





Abstract
Myocarditis is an inflammatory heart disease that mostly affects young people. Myocarditis involves a complex immune network; however, its detailed pathogenesis is currently unclear. The diversity and plasticity of immune cells, either in the peripheral blood or in the heart, have been partially revealed in a number of previous studies involving patients and several kinds of animal models with myocarditis. It is the complexity of immune cells, rather than one cell type that is the culprit. Thus, recognizing the individual intricacies within immune cells in the context of myocarditis pathogenesis and finding the key intersection of the immune network may help in the diagnosis and treatment of this condition. With the vast amount of cell data gained on myocarditis and the recent application of single-cell sequencing, we summarize the multiple functions of currently recognized key immune cells in the pathogenesis of myocarditis to provide an immune background for subsequent investigations.
1. Introduction
Myocarditis, an inflammatory heart disease (i.e. mainly observed in young people), can cause sudden death, secondary dilated cardiomyopathy (DCM), and heart failure.1 Although myocarditis has a clear definition, its diagnosis is far from simple. Clinical symptoms, laboratory tests, electrocardiography, echocardiography, cardiac magnetic resonance imaging (cMRI), and endomyocardial biopsy (EMB) are all used to diagnose myocarditis.2,3 Several previous studies4–7 have revealed susceptible strains of mice and genetically susceptible individuals8–11 that progress to DCM after myocarditis.
Even though the detailed pathophysiology of myocarditis is still unknown, immune cells have attracted much interest as playing key roles. The pathogenesis of myocarditis is thought to primarily involve neutrophils, eosinophils, mast cells, innate lymphoid cells (ILCs), monocytes, macrophages, dendritic cells (DCs), T lymphocytes, and B lymphocytes.12–15 In the steady state, the heart contains a population of intrinsic immune cells. During illness, these cells act as the body's first line of defense,12 and along with subsequent activation of adaptive immunity, they form a complex immune network that plays a dual role comprising both protection and destruction. In addition to immune system dysregulation, myocarditis may also involve an imbalance within immune cells.15
Single-cell sequencing techniques16 have been used to gain deeper insights at the single-cell level. Several studies17–20 based on single-cell RNA sequencing (scRNA-seq) have suggested that scRNA-seq has been regarded as the more suitable tool to find out some molecules specifically expressed within certain cell classes because these molecules may be easily overlooked because they are likely to be expressed sightly on bulk RNA-seq.21
In this review, we comprehensively describe the diversity and plasticity in the function of major immune cells and their associated cellular contents in the context of myocarditis, laying the foundation for subsequent in-depth studies based on the vast amount of immunological data.
2. Myocarditis
2.1 Definition, prevalence, diagnosis, and etiology
Myocarditis is an inflammatory disease of the myocardium that appears without an ischemic event. Myocarditis is a frequent contributor to sudden death, secondary DCM, and heart failure in the young population.1
In 2021, myocarditis was estimated to be the cause of 33,600 deaths (95% uncertainty interval [UI] 27,100–38,000), and the number of disability-adjusted life-years was 962,000 (95% UI 810,000–1,090,000).22 Males make up a larger proportion of adult myocarditis patients than females, and mortality follows a similar trend.22,23
With the increased use of immune checkpoint inhibitors (ICIs), which target programmed cell death 1 (PD-1), CTLA-4 (cytotoxic T lymphocyte-associated protein 4), and programmed cell death ligand 1 (PD-L1) in oncology therapy, ICI-associated myocarditis (ICI-MC) is becoming more common. The incidence of ICI-MC ranges from 0.04% to 1.14%, but the associated mortality rate is up to 25% to 50%, which is much greater than that of other immune-related adverse events.24
With the recent appearance of COVID-19, myocarditis after COVID-19 and messenger RNA (mRNA) COVID-19 vaccination has attracted more attention. Research based on the Premier Healthcare Database Special COVID-19 Release found that patients with COVID-19 had an approximately 16 times greater risk of myocarditis than patients without.25 According to many previous reports, myocarditis is regarded as a rare complication of COVID-19 vaccination.26–30 Based on a self-controlled case series study of individuals 16 years of age or older who received COVID-19 vaccination in England between December 1, 2020, and August 24, 2021, researchers31 found that increased risks of myocarditis within 1 to 28 d postvaccination were related to the first dose of adenovirus ChAdOx1 vaccines (incidence rate ratio [IRR] 1.29, 95% confidence interval [CI] 1.05–1.58) and mRNA-based BNT162b2 vaccines (IRR 1.31, 95% CI 1.03–1.66), as well as the first (IRR 2.97; 95% CI 1.34–6.58) and second (IRR 9.84, 95% CI 2.69–36.03) doses of the mRNA-1273 vaccine. In the meantime, the risk of myocarditis was increased within 1 to 28 d after COVID-19 (IRR 9.76, 95% CI 7.51–12.69).
Medical history, clinical manifestation, electrocardiography, echocardiography, and laboratory tests are now used to support the clinical diagnosis of myocarditis.3 Nowadays, histological or immunohistological evidence of inflammatory cell infiltration, with or without myocardial cell damage in the myocardium as revealed by EMB, is thought to be the gold-standard diagnostic method for myocarditis.32 When EMB is performed by a skilled technician, complications are quite rare.33 However, EMB can only be conducted locally, is intrusive, and has risks that are correlated with the operator's degree of competence. As a result, its performance is constrained by the patient's preference and the operator's skill.
cMRI is gradually being used as a noninvasive imaging test for the diagnosis and dynamic detection of myocarditis. Only symptomatic patients, patients with significant clinical evidence of myocarditis, and patients whose cMRI results are likely to have an impact on clinical therapy should undergo cMRI examination.34 The initial Lake Louise Criteria34 for myocarditis were published in 2009, which identified 3 signs of myocardial inflammation and their corresponding markers on cMRI, including (1) hyperemia, which presents as an intense signal on early gadolinium enhancement images; (2) tissue edema, which manifests as an increased myocardial T2 relaxation time or an elevated signal intensity on T2-weighted images; and (3) necrosis/fibrosis, which appears as a late gadolinium enhancement image. The diagnosis of acute myocarditis is possible if 2 of these 3 criteria are fulfilled. In 2018, the updated Lake Louise Criteria2 were published, including (1) T2-based markers, which include a regional high T2 signal intensity, a global T2 signal intensity ratio of ≥2.0 on T2-weighted images, or a regional or global increase in myocardial T2 relaxation time; and (2) T1-based markers, which include a regional or global increase in the native myocardial T1 relaxation time or extracellular volume mapping, or areas with a high signal intensity in a nonischemic distribution pattern on late gadolinium enhancement images. The specificity for identifying acute myocarditis may increase if both a positive T2-based marker and a T1-based marker are present.2 In 2020, the Society for Cardiovascular Magnetic Resonance included myocarditis as a primary indication for cMRI, revealing the importance of this imaging modality in the evaluation of myocarditis.35
An additional noninvasive imaging test is echocardiography, which is often useful for ruling out heart failure not due to myocarditis and in detecting ventricular thrombi. Echocardiography may reveal ventricular enlargement, ventricular wall thickening, decreased myocardial segmental mobility, and lower ejection fraction.3
The main way to identify changes in the electrical activity of the heart in patients with myocarditis is electrocardiography. Nonspecific T-wave alterations are the most frequent modifications; however, ST-segment dynamic modifications and newly developed pathogenic Q waves resembling myocardial infarction may also be present. Many arrhythmias, such as ventricular tachycardia, ventricular fibrillation, premature contraction, atrioventricular block, and (in severe cases) cardiac arrest, are commonly present in patients with myocarditis.3,36
Laboratory tests in patients with myocarditis may show elevated myocardial injury markers, such as cardiac troponin T, cardiac troponin I, creatine kinase isoenzyme, and N-terminal pro–brain natriuretic peptide, as well as nonspecific serum markers associated with inflammation, such as elevated white blood cell counts, C-reactive protein, and erythrocyte sedimentation rate.1,3,32 However, it is worth noting that the troponins are not often increased in mild cases of myocarditis.
Patients with myocarditis may have a variety of clinical manifestations, including chest pain, dyspnea, syncope, palpitations, and prodromal respiratory and gastrointestinal infections.3
The etiology of myocarditis is mainly divided into infectious and noninfectious factors. Infectious factors include a variety of pathogens, such as viruses, bacteria, chlamydia, Rickettsia, fungi, and parasites, with viral infections predominating in developed countries.37 Noninfectious factors include systemic autoimmune diseases, such as systemic lupus erythematosus and rheumatoid arthritis; drugs, such as aminophylline, amphetamines, anthracyclines, catecholamines, and ICIs; hypersensitivity reactions; and tumors.1
2.2 Pathogenesis of myocarditis
Previous studies have examined the pathogenesis of myocarditis, summarizing the classic 3-phase model,38,39 comprising the acute infection phase, the subacute immune response phase, and the chronic myopathy phase, using viral myocarditis as an example.
