, Vieira, and Charlesworth 1999 ). The age of the split between Antirrhinum and Verbascum is unknown, since fossils are unavailable for Scrophulariaceae, but the high synonymous-site divergence in three chloroplast genes (see above and Olmstead et al. 2001 ) rules out recent origin. The expected synonymous-site divergence between Antirrhinum and Verbascum for nuclear genes, predicted from the chloroplast data (see above), is somewhat greater than the mean synonymous divergence between Drosophila melanogaster and Drosophila pseudoobscura (Zeng et al. 1998 ). In the data from these Drosophila species, the range (for 24 loci) is 0.28–1.7, whereas our data show extraordinarily low divergence for several genes (table 2 ). If the Adh gene divergence is not due to paralogy, our results also indicate high heterogeneity in synonymous divergence.
Recent gene duplication cannot explain our findings of highly similar sequences in such distant relatives as Antirrhinum and Verbascum. Concerted evolution due to gene conversion can retard divergence among paralogous sequences within a genome. This could explain the similarities we find among some of the genes within species (Vieira, Vieira, and Charlesworth 1999 ) but should not affect divergence of orthologous genes (Ohta 1981, 1984 ; Nagylaki and Petes 1982 ; Arnheim 1983 ).
In the case of the intronless cyc-like genes, only the coding region was analyzed, and it was therefore possible that the low diversity and divergence observed was due to an unusually high level of selective constraint on the coding sequences (Vieira, Vieira, and Charlesworth 1999 ). An unusual level of purifying selection cannot, however, readily explain the low diversity and divergence for the genes studied here, since similar results were obtained for introns, nor that for the fil1-like genes, for which our sequences include introns and the 3′ noncoding regions (Vieira and Charlesworth 2001 ). In Drosophila and other species, synonymous and nonsynonymous divergence rates are positively correlated (reviewed in Dunn, Bielawski, and Yang 2001 ), so our results could possibly be explained by strong constraints acting on the amino acid sequences of the genes studied, along with correlated low sequence divergence rates at synonymous sites. However, as already mentioned, the heterogeneity of divergence values between our plant taxa is much more extreme than that observed between Drosophila taxa with synonymous site-divergence similar to that expected based on chloroplast gene divergence between our study taxa.
Codon usage bias is also unlikely to explain the low divergence values we found. Most of the Antirrhinum genes we have studied, including those described here, have low codon usage bias; ENC values (Wright 1990 ) are above 48 (Vieira and Charlesworth 2001 ). There is therefore no evidence suggesting severe constraints of any kind, although codon usage analyses will not detect every type of constraint. For the fil1 genes, we also found no evidence for constraints imposed by the mRNA structure (Vieira and Charlesworth 2001 ).
Another possible explanation for low divergence of nuclear gene sequences is hybridization and introgression in an ancestor of Antirrhinum and Digitalis, resulting in some nuclear genes of these species being similar to Verbascum, even though different chloroplast gene sequences are retained. The New World cotton Gossypium gossypoides has A genome (Old World) nuclear ribosomal DNA sequences, although chloroplast restriction sites and other evidence clearly group it with other New World species, suggesting such an event (Wendel, Schnabel, and Seelanan 1995 ), and in Heuchera and Tellima, populations have been found with similar allozymes but differing chloroplast genomes (Soltis and Kuzoff 1996 ). However, the chloroplast gene differences between our species are large compared with those between those involved in these examples (estimated to be about 1% for New and Old World cottons; Wendel 1989 ) or other documented plant hybrids (Sang, Crawford, and Stuessy 1995 ; Rieseberg and Carney 1998 ), making exchange less plausible. Moreover, the low nuclear sequence divergence between Antirrhinum and Digitalis remains puzzling if the putative hybridization event is quite old. If this is the explanation for the incongruity, moreover, the relationships of these taxa may need to be reevaluated.
Another possible explanation for the low divergence observed for the fil1, fil2, far, and globosa genes between Antirrhinum and Verbascum is a low rate of nucleotide substitution. Substitution rate estimates for the cyc-like genes (Vieira, Vieira, and Charlesworth 1999 ) and fil1A genes (Vieira and Charlesworth 2001 ) are lower than most other estimates for nuclear genes in monocotyledons (Wolfe, Sharp, and Li 1989 ; Gaut et al. 1996 ) and dicotyledons (Small, Ryburn, and Wendel 1999 ; Small and Wendel 2000 ). Moreover, our estimates assumed implausibly recent origins. On the same basis, at least four of the genes studied here must have equally extreme low substitution rates.
At present, it remains unclear why such low divergence is found between Antirrhinum and Verbascum genes. Many of the genes we have studied are involved in flower development, and it is possible that they evolve much slower than other genes (Purugganan 1998 ). Alternatively, it may have to do with their belonging to large gene families. Our data on Adh differ from the results for the other loci. Either there is more gene turnover, so that (unlike the other loci) orthologs are rarely found, or the Adh sequences evolve faster than those of the other loci. More comparative studies using nondevelopmental genes, including allozyme loci, are needed to clarify the situation.