2.2.1 Acute infection phase
The acute infection phase is mainly caused by the first invasion of the pathogen40,41 or reactivation of the pathogen in a dormant state.40,42 When the virus reaches the cardiomyocyte, it uses cell membrane surface-specific receptors or receptor complexes to enter the cell. For example, coxsackievirus (CVB3) uses the coxsackievirus-adenovirus receptor (CAR) to enter the cell and then proliferates, leading to direct cellular damage.41,43,44
2.2.2 Subacute immune response phase
An appropriate immune response contributes to pathogen clearance, whereas an excessive immune response can cause tissue damage.40 Pattern recognition receptors (PRRs), such as Toll-like receptors and nucleotide oligomerization domain-like receptors,45 are present on the surface or in the cytoplasm of most cells in the cardiovascular system.46 PRRs recognize and bind to pathogen-associated molecular patterns, such as lipopolysaccharides, lipoproteins, and nucleic acids, as well as to damage-associated molecular patterns, activating47 a series of downstream signaling pathways, such as nuclear factor (NF-κB) signaling, to produce proinflammatory cytokines.47 The combination can also induce NLRP3 inflammasomes, subsequently causing pyroptosis.48
Complement (C), which is one of the key components of innate immunity, can be activated by bound antibodies through either the alternative pathway or the classical pathway.49 In response to activation, C split products are produced, which have strong proinflammatory effects and enhance both the innate and adaptive immune responses by binding to C receptors (CRs).49 Upregulation of CD11b/CR3 in the majority of infiltrating immune cells has been reported, both in male and female BALB/c mice using the new model of CVB3 myocarditis.50–53 The depletion of C3 or the blockade of CR1/2 can prevent experimental autoimmune myocarditis (EAM) in A/J mice by impairing the ability to form the membrane attack complex.5,54 The lack of CR1/2 promotes the severity of CVB3 myocarditis in male C57BL/6 backcrossed to A/J mice by increasing macrophage, interleukin (IL)-1β, and immune complex deposition in the myocardium.51
Subsequently, T lymphocytes, otherwise known as T cells, are activated by the binding of antigen peptide–major histocompatibility complexes (MHCs) to T cell receptors,55 which subsequently participate in B-lymphocyte (B cell) activation.56 The activation of adaptive immunity and the production of cytokines and antibodies lead to pathogen elimination and/or tissue damage.56
2.2.3 Chronic myopathy phase
When there is persistent overreaction of the immune response, myocardial tissue undergoes matrix remodeling and fibrosis, which can lead to heart failure and secondary DCM. Several susceptible strains of mice4–7 and genetically susceptible individuals8–11 progress to this phase. It has been reported that more males develop DCM and have a worse outcome after acute myocarditis than females.57,58 Although the presence of enteroviral genomic RNA has been examined in EMB tissues obtained from patients with DCM,59–61 non-DCM populations have parvovirus B19 DNA detected in heart tissues.62 Not only viruses, but also overreactive immune responses are necessities of the progress to DCM.58,63 Matrix remodeling and fibrosis are mainly triggered by the dysregulation of collagen production and degradation. In male BALB/c mice with CVB3 myocarditis, the imbalance in the matrix-degrading system, which refers to increased expression of matrix metalloproteinases (MMPs) and decreased expression of tissue inhibitors of MMPs, causes a pathologic collagen turnover that compromises left ventricular function and causes the structural integrity of the heart to deteriorate.64–67 Some proinflammatory cytokines and enzymes have been reported to induce this imbalance.58,67–69
2.3 Sex differences in myocarditis
According to previous reports,70–74 the incidence of myocarditis and the cardiac expression of proteins related to apoptosis and fibrosis are higher in men, which reveals that there may be sex differences in the pathogenesis of myocarditis. Male rodents, such as BALB/c mice and Lewis rats, show more acute and chronic inflammation in CVB3-induced myocarditis52,75 and EAM,53,76 and these differences are likely due to sex hormones.72 During CVB3 myocarditis, testosterone contributes to the predominant T helper 1 (Th1) immune response in the myocardium and more viral replication, but estradiol tends to develop the Th2 immune response and promotes the generation of regulatory T (Treg) cells and anti-inflammatory M2a macrophages.77–82 Greater testosterone concentrations are linked to a number of cardiac pathologies,58,74 which encourage higher collagen synthesis, fibrosis development, and extracellular matrix (ECM) remodeling.
In terms of the immune response, 17β-estradiol (E2) enhances the expression of TLR4 in rodent peritoneal macrophages83 and upregulates TLR7 and TLR9 signaling in human monocytes exposed to granulocyte-macrophage colony-stimulating factor (GM-CSF).84 In contrast, testosterone tends to deregulate the expression of TLR4 in rodent macrophages.85 E2 exhibits bimodal effects on human monocytes/macrophages, boosting the production of proinflammatory cytokines (including IL-1, IL-6, and tumor necrosis factor α [TNF-α]) at low concentrations, while decreasing the production of these cytokines at high concentrations.86 Increased levels of inflammatory cytokines, such as IL-1, IL-2, and TNF-α, have been observed in men with androgen deficiencies.87–89
3. Immune cells in myocarditis
3.1 Neutrophils
Neutrophils are the specialized phagocytic cells of innate immunity and are the first line of defense against invading pathogens. Neutrophils perform antipathogen functions, including phagocytosis, degranulation, production of neutrophil extracellular traps (NETs), and immunomodulation.90 Neutrophils express a variety of surface receptors, including PRRs, cytokine receptors (TNF-α, IL-1, and IL-6 receptors), and chemokine receptors (CXCR2 [C-X-C motif chemokine receptor 2]).91 In autopsies of patients with severe COVID-19, researchers found extensive neutrophil infiltration in the myocardium of patients who died of active myocarditis.92 Advanced depletion of neutrophils with anti-Ly6G antibodies in male C57BL/6 mice of CVB3-induced myocarditis led to a reduction in myocardial tissue inflammation and viral load.93 These studies suggest a possible role for neutrophils in the pathogenesis of myocarditis during the immune response phase.
In myocarditis, neutrophils engulf invading pathogens, forming phagocytic vesicles. The phagocytic vesicles come into contact with intracytoplasmic toxic particles, and the pathogens within the vesicles can be eliminated. Reactive oxygen species generation mediated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activation can also enter the phagocytic vesicles containing pathogens and participate in their elimination. Neutrophil infiltration and microabscesses were found in the myocardium of a patient with nonrheumatic streptococcal fulminant myocarditis, in which neutrophils that had phagocytosed streptococci were observed.94 Phagocytosis of pathogens or cellular debris activates neutrophils, which in turn release proinflammatory cytokines and chemokines to amplify inflammation and recruit more proinflammatory cells.93 In male C57BL/6 mice with CVB3-induced myocarditis, neutrophils recognize CVB3 mainly through intracellular TLR8,93 which triggers certain signaling pathways, such as the NF-κB pathway, further mediating the release of proinflammatory factors, such as IL-6, IL-1β, TNF-α, and IL-12, as well as chemokines. After phagocytosis of CVB3, CD11b expression in neutrophils is upregulated, enhancing their adhesion to the ECM and contributing to their residence at the site of inflammation. Neutrophils infected with CVB3 have an extended lifespan, but CVB3 does not replicate massively in neutrophils; thus, whether neutrophils can act as vectors for viral transmission needs further investigation.93 The phagocytosis of neutrophils plays both a protective role in the elimination of pathogens and a pathogenic role through the production of proinflammatory cytokines, chemokines, and adhesion factors in response to pathogens or dead cells.12
The cytoplasm of neutrophils contains a variety of granules that can be classified as primary, secondary, and tertiary granules containing myeloperoxidase and elastase, which are involved in killing pathogens and digestion, and containing lactoferrin and gelatinase proteins that degrade the ECM.95 The excreted granules also contain preformed cytokines.95 The degranulation process is dependent on adhesion signals and receptor–ligand binding signals, such as the binding of formylated peptides to formylated peptide receptor 1.91,96 The dual signals are activated to avoid random degranulation of neutrophils,95 while the gradual release of granules contributes to the control of inflammation.97 CVB3-infected neutrophils are stimulated by TNF-α to release myeloperoxidase and trigger NET formation.93 Neutrophil granules are essentially highly cytotoxic, so when the contents of these granules leak or are excreted excessively, they can damage the normal tissues that surround the inflammation and drive neutrophil recruitment.95 However, the released particles are not always detrimental. It has been suggested that human CAR may be a target for neutrophil elastase shedding and that the elastase released by neutrophils may protect cardiomyocytes from viral infection by cleaving CAR from the surface98 during the acute infection phase.
In recent years, the role of NETs, which are extracellular reticulated complexes that consist of DNA, histones, and neutrophil granule proteins, has been increasingly recognized.99,100 This unique cell death program in which neutrophils abandon their integrity and release DNA is called NETosis.101 Not only do NETs help to fix and remove pathogens, but also they cause direct or indirect damage to surrounding tissues due to the combination of various proteases and cytotoxic proteins.102 NETs are found in myocardial tissues from patients with myocarditis, as well as in male mice with CVB3-induced myocarditis and EAM, and they exert their pathogenic effects mainly in the acute phase.103,104 Inhibition of NET formation during the acute phase helps to reduce the inflammatory response in the heart.103,104 Cytokine intermediate factor (MK) can induce neutrophil infiltration and NET formation, a process that may be achieved through interaction with low-density lipoprotein receptor–related protein 1.104 IL-37 attenuates CVB3-induced myocardial injury by inhibiting NET formation.105 In the lung, NETs induce activation of lung fibroblasts to the myofibroblast phenotype and exhibit increased production of connective tissue growth factor and collagen.106 In male BALB/c mice with EAM, NET formation can be reduced by inhibiting MK,104 which also reduces fibrosis and improves cardiac contractile function. Thus, NET formation in the myocardium may contribute to fibrosis. Moreover, NETs activate CD4+ T cells by lowering their activation threshold and promoting the adaptive immune response phase.107
Lipocalin-2 (LCN2), also known as neutrophil gelatinase-B-associated lipid transport protein, is involved in the antimicrobial response, iron transport, apoptosis regulation, and resistance to oxidative stress.108,109 Recently, Sun's team110 showed that LCN2 exacerbates lupus nephritis by promoting Th1 cell differentiation. Previously, Ding et al.111 found that neutrophils in the hearts of patients with myocarditis and male Lewis rats with EAM express LCN2, while its role in myocarditis has not been revealed.
Neutrophils are also involved in the regulation of other immune cells. On the one hand, neutrophils recruit themselves and monocytes by secreting several proinflammatory mediators mentioned previously, which promotes the formation of inflammatory macrophages and the differentiation of Th1 and Th17 cells.103 On the other hand, neutrophils contribute to the maintenance of the cardiac CD8+ T cell inflammatory response.112 Moreover, neutrophils produce the cytokines B cell activating factor and proliferation-inducing ligand, both of which are required for B cell survival and activation.113 Further studies are needed to determine whether it is necessary to modulate B cell function in myocarditis.
3.2 Eosinophils
Eosinophils are a special type of leukocyte that are usually considered as cytotoxic effector cells because they contain a variety of cytotoxic granules. They contain major basic protein (MBP), eosinophil cationic granule protein (ECP), and eosinophil peroxidase, which can be toxic to pathogens and cells, either directly or by promoting the production of reactive oxygen species. Eosinophils also contain cytokines (IL-2, IL-4, IL-5, IL-6, IL-13, and TNF-α), chemokines (C-C motif chemokine ligand 5 [CCL5] and CCL11), transforming growth factor (TGF), and lipid mediators, which are selectively released in response to different stimuli.114–116 Recently, it has been shown that eosinophils are involved in the regulation of immunity and homeostasis.117
Myocarditis with predominant eosinophil infiltration on EMB is known as eosinophilic myocarditis. According to a previous summary, eosinophilic myocarditis is mainly associated with hypersensitivity reactions, infections (parasites, fungi), malignancies, drugs, hypereosinophilic syndrome, eosinophilic granulomatous polyangiitis, systemic lupus erythematosus, and Churg-Strauss syndrome, and is usually treated with strong immunosuppressive agents.118–120 Recently, in autopsies of deceased individuals who developed fulminant myocarditis after the COVID-19 mRNA vaccination, cardiac tissue demonstrated eosinophil infiltration.121
The pathogenesis of eosinophilic myocarditis is commonly described as follows. First, eosinophils infiltrate into the heart and recognize invading parasites, fungi, certain bacteria, and damage-associated molecular patterns signals in vivo through PRRs during the immune response phase. Then, eosinophils enter an activated state, achieve degranulation, and kill pathogens, mainly through piecemeal degranulation, exocytosis, and cytolysis, causing myocardial injury and dysfunction.122 This can be confirmed by the detection of ECP, MBP, and eosinophil peroxidase deposition on EMB.123
Subsequently, the thrombogenic phase occurs, in which the formation of the thrombomodulin-thrombin complex is blocked due to the binding of ECP to the anionic site on thrombomodulin, resulting in an elevated level of circulating thrombin and putting the body in a hypercoagulable state with the release of tissue factor stored in specific granules by eosinophils. In addition, MCP activates platelets. These processes contribute to the formation of cardiac thrombosis.124
The final stage is dominated by fibrosis. In male BALB/c mice with EAM, eosinophil depletion prevents the transition from myocarditis to DCM, while high eosinophil counts promote the development of more severe DCM.125 This may be due to endothelial cell proliferation stimulated by ECP and MBP, fibroblast activation, and proliferation promoted by secreted TGF-β and IL-1, and the unknown function of eosinophil-derived IL-4.116,118,125
IL-5 is a key biological factor (i.e. involved in the activation, survival, and migration of eosinophils in tissues), and it is the most potent activator of eosinophils, produced by both Th2 cells and type 2 ILCs.126 Researchers have found that hypereosinophilic mice with IL-5 transgene expression have spontaneous eosinophilic myocarditis, and that more than 60% of cardiac infiltrating cells are eosinophils in IL-5 transgene male BALB/c mice with EAM.115,125 IL-33, which is a cytokine from the IL-1β family,127 significantly induces eosinophilic myocarditis during CVB3 infection.128 The recruitment of eosinophils is influenced by Th2 cells, macrophages, fibroblasts, and eosinophils themselves, by producing chemokine ligands in the myocardium.116,129
Similar to neutrophils, eosinophils produce extracellular traps called eosinophil extracellular traps (EETs), and the form of eosinophil death is called EETosis. The nuclear and plasma membranes of eosinophils rupture under the mediation of reactive oxygen species generated by NADPH oxidase, releasing nuclear or mitochondrial DNA and intact eosinophil granules, mediating the killing of pathogens and tissue damage.130,131 EETs are more stable than NETs.122 In eosinophilic airway inflammation, activated platelets and their products induce EET formation, which enhances their inflammatory effects.132 Therefore, we hypothesized that MCP activates platelets, which induces EET formation and MCP release, and it may enter a vicious MCP–platelet–EET–MCP cycle.
In addition, ECP and MBP activate human cardiac mast cells. Then, mast cells release histamine, trypsin, and prostaglandin D2.133 Whether this process plays a definite role in the pathogenesis of myocarditis remains to be explored.