Brandon Gaut, Reviewing Editor
Present address: Instituto de Biologia Molecular e Celular, Universidade do Porto, Porto, Portugal.
Keywords: AdhAntirrhinumfarfil2globosaVerbascum
Address for correspondence and reprints: Deborah Charlesworth, Institute of Cell Animal and Population Biology, University of Edinburgh, Ashworth Laboratories, King's Buildings, West Mains Road, Edinburgh EH9 3JT, United Kingdom. [email protected] .
Table 1 Primers Used in this Study
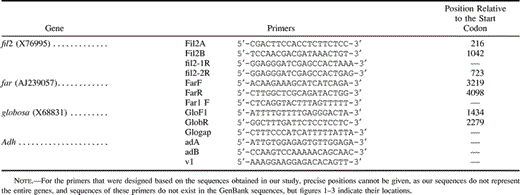
Table 1 Primers Used in this Study
Table 2 Divergence Between Antirrhinum majus ssp. cirrhigerum and Verbascum nigrum for Several Orthologous Genes
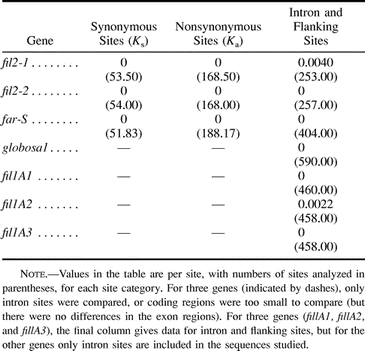
Table 2 Divergence Between Antirrhinum majus ssp. cirrhigerum and Verbascum nigrum for Several Orthologous Genes

Fig. 1.—A, Schematic diagram of the fil2 GenBank sequence (Antirrhinum majus; modified from Steinmayr et al. 1994 ). Boxes represent exons, and the thin line represents the single intron. The 845-bp region analyzed is indicated by a line, and the primer names are indicated. B, Variable sites in the fil2-like sequences. Absence of symbols means that sequence was not obtained for the region in question. Dots indicate that a nucleotide is the same as in the first sequence, and dashes indicate deletions. “i” indicates an insertion (with the sizes of insertions in brackets); at position 228, the inserted sequence is GC; at position 236, it is GG; at position 296, it is ATATTAA; and at position 366, it is TAAG. Sequence names are population codes for Antirrhinum majus subsp. cirrhigerum followed by sample numbers (Vieira, Vieira, and Charlesworth 1999 ; Vieira and Charlesworth 2001 )
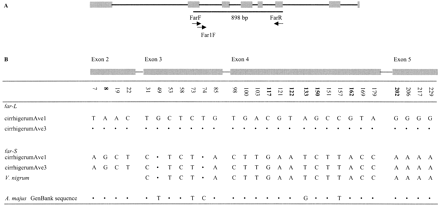
Fig. 2.—A, Schematic diagram of the far GenBank sequence (Antirrhinum majus; modified from Davies et al. 1999 ). Symbols are as for figure 1 . B, Variable sites in the putative coding region within the 898-bp region of far-like sequences analyzed
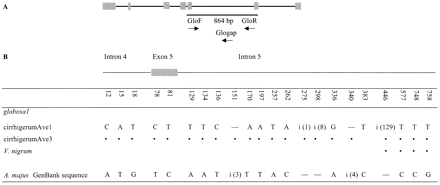
Fig. 3.—A, Schematic diagram of the globosa GenBank sequence (Antirrhinum majus; Tröbner et al. 1992 ). Symbols are as for figure 1 . B, Variable sites of the 864-bp region of globosa-like sequences analyzed. “i” indicates insertions; at position 151, the inserted sequence is ATA; at position 275, it is A; at position 298, it is CTAACACA; at position 340, it is TTAA; and at position 446, there is a 129-bp duplication identical to positions 313–445 except for a 4-bp deletion
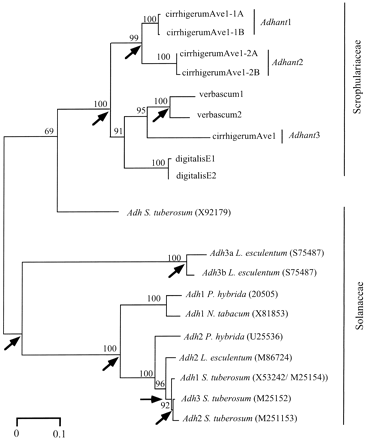
Fig. 4.—Unrooted neighbor-joining tree, using Kimura two-parameter distances, showing the relationships among the Adh genes of Scrophulariaceae and Solanaceae. The Antirrhinum majus sequences are labeled “cirrhigerum,” to indicate the subspecies, and “Ave,” to indicate the Aveiro population of origin. Bootstrap replicates supporting the branches are shown for values greater than 68%. Arrows indicate duplication events in the Scrophulariaceae and the Solanaceae, respectively
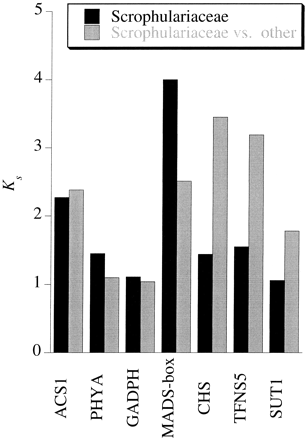
Fig. 5.—Comparison between GenBank sequences of nuclear genes from Antirrhinum, other species of Scrophulariaceae, and more distantly related plant species
We thank Jorge Vieira for helpful comments on the manuscript. C.P.V. was supported by the Commission of the European Communities (Grant ERBFMBICT 972455), and D.C. was supported by an NERC Senior Research Fellowship.