3.3 Mast cells
Mast cells are immunomodulatory cells that play an important role in both innate and adaptive immunity. Depending on their contents, mast cells can be divided into 3 subpopulations in humans: tryptase-positive mast cells, chymase-positive mast cells, and tryptase/chymase-positive mast cells.134 Mast cells are mainly found in contact with the environment and are thought to be closely associated with allergic reactions.135
A previous study showed that in encephalomyocarditis virus myocarditis, mast cell–deficient male mice had a higher survival rate, significantly less myocardial necrosis, and less inflammatory infiltration than control mice.136 This suggests that mast cells play a pathogenic role. In 2022, Luo et al.137 found that in male C57BL/6 mice with CVB3 myocarditis, fibroblasts triggered mast cell degranulation and CCL2 and TNF-α release by producing early stem cell factor, and mast cell–derived TNF-α in turn stimulated fibroblasts to increase CCL2, α-smooth muscle actin, collagen, and TGF-β, thereby exacerbating cardiac fibrosis. In addition, mast cell–derived CCL2 recruits monocytes, which differentiate into inflammatory macrophages, promoting inflammation. The stem cell factor/mast cell/CCL2/monocyte/macrophage axis was first described in CVB3-induced myocarditis137; however, mast cells are not simple. The subpopulations and enzymatic profiles of mast cells are related to the tissue.138 Under different stimuli in vivo or in vitro, mast cells produce a variety of ILs (IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, and IL-13, among others), interferon-γ (IFN-γ), TNF, TGF-β, vascular endothelial growth factor (VEGF), and chemokines.139 All of these molecules exert proinflammatory, anti-inflammatory, and regulatory fibrosis, as well as angiogenic effects.139 Mast cells, like macrophages, have considerable plasticity and may play unique roles in different local conditions through different mechanisms.138
A detailed summary of cardiac mast cells (CMCs) was previously presented by Varricchi et al.140 Mast cells are found in both human and animal hearts, mainly between the myocardium, in the epicardium, and near blood vessels.141 Varricchi et al.140 concluded that activated human CMCs can release preformed histamine, trypsin, and chymotrypsin. Histamine can lower blood pressure. In addition, Stasiak et al.142 found that inhibition of histamine receptor 4 (H4R) in Lewis male rats with EAM inhibited cardiac remodeling, improved cardiac systolic function, and increased survival. However, blocking histamine receptor 1 inhibited cardiac dilation, but it did not prolong life. Chymotrypsin has different effects in different models of heart disease. Chymotrypsin activates the renin–angiotensin system and promotes the release of local angiotensin II and norepinephrine, which are closely associated with arrhythmia.140
Varricchi et al.140 concluded that activated CMCs also synthesize secreted renin, which is involved in RAS activation; release cysteine leukotrienes to promote inflammation and proangiogenesis; possibly secrete PGD2 to reduce allergic reactions and fibrosis; synthesize VEGF-A to promote angiogenesis and VEGF-C to promote lymphatic growth; and reduce tissue edema and fibrosis.
Human mast cells express PD-L1 and PD-L2. Varricchi et al.140 postulated that ICIs may cause myocardial damage by killing peripheral CMCs, which releases cytotoxic factors and recruits inflammatory cells.
Mast cells actively form mast cell extracellular traps in response to specific stimuli that surround and clear pathogens, possibly limiting the inflammatory response.143 However, it is unclear whether CMCs form mast cell extracellular traps to function in myocarditis.
Varricchi et al.140 analyzed previous studies on mast cells and cardiac fibrosis. They summarized the possible reasons for the conflicting results among these studies, including the different phenotypes between mouse CMCs and human CMCs, the existence of models that eliminate mast cells while causing a lack of other cell types or incomplete elimination of mast cells, and the fact that the mast cell degranulation inhibitors used for the experiments were not specific. In 2021, Bruno et al.144 found that during CVB3-induced myocarditis, feeding female BALB/c mice in plastic cages or with water containing bisphenol A increased the number of degranulated mast cells in the myocardium and pericardium, and an increase in myocardial fibrosis was observed. However, these manifestations were not present in male BALB/c mice under the same conditions.144 We speculate that the effect of mast cells on myocardial fibrosis is affected by sex; however, this requires further experimental investigation.
3.4 Innate lymphoid cells
ILCs can be divided into natural killer (NK) cells, ILC1, ILC2, and ILC3. NK cells are the only cytotoxic cell ones that originate from the bone marrow and migrate continuously through the circulation to peripheral tissues.145 Human NK cells can be divided into 2 subpopulations based on the differential expression of CD56 and CD16.146 CD56loCD16hi NK cells account for approximately 90% of human peripheral blood NK cells and exhibit classic cytotoxic effects. The remaining 10% are CD56hiCD16−/lo NK cells, which are involved in the secretion of cytokines for immune regulation.147,148
NK cells are cytotoxic and directly lyse pathogen-infected cells, autoreactive T cells, overactivated inflammatory cells, and tumor cells, and the choice of target is related to abnormalities in MHC class I molecules in target cells.149 Activated NK cells express the Fas ligand or TNF-related apoptosis-inducing ligand, and binding of these ligands to the corresponding receptors induces target cell apoptosis.150 Moreover, NK cells release cytotoxic proteins (perforin, granzyme, etc.).151,152 Perforins undergo oligomerization in the target cell membrane and form transmembrane pores with the transmembrane structural domain–MACPF structural domain. Granzymes and proapoptotic serine proteases are released through these pores and initiate target cell apoptosis, leading to cell death.153 This suggests a role for NK cells in pathogen clearance, but uncontrolled perforin expression can also lead to autoimmune diseases, including cerebral myocarditis.153 Pores induced by perforins on the surface of virus-infected cardiomyocytes have been found in both patients with myocarditis and animal models of viral myocarditis,154 and attacking virus-infected cardiomyocytes also inevitably leads to the release of autoantigens and subsequent induction of the autoimmune response.14
In addition to cytotoxicity, activated NK cells secrete a variety of cytokines (IFN-α, IFN-β, IFN-γ, TNF-α, and ILs, among others)155 that function in antiviral responses, induction of the inflammatory response, and activation or suppression of other immune cells.156 Estrogen downregulates the expression of Th1 response–specific T box transcription factor, a key transcription factor for IFN-γ production, in CVB3-stimulated NK cells, thereby reducing IFN-γ-secreting NK cell infiltration and myocardial injury in myocardial tissues.157 Loebel et al.158 found that in viral myocarditis, the forkhead transcription factor negatively regulates IFN-γ production and cytotoxic degranulation in NK cells, which is detrimental to the control of early viral infection, but it has the potential to play a protective role in the subsequent chronic inflammatory process.
According to previous studies, NK cells control the number of eosinophils to play a protective role in humans with myocarditis, EAM models, and cellular assays in vitro.159–161 This protective effect appeared to be dependent on the presence of eosinophils, as depletion of NK cells in eosinophil-deficient mice did not increase the severity of myocarditis.159 The mechanism may be related to the increase in reactive oxygen species and apoptosis in eosinophils induced by NK cells.161 Moreover, NK cells can reduce cardiac eosinophil recruitment by producing IFN-γ, which downregulates the release of Th2-related cytokines in ILC2.160,162
The finding that NK cells exert a protective effect by inhibiting fibrosis has been demonstrated mainly in liver disease,163 mainly by directly lysing activated fibroblasts or promoting the inactivation of hepatic stellate cells, which reduces collagen production and deposition.164,165 However, the functions of the different human NK cell subpopulations in different tissues are not identical.146 In Ong et al.'s study,159 NK cells reduced fibrosis during myocarditis, but when NK cells were cocultured with activated primary cardiac fibroblasts in vitro, cardiac fibroblasts did not show cytolysis similar to that of liver fibroblasts. Further study159 in male BALB/c mice with EAM revealed that NK cells, by directly limiting collagen formation in cardiac fibroblasts, may protect against fibrosis and DCM development by preventing the accumulation of certain inflammatory cells in the heart and negatively regulating profibrotic factors. However, the exact mechanism has not been well revealed.
In addition to NK cells, the remaining noncytotoxic ILCs include ILC1, ILC2, and ILC3. ILC1 is stimulated by IL-12 to express IFN-γ and TNF-α, which assist in the antiviral response in a noncytotoxic manner and promote the Th1 response.166,167 ILC2 participates in the Th2 response mainly by secreting IL-5 and IL-13, which contribute to eosinophil maturation and recruitment.168 ILC3 responds to IL-23 and IL-1β and promotes the Th17 response by producing IL-17A and GM-CSF, and is associated with antipathogen and autoimmune diseases.169 Under normal conditions, the major cardiac ILC population is in an undifferentiated state with the potential to differentiate toward ILC2. Studying samples from myocarditis patients and BALB/c mice with EAM has revealed that cardiac ILCs differentiate into conventional ILC2, but they do not differentiate into ILC1 and ILC3 during myocarditis. Their differentiation is positively associated with increased IL-33 production by cardiac fibroblasts. Unlike NK cells, circulating noncytotoxic ILCs are unable to enter the heart during adulthood. Thus, cardiac noncytotoxic ILCs are strictly cardiac-resident cells.170 However, the findings may need further verification because of the limitations of cardiac tissue sampling. Subsequent tissue-specific single-cell spatial transcriptome sequencing may provide a better understanding.
3.5 Monocytes
Monocytes are circulating leukocytes that are released into the circulatory system from the bone marrow or extramedullary hematopoietic tissues (spleen). They are then transported through the circulatory system to peripheral tissues. During inflammation, circulating monocytes are recruited by monocyte chemoattractant protein 1, leave the bloodstream to migrate to inflamed tissues, kill a variety of pathogens, promote or inhibit inflammation by secreting mediators (e.g. nitric oxide, TNF, ILs, reactive nitrogen intermediates, and reactive oxygen intermediates, and phagocytic lysosomal enzymes, among others), and differentiate into macrophages or DCs that remain in the tissues and continue to function.171
In general, monocytes are divided into 3 major categories, the first being classical or inflammatory monocytes,172 which in mice are characterized by high expression of Ly6C and C-C chemokine receptor 2 (CCR2), and low expression of C-X3-C chemokine receptor 1, named Ly6chi monocytes. In humans, they are named CD14++CD16− monocytes based on high expression of CD14 and lack of CD16.173 Although their markers vary by species, they also exhibit higher peroxidase activity and can produce more chemokine receptors, suggesting that they are associated with phagocytosis to destroy pathogens and migrate more easily to sites of inflammation or injury. It has been shown that in human myocarditis and male BALB/c mice with EAM, inflammatory monocytes are the main precursors of cardiac monocyte-derived macrophages.174 The second category, nonclassical monocytes, whose function differs from that of classical monocytes in that they play a patrolling role along the vessel wall, promote healing and respond to viral infection. Nonclassical monocytes include Ly6clo monocytes in mice and CD14 + CD16++ monocytes in humans.175 For the question of whether nonclassical monocytes can be transformed into macrophages in tissues, Hou et al.174 demonstrated that in male BALB/c mice with EAM and patients with myocarditis, nonclassical monocytes have a limited ability to convert into macrophages, possibly due to IL-17A signaling through cardiac fibroblasts to inhibit conversion. In the myocardium of EAM mice lacking IL-17A, approximately 6% of nonclassical monocytes converted into macrophages and exhibited different molecular features that may have a unique role compared with inflammatory monocyte–derived macrophages in the pathogenesis of myocarditis.174 The third category is intermediate monocytes, including Ly6cint monocytes in mice and CD14++CD16+ monocytes in humans, which are intermediate between classical and nonclassical monocytes. They display inflammatory properties and share low peroxidative activity with nonclassical monocytes, and they also secrete TNF and IL-1β.176
Unlike other innate immune cells, monocytes can differentiate into macrophages or DCs in the periphery and continue to function in the proinflammatory and anti-inflammatory responses, as well as in antigen presentation,177 as detailed subsequently. In addition, monocytes are directly involved in regulating T cell differentiation, including differentiation of Th1, Th2, Th17, and Treg cells, among others, and they promote or inhibit T cell differentiation through antigen presentation, secretion of cytokines or killing mediators, regulation of cysteine metabolism, and expression of multifunctional transmembrane receptors.178,179 By inhibiting myocardial inflammatory monocyte recruitment in A/J mice with EAM using silencing RNA targeting CCR2, myocardial inflammation and fibrosis were attenuated.180 In male and female C57/BL6 mice with CVB3-induced myocarditis, myocardial monocyte/macrophage infiltration was reduced, and the Th1 response was increased in the absence of stabilin-1 (STAB1). The authors considered that STAB1 deletion leads to a block in fibronectin-mediated recruitment of STAB1+ monocytes, which play a role in suppressing the T cell response.181 We hypothesized that monocytes themselves can exert opposite regulatory effects through different mechanisms, and that the final outcome depends on the type of disease and the strength of different triggers or maintenance factors.
We realize that the definition of monocytes is by no means monotonous. When monocytes enter tissues, they exhibit changes in their transcriptional profile, especially when they enter inflammatory tissues and begin to synthesize some proinflammatory or anti-inflammatory factors. To a large extent, the diversity of monocytes depends on the local microenvironment.171,177 Using scRNA-seq technology, researchers have shown that in patients who develop myocarditis after BNT162b2 (Pfizer COVID-19 vaccine) vaccination, the transcriptome profile of classical monocytes showed upregulation of genes related to fatty acid metabolism and downregulation of transcription factor AP-1.182 These findings revealed the importance of classical monocytes in the pathogenesis of myocarditis after BNT162b2 vaccination, as well as the role of vaccination on monocytes leading to myocarditis. In addition, Jimenez et al.183 concluded that at least 7 types of monocytes with different morphological and transcriptional characteristics were identified in the infarcted myocardium and may play their own unique roles. Therefore, further studies are needed to demonstrate whether the inflamed myocardium may also have a variety of monocytes with different characteristics that play as yet unspecified roles.
3.6 Macrophages
Macrophages were first recognized as phagocytes, which represents their most basic role of pathogen removal. In CVB3-induced myocarditis, macrophage-depleted male C3H/HeJ mice had higher viral loads, suggesting that reduced macrophages may lead to reduced pathogen clearance.184 However, macrophage-depleted mice exhibited milder cardiac inflammation and fibrosis. Thus, the function of macrophages is never simply limited to phagocytosis. With further studies, macrophages began to be recognized as immune cells.
Traditionally, macrophages are divided into 2 types according to their activating factors. The first type is classical (M1) macrophages activated by TLR ligands or IFN-γ, which mainly mediate oxidative stress, inflammasome formation, and proinflammatory factor secretion (TNF-α, IL-1β, IL-6, and IL-12, among others) to produce costimulating factors and chemokines that promote inflammatory cell infiltration and proliferation. They subsequently cause cardiac injury. The other type are nonclassical (M2) macrophages, which form under the stimulation of anti-inflammatory signals, such as IL-4. M2 macrophages secrete anti-inflammatory cytokines, chemokines, and growth factors, mainly playing an anti-inflammatory role.185 In BALB/c male mice with CVB3-induced myocarditis, the myocardial macrophage phenotype changes from the classical M1 phenotype to the nonclassical M2 phenotype at approximately days 7 to 10 when the disease enters a phase characterized by fibrotic repair186 and conditional knockdown of CAPN4 could reduce M1 macrophage transformation, and moderately increase M2 transformation via inhibiting the C/EBP homologous protein-STAT1/STAT3 signaling pathway and could ameliorate myocardial inflammation and injury.187 Besides, macrophage-specific CAR depletion and IL-13 knockout have been reported to increase M1 transformation and induce T cell activation in CVB3-infected mice188 or EAM mice,189 along with enhanced heart inflammation and injury. In contrary, treating with CD73 inhibitor prevented the premature transformation from M1 to M2 and decreased the parasite burden in heart during the acute phase in BALB/c mice infected with Trypanosoma cruzi, leading to a good outcome, because the persistence of parasites is a necessary condition for the development of Chagas cardiomyopathy.190 It seems that dysregulated transformation may be the key point in myocarditis. Macrophages also secrete several types of MMP.185 For example, Amoah et al.191 concluded that the release of MMPs not only hydrolyzes ECM proteins and non-ECM proteins to amplify the inflammatory response, but also plays a protective role in viral myocarditis by cleaving chemokines and reducing inflammatory cell infiltration.
In addition to traditional M1/M2 macrophage polarization, the understanding of cardiac macrophages has been further improved. There are native tissue-resident macrophages in the heart, which are derived from embryonic (yolk sac or fetal liver) precursor cells. Tissue-resident macrophages in the heart lack CCR2 on their surface and rely on replication in the tissue for renewal.192 Circulating monocyte–derived macrophages are also present in the heart and are distinguished from the former type by their expression of CCR2. The roles of these 2 types of macrophages are different. It is currently believed that cardiac-resident macrophages play an important role in tissue developmental remodeling and maintenance of homeostasis by removing abnormal cells or dysregulated mitochondria,193 secreting anti-inflammatory factors, regulating lymphangiogenesis, and avoiding the excessive inflammatory response.183 Circulating monocyte–derived macrophages are thought to play a potent inflammatory role. Upon activation, they generate an inflammatory cascade by secreting proinflammatory factors and chemokines, and further recruiting neutrophils and monocytes.137,183 Their abundance may be associated with a poor prognosis in cardiac diseases.183 Researchers found that reduction of CCR2+ macrophage infiltration in the heart by CCR2-siRNA attenuated myocardial inflammation and fibrosis in EAM mice.180 However, impeding CCR2-macrophage development and aggregation by knocking down CX3CR1 exacerbated cardiac inflammation and fibrosis in CVB3-infected mice.194
It appears that macrophages respond differently to different signals, and the macrophage phenotype can be modulated by external signals. Mosser et al.195 proposed a new concept in which macrophages act as cellular transducers, receiving different input signals from the environment and translating them into a corresponding response. Hulsmans et al.196 revealed that cardiac macrophages expressing connexin 43 facilitated electrical conduction in the distal atrioventricular node by altering the membrane potential, and that connexin 43 macrophage deficiency delays atrioventricular conduction. Whether connexin 43 macrophages are associated with the development of atrioventricular block in myocarditis needs to be clarified. Matheis et al.197 showed that intestinal myocardial macrophages upregulate neuroprotective programs via β2-adrenergic receptor signaling in response to intestinal infection and complete neuronal protection via the arginase 1–polyamine axis. Whether similarly acting macrophages reside in the heart to exert neuronal protection against myocarditis needs to be explored in future studies.