References
Altschul S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, D. J. Lipman,
Arnheim N.,
Clegg M. T., M. P. Cummings, M. L. Durbin,
Crawford D. J.,
Cubas P., C. Vincent, E. Coen,
Davies B., P. Motte, E. Keck, H. Saedler, H. Sommer, Z. Schwarz-Sommer,
Dunn K. A., J. P. Bielawski, Z. Yang,
Durbin M. L., B. McCaig, M. T. Clegg,
Elisens W. J.,
Elisens W. J., D. J. Crawford,
Elisens W. J., A. D. Nelson,
Futamura N., H. Mori, H. Kouchi, K. Shinohara,
Gaut B. S.,
Gaut B. S., B. R. Morton, B. C. McCaig, M. T. Clegg,
Gaut B. S., A. S. Peek, B. R. Morton, M. T. Clegg,
Hadfield K. A., A. B. Bennett,
Ingram G. C., S. Doyle, R. Carpenter, E. A. Schultz, R. Simon, E. S. Coen,
Jukes T. H., C. R. Cantor,
Koch M., B. Haubold, T. Mitchell-Olds,
Kramer E. M., R. L. Dorit, V. F. Irish,
Kumar S., K. Tamura, M. Nei,
Mateu-Andres I.,
Mateu-Andres I., J. G. Segarra-Moragues,
Mather K.,
Meyers B. C., A. W. Dickerman, R. W. Michelmore, S. Sivaramakrishnan, B. W. Sobral, N. D. Young,
Morton B. R., B. S. Gaut, M. T. Clegg,
Nagylaki T., T. D. Petes,
Oberholzer V., M. L. Durbin, M. T. Clegg,
———.
Olmstead R. G., C. W. DePamphilis, A. D. Wolfe, N. D. Young, W. J. Elisens, P. A. Reeves,
Olmstead R. G., P. Reeves,
Pan Q., Y.-S. Liu, O. Budai-Hadriana, M. Sela, L. Carmel-Gorenc, D. Zamirc, R. Fluhra,
Ritland K.,
Rounsley S. D., G. S. Ditta, M. F. Yanofsky,
Rozas J., R. Rozas,
Sang T., D. J. Crawford, T. F. Stuessy,
Schoen D. J., A. H. D. Brown,
Small R. L., J. A. Ryburn, J. F. Wendel,
Small R. L., J. F. Wendel,
Soltis D. E., R. K. Kuzoff,
Soltis P. S., D. E. Soltis,
Soltis P. S., D. E. Soltis, M. W. Chase,
Soltis D. E., P. S. Soltis, D. L. Nickrent, et al. (16 co-authors)
Steinmayr M., P. Motte, H. Sommer, H. Saedler, Z. Schwarz-Sommer,
Theissen G., A. Becker, A. Di Rosa, A. Kanno, J. T. Kim, T. Munster, K. U. Winter, H. Saedler,
Thompson J., T. J. Gibson, F. Plewniak, F. Jeanmougin, D. G. Higgins,
Torki M., P. Mandaron, F. Thomas, F. Quigley, R. Mache, D. Falconet,
Tröbner W., L. Ramirez, P. Motte, I. Hue, P. Huijser, W. E. Lonnig, H. Saedler, H. Sommer, Z. Schwarz-Sommer,
Vieira C. P., D. Charlesworth,
Vieira C. P., J. Vieira, D. Charlesworth,
Wendel J. F.,
Wendel J. F., A. Schnabel, T. Seelanan,
Wolfe A. D., C. W. dePamphilis,
Wolfe K. H., P. M. Sharp, W.-H. Li,
Yu D., M. Kotilainen, E. Pollanen, M. Mehto, P. Elomaa, Y. Helariutta, V. A. Albert, T. H. Teeri,
Zeng L. W., B. Chen, J. Comeron, M. Kreitman,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}