3.7 Dendritic cells
DCs, specialized antigen-presenting cells with a complex subpopulation, are considered to be the body's outpost cells, linking innate and adaptive immunity.198,199 According to the current understanding of DCs, they are classified into 3 categories. Conventional DCs (cDCs) recognize signals from intracellular and extracellular antigens in an inflammatory environment through PRRs, upregulate costimulatory molecules, present antigens to naïve T cells and activate them200 in lymph nodes. In the steady state, cDCs also present innocuous antigens and mediate T cell incompetence to complete the body's immune tolerance to autoantigens, inhaled antigens, and food antigens.201,202 cDCs include type 1 cDCs, characterized by their strong ability to activate cytotoxic CD8+ cells through cross-antigen presentation, and type 2 cDCs, which express a wide variety of TLRs and NLRs in response to different danger signals, and they may be more inclined to present MHC-II-antigenic peptides to CD4+ T cells.203 Second, plasmacytoid DCs (pDCs) are important for host antiviral immune response and autoimmunity as efficient producers of IFN-α following stimulation of the nucleic acid-sensitive receptors TLR7 and TLR9,203 not as potent in antigen presentation as cDCs. Finally, monocyte-derived DCs are a DC subpopulation that differentiate from tissue-infiltrating monocytes in response to inflammatory stimuli.203 Monocyte-derived/myeloid DCs are mainly considered to be a subpopulation of inflammatory DCs that can elicit local Th1 or Th2 responses.201,202
Following the viral infection in the myocardium, the number of myocardial DCs increased rapidly relying on local proliferation and recruitment from circulation.204,205 During subclinical myocarditis in mice infected with encephalomyocarditis variant-D virus, CD103+ resident type 1 cDCs facilitated the development and infiltration of encephalomyocarditis variant-D virus–specific CD8+ T cells, alleviated cardiac damage, and prevented the progression to heart failure.206 Abnormalities in the maturation/activation and MHC class I antigen processing of DCs were proved to be responsible for persistent chronic CVB3-induced myocarditis in A.BY/SnJ mice caused by a failure to remove the virus.207 The activation of antiviral CD8+ T cells by DCs in the early stage is conducive to viral clearance and prevent cardiac remodeling from sustained immune activation by hidden viruses. In addition, it has been reported that the number of circulating myeloid DCs and pDCs increased in the patients with acute myocarditis, and increased myeloid DCs exhibited a proinflammatory phenotype with higher costimulatory molecules and lower inhibitory receptor expression linked to the worser clinical outcome.208 Besides, increased pDCs could be the source of peripheral cytokine storm in myocarditis. In CVB3-infected mice, myeloid DCs accumulated following the increased level of CCL4 and CCL19 in the heart.209 Overaccumulation of DCs might mean loss of the body's immune tolerance to myocardial tissue. Activated by IL-1210 and GM-CSF211 in the proinflammatory environment, DCs, especially type 2 cDCs,212 have the potential to take up self-antigens released from damaged cardiomyocytes or newly generated ECM, activate self-reactive CD4+ T cells, leading to cardiac injury.202,213 TN-C, a nonstructural ECM glycoprotein (i.e. strongly expressed during acute inflammation in mice with EAM) can activate DCs to generate pathogenic autoreactive T cells that attack cardiac tissue.214
The body responds to the inflammatory cascade by generating a corresponding anti-inflammatory response to maintain homeostasis.215 In an anti-inflammatory environment, DCs show phenotypic changes. IL-10–induced human regulatory DCs express a series of inhibitory receptors (PD-L1 and PD-L2, among others) that suppress harmful T cell responses.216 PD-L2–deficient DCs enhance the proliferation of self-reactive CD4+ T cells in the presence of T cell receptors and CD28 transduction signals,217 which exacerbated EAM. This also indirectly suggests that reprograms of DCs induced by IL-10 express PD-L2 and prevent self-reactive CD4+ T cell expansion which alleviates the severity of myocarditis, indicating the multiple roles of DCs due to different environments.
Furthermore, with the emergence of immune-targeted therapies, investigators have recognized a new DC type, namely tolerogenic DCs (tDCs). The mechanism of tDCs formation in vivo is currently unclear, but it is associated with stimulation of immunosuppressive factors, including anti-inflammatory factors, such as IL-10 and TGF-β, as well as activation of vitamin A receptor ligands.218,219 In addition, knockdown of NEAT1 induced the formation of tDCs by inhibiting activation of the NLRP3 inflammasome.220 Several studies221,222 have shown that injecting IL-10 gene–modified tDCs into EAM mice could regulate Th1/Th2 unbalance and cardiac inflammation has been alleviated. Antigen-specific tDCs generated by exposing immature DCs to cardiac myosin with TNF-α223 or protosappanin A224 were found to significantly reduce myocardial inflammation in treated EAM mice by enhancing the induction of myosin-specific Treg cells and anti-inflammatory cytokine secretion. These tDCs had switched phenotype with lower levels of surface DC–related markers and higher levels of indoleamine 2,3-dioxygenase compared with mature DCs, or reprogrammed glycolytic phenotype. Therefore, tDCs may be a novel therapy direction in myocarditis and exert immunosuppressive effects by secreting anti-inflammatory factors, suppressing effector T cells, regulating the Th1/Th2 response, and inducing Treg cell activation.
In summary, the various roles of DCs are closely related to their phenotypes and surroundings. We recognize that their phenotypes also go far beyond the species identified. Villar et al.225 combined single-cell sequencing to provide new insights into the classification of human DC subpopulations, suggesting that single-cell transcriptome sequencing could theoretically provide specific and unique molecules for the identification of each DCs subpopulation. This is expected to make it possible to isolate specific DCs for functional characterization beyond transcriptome profiling in healthy and disease states. This also provides some ideas for subsequent studies on myocarditis.
3.8 T lymphocytes
In the bone marrow, pluripotent hematopoietic stem cells differentiate into lymph-like progenitor cells, which follow the blood into the thymus and undergo positive and negative selection before differentiating into mature T cells. Depending on the expression of CD4 and CD8, T cells can be divided into CD4+ T cells and CD8+ T cells. CD4+ naïve T cells (Th0 cells) migrate to peripheral immune organs and are activated into Th cells upon stimulation by antigenic peptide–MHC class II complexes and co-stimulatory signals. They can differentiate into Th cell subsets with different functions (Th1, Th2, Th9, Th17, Th22, follicular helper T, and Treg cells) under different cytokine environments. In contrast, CD8+ T cells are activated into cytotoxic T cells or CD8+ Treg cells upon stimulation by antigenic peptide–MHC class I complexes. The roles of T cells and their secreted cytokines in myocarditis have been summarized in more detail in previous reviews.15,191,226 Here, we make a brief summary and add details on the regulation or crosstalk. We also briefly summarize other cell subsets that have not yet been mentioned to provide a comprehensive understanding.
3.8.1 CD4+ T cell subsets in myocarditis
Th1 cells could be a double-edged sword in the pathogenesis of myocarditis by secreting IL-12, IFN-γ, IL-1β, and TNF-α. IL-12 has been reported to be proinflammatory, and the knockout of IL-12R showed the resistance to EAM, while exogenous usage of IL-12 leads to aggravated heart inflammation. IFN-γ shows strong abilities of antivirus, activating immune response, and balance the excessive inflammation.227 In the acute phase, knockout of IFN-γ receptor in T cell receptor transgenic mice with EAM,228 depleting IFN-γ in viral myocarditis,229 or lowering IFN-γ production in T. cruzi–related myocarditis230 reduced myocarditis. However, IFN-γ–deficient mice showed more severe chronic myocarditis after CVB3 infection50 or high levels of T. cruzi hidden in heart and reactivation,231 leading to bad prognosis. Besides, knockout of IFN-γ receptor in cardiac α-myosin heavy chain–immunized EAM mice leaded to enhanced CD4+ T cell response and persistent myocarditis.227
Th2 cells secrete IL-4, IL-10 and IL-13, and also exhibit reverse function in myocarditis. Th2-biased inflammatory phenotype was observed in vitamin D receptor knockout mice with spontaneous myocarditis232 and A/J mice with EAM.233 While anti-IL-4 treatment could reduce inflammation.233 Besides, the amount of Th2 cells and related cytokines were higher in the heart from patients with myocarditis than them with DCM.234 In contrary, hepatocyte growth factor has been reported to promote IL-4 and IL-10 production by Th2 cells, inhibit cardiomyocyte apoptosis, and attenuate the severity in Lewis rats with EAM.235
Th17 cells are novel proinflammatory subset characterized by the production of IL-17. In the early phase of CVB3-induced myocarditis, Th17 cell differentiation was activated directly by CVB3236 and regulated positively by the IL-17-NLPR3 positive feedback.237 The inhibition of Th17 differentiation and IL-17 production has been reported to ameliorate viral myocarditis.238,239 In addition, Gergely et al.240 found that anti-PD-1 treatment led to cardiac insufficiency and left ventricular dilation in male C57BL/6J mice and resulted in a significant increase in thymic inflammatory IL-17A signaling, and IL-17A inhibition prevented anti-PD-1-induced cardiac dysfunction in male C57BL/6J mice. This study revealed that IL-17A plays a promoting role in ICI-MC, but the source of IL-17A needed to be verified. In terms of EAM, neutralization of IL-17241 or inhibiting myocardial-tropic Th17 responses242 could alleviate the production of antimyocardial antibodies and cardiac inflammation. Interestingly, IL-17 exhibited strong association with myocardial fibrosis, cardiac remodeling, and DCM242 in the later stages of myocarditis. Both inducing EAM in IL-17–deficient mice and treatment EAM mice with anti-IL-17A monoclonal antibodies reduced cardiac fibrosis and improved cardiac function, indicating the essential role of IL-17 in the progression to DCM.243 A recent study244 used single-cell sequencing analysis to show that PGK1 in mice with EAM promotes Th17 cell differentiation by increasing glycolytic activity and promoting pyruvate dehydrogenase kinase 1 phosphorylation, which reduces mitochondrial reactive oxygen species production, leading to increased myocardial inflammation. This reveals the dysregulated metabolism in Th17 cells in myocarditis and provides a new direction for further exploration.
Treg cells, characterized by CD4, CD25, and FOXP3 (forkhead box protein 3), have been reported to execute immunosuppressive function via anti-inflammatory cytokines IL-10 and TGF-β, inhibitory receptors. Patients with myocarditis/DCM showed the lower percentage of Treg cells in peripheral blood.245 Depletion of GITRhighFOXP3+ natural Treg cells led to fatal autoimmune myocarditis in nude mice.246 Before CVB3 infection, adoptive transfer of Treg cells protected from the excessive inflammatory response and promote virus clearance via TGF-β-CAR pathway.247 Besides, adoptive transfer after CVB3 infection alleviated fibrosis via secreting more IL-10.248 Heart stromal/progenitor cell–derived extracellular vesicles promoted Treg cell differentiation and the production of IL-10, resulting in the reduced myocardial inflammation and functional failure in mice with EAM.249 However, TGF-β produced by Treg cells in the chronic phase exhibited profibrotic effect.250 Pre-exposure to particulate matter 2.5 activated Treg cells in spleen and aggravated CVB3-induced myocarditis,251 indicating that the premature or long-term activation of Treg cells had a bad influence in myocarditis.
The latest finding revealed that the previous Th cell differentiation could be regulated by autonomic nerves. Li et al.252 found that vagotomy in BALB/c male mice with CVB3-induced myocarditis increased the percentage of Th1 and Th17 cells, but it decreased the percentage of Th2 and Treg cells in the spleen. It also increased the expression of proinflammatory mediators and amplified inflammatory infiltration, as well as myocardial lesions in the acute and subacute phases. The authors hypothesized that this might be related to the fact that vagotomy inhibited Janus kinase 2/signal transducer and activator of transcription 3 activation and enhanced NF-κB activation of splenic CD4+ T cells. We realize that there is complicated communication between the autonomic nerves and CD4+ T cells. Vagus nerves may have a potential protective effect on inflammation, which requires more rigorous experiments in the future.
In addition to the common Th cells, there is increasing interest in the role of cytotoxic CD4+ T cells in different pathologies. With granzyme B and perforin, CD4+ T cells exert specific cytotoxicity on cells presenting pathogenic antigens and express immunomodulatory or exhaustion-related molecules, which is controlled through intrinsic IL-18R/MyD88 signaling.13,253,254 A previous study253 found that cytotoxic CD4+ T cells predominated among CD4+ T cells in the cardiac tissue of T. cruzi–infected mice and that the number correlated with the severity of myocardial inflammation.
3.8.2 CD8+ T cell subsets in myocarditis
CD8+ T cells, recognized primarily as cytotoxic T cells, usually kill infected cells via granzymes, perforins, and FAS-induced apoptosis.255 CD8+ T cells also take part in viral clearance by producing IFN-γ256 and TNF-α.257 Deficiency of CD8+ T cell with reduced IFN-γ impaired early CVB3 clearance and promoted the progression to chronic myocarditis.258 While type I IFN DNA therapy relieved chronic myocarditis by reducing CD8+ T cell aggregation in the myocardium after cytomegalovirus infection.259 These results indicated that CD8+ T cells are conducive to early viral clearance while appropriate reduced aggregation prevent the progression to chronic myocarditis. CD8+ T cells are also involved in autoimmunity. One previous study260 sequenced single-cell RNA and T cell receptors in the context of cardiac immune infiltration in Pdcd1Ctla4 mice (an ICI-MC animal model), and the investigator identified CD8+ T cells with clonal effects as the predominant cell population. Treatment with anti-CD8–depleting antibodies improved survival in Pdcd1Ctla4 mice. In both mice and patients with ICI-MC, this study suggested that α-myosin may be an important autoantigen in ICI-MC and highlights the key pathogenic role of cytotoxic CD8+ T cells. Similar immune intolerance also happened in A/J mice with EAM.261 Cytotoxic CD8+ T cells attack tissues that have lost their immune tolerance, just as they attack allografts. This process may be regulated by the surrounding tissues that express PD-L1. In cardiac transplantation,262 deletion of PD-L1 expression in graft endothelial cells may play a role in regulating CD8+ T cell infiltration. Results from mouse models suggest that loss of PD-L1 in graft endothelial cells may promote the infiltration of CD8+ T cells, thereby promoting allogeneic responses and rejection.
3.8.3 Other phenotypic T cells in myocarditis
Komarowska et al.263 described that T cells expressing c-mesenchymal epithelial transition factor (c-Met) mediated T cell cardiophilia during lymph node initiation by interacting with cardiac-produced hepatocyte growth factor. c-Met signaling facilitated T cell recruitment to the heart by inducing the release of autocrine CCL5. The authors found that autoantigen-specific c-Met T cells were increased in the circulation and myocardium of patients with inflammatory cardiomyopathy, representing loss of cardiac immune tolerance. Unlike c-Met− T cells, c-Met+ T cells preferentially proliferate in the myocardium and produce multiple cytokines (IL-4, IL-17, and IL-22) to exert pathogenic effects.264 During the immune response of myocarditis, is it possible to protect the myocardium from excessive inflammatory damage by temporarily targeting c-Met inhibition, thereby reducing excessive T cell migration to the myocardium. This provides a new idea for the treatment of myocarditis.
In addition, existing studies on the role of T cells in myocarditis are mainly directed at αβT cells. Here, we briefly summarize the role of γδT cells, which are named according to the T cell receptor type. These cells mainly recognize CD1-presenting molecules and have anti-infective and antitumor effects, but they also secrete a variety of cytokines, such as IFN-γ, ILs, and TNF, upon activation to exert immunomodulatory effects.265 Several studies266–268 in viral myocarditis models have shown that γδ T cells promote the Th1 cell inflammatory response or memory function through Fas-Fas ligand interaction, restriction of Treg cell activation, and IFN-γ secretion, which also subsequently promotes the effects of self-reactive CD8+ cells. The effects of limiting Treg cells may be through the killing of Treg cells by Vγ4+γδ T cells via recognition of CD1d expressed on Treg cells using caspase-dependent apoptosis. However, these studies are far from sufficient to reveal the veil of γδT cells.
3.9 B lymphocytes
B lymphocytes, otherwise known as B cells, can be divided into 2 groups depending on their origin: B1 cells, which originate mainly from the yolk sac or fetal liver, and B2 cells, which originate mainly from the bone marrow. Both B1 and B2 cells participate in adaptive immunity, differentiating into memory B cells or secreting plasma cells that produce specific antibodies, provided that BCR and costimulatory signals are activated.269 A recent study270 used the term “regulatory B” (Breg) cell to describe the ability of B cells to suppress the immune response, and this ability to regulate immunity was present in almost all B cell subsets.
Another study showed that B cells act as a dissemination vector271 in the early stages of CVB3-induced myocarditis, and that CVB3 can directly invade and replicate in mouse B cells. Using CVB3-infected B cells as a vector for CVB3 transfer to various tissues of the body, CVB3-infected B cells were detected in the hearts of mice with CVB3-mediated myocarditis. When the virus expands substantially, leading to host cell death, CVB3 is released into local tissues and continues to infect other cells.
The main function of B cells, as an important part of humoral immunity, is antibody production. The roles of these antibodies are to neutralize toxins through antigen binding by the Fab segment, activate complement, exert regulatory effects through the Fc segment, and exert antibody-dependent cell-mediated cytotoxic effects in concert with NK cells and macrophages, among others. However, antibodies are a double-edged sword, as some low-neutralizing activity or non-neutralizing virus-specific antibodies promote viral entry into, and massive replication in, immune cells through the antibody-dependent enhancement effect, an effect that has been demonstrated in CVB3272 and COVID-19.273 Thus, the pathogenic role of antibody-dependent enhancement cannot be ignored in the pathogenesis of this virus-associated myocarditis. Anticardiac antibody–mediated myocardial injury also plays a pivotal role in the pathogenesis of myocarditis. A variety of anti-heart antibodies (AHAs) have been identified in patients with myocarditis and in animal models of CVB3-induced myocarditis and EAM, including antimyocardial contractile protein antibodies, antimyocardial membrane protein receptor antibodies, antimyocardial mitochondrial antibodies, and antimyocardial structural protein antibodies.274,275 Part of the production of these antibodies may be related to the presence of highly homologous sequences between certain viral proteins and myocardial proteins that can be mimicked by antigenic molecules to produce anticardiac antibodies. Alternatively, autoantigens that leak from cardiomyocytes after damage can cause B cells to produce AHA-mediated myocardial injury. Recently, researchers214 also identified autoreactive T cells that target TN-C, a component of the cardiac ECM, so further studies are needed to determine whether anticardiac ECM antibodies play a role in the pathogenesis of myocarditis.
B cells internalize antigens bound to B cell receptors and process them toward the presentation of T cell antigenic peptide-MHC class II molecular complexes to activate T cells. By targeting disruption of the IgM transmembrane structural domain or using anti-IgM antibodies from birth to establish a mouse model of B cell deficiency, a previous study observed comparable myocardial inflammation in B cell–deficient mice immunized simultaneously with cardiac myosin vs in wild-type mice. They inferred that macrophages or DCs, but not B cells, were the key antigen-presenting cells in the pathogenesis in EAM.276 In addition, Cen et al.277 found that myocardial pathology scores and cardiac troponin T levels in B cell–deficient mice and severe combined immunodeficient mice were lower than in wild-type mice and B cell–reconstituted severe combined immunodeficient mice, respectively, in CVB3-induced myocarditis. B cells demonstrate more powerful antigen presentation and the ability to function independent of T cells. In addition, trehalose278 alleviates myocardial inflammatory injury in CVB3-induced myocarditis by inducing B cell autophagy mediated by the AMP-activated protein kinase/ULK1 signaling pathway.
B cells secrete a variety of cytokines, including IL-2, IFN-γ, IL-12, IL-6, IL-4, TNF-α, IL-10, and IL-17, which play antipathogenic, proinflammatory, profibrotic, anti-inflammatory, and immunomodulatory roles. Here, we focused on the role of B cells in regulating the activity of other immune cells. Cen et al.277 observed a decrease in the number of Th1 and Th17 cells, no difference in Th22 cells, and an increase in Th2 cells in the spleen of B cell knockout mice compared with wild-type mice in CVB3-induced myocarditis. This trend was also demonstrated in the quantitative polymerase chain reaction results for Th1, Th2, Th17, and Th22 cell–specific transcription factors, and was also verified in in vitro cellular assays. This may be related to the fact that B cells overexpressing CD80 activate CD4+ T cells, which in turn promotes Th1 and Th17 cell differentiation.279 In contrast, Wei et al.280 found that IL-10–producing B cells are T cell dependent in the pathogenesis of CVB3-induced myocarditis. They mainly downregulate T-bet and RORγt mRNA early in the pathogenesis, and they reduce the proportion of Th1 and Th17 cells, thereby suppressing inflammation and injury in the myocardium.
In conjunction with the proposed concept of Breg cells, we should not focus only on the pathogenic role of B cells; rather, we should consider their potential protective role. In addition to secreting IL-10 to exert immunosuppressive effects, Breg cells also act by affecting Treg cells. Lu et al.281 found that B cell knockout, although alleviating the severity of myocardial inflammation in CVB3-induced myocarditis, significantly reduced the number and function of Treg cells in the spleen, blood, and heart of infected mice. This situation was reversed by B cell transmigration. Furthermore, coculture of purified B cells from wild-type mice with effector T cells in vitro allowed their conversion to Treg cells. It can be concluded that B cells contribute to the homeostasis of Treg cells in myocarditis.
In addition to interacting with T cells, B cells can increase myocardial inflammation in CVB3 mice by inhibiting M2 macrophage polarization. Li et al.282 found that in CVB3-induced myocarditis, the number of M2 macrophages was increased in the B cell knockout group compared with the wild-type group, whereas after B cell reconstitution, the number of M2 macrophages tended to decrease, and myocardial pathology scores were significantly higher. This may be related to the increased production of certain cytokines, such as IFN-γ, which promote the polarization of M1 macrophages, and the relatively low production of other cytokines, such as IL-4, which promote the polarization of M2 macrophages.277
4. Single-cell sequencing in myocarditis
scRNA-seq is a high-resolution transcriptomics sequencing technology that maps gene expression profiles and differential profiles at the single-cell level through novel technologies and probes tissue heterogeneity issues, cell development issues, and the discovery of new cell populations through in-depth and detailed analysis of complex cell populations. In addition, there is single-cell T cell receptor sequencing and single-cell B cell receptor sequencing based on the expansion of scRNA-seq. Single-cell sequencing studies in the context of myocarditis have led to a deeper understanding of these major immune cells. Hua et al.20 used scRNA-seq on the hearts from BALB/c male mice with EAM to show that macrophages were the predominant immune cell population at all disease stages and identified an inflammation-associated population of macrophages that highly expressed hypoxia-inducible factor 1-α (HIF-1α). In terms of neutrophils, they found that neutrophils increased and released IL-1 to participate in early pathogenesis. Moreover, Th17 cells with upregulated HIF-1α expression constituted the major T cell population in the acute inflammatory phase, Treg cells were the major T cell population in the subacute inflammatory phase, and γδT cells releasing IL-17 were the major T cell population in the chronic cardiomyopathy phase. They proposed for the first time that the expression of HIF-1α correlates with the degree of inflammation.
In addition, Yoon et al.19 described for the first time the peripheral immune landscape of patients with myocarditis after BNT162b2 vaccination based on the analysis of peripheral blood mononuclear cells. They found the greatest change in the transcriptome profile of monocytes and detected highly increased mRNA expression of IL-7R in a T cell subpopulation, but it was not increased in terms of protein expression. This may give us a warning that scRNA-seq detects changes only at the transcriptional level. However, this may not necessarily reflect a change at the translational level (i.e. protein expression). The different results between transcription and translation remind us that something may happen during posttranscriptional modification that prevents the protein from being expressed, and noncoding RNAs may contribute to this outcome.15
Zhu et al.18 found a significant increase in clonal cytotoxic Temra CD8+ T cells in the blood of patients with ICI-MC, while a similar phenotype was found in the blood and myocardium of relevant animal models. This study described unique transcriptional changes in Temra CD8+ T cells, including chemokine upregulation (CCL5/CCL4/CCL4L2). This subpopulation of T cells has been described in myocarditis for the first time, which may provide a new interpretation in terms of myocarditis pathogenesis.
Lasrado et al.17 studied the hearts of male A/J mice with CVB3 myocarditis and found that M2 phenotype myeloid cells and T cells exhibited increased expression of unique transcription factors that regulate cardiac remodeling, while fibroblasts exerted immunomodulatory effects due to their distinct transcriptomic profiles and subtypes. In addition, this study revealed that the composition of cardiac ILCs includes NK cells, ILC2, and ILC3, confirming our suspicions in the previous section on ILCs regarding the composition of the ILC population in myocarditis. All of the studies described previously provide an immune and transcriptome landscape of the circulation and the heart for further exploration.
5. Conclusions and expectations
Different types of innate immune cells (Fig. 1) and adaptive immune cells (Fig. 2) are involved in myocarditis development and they form a complex immune network. They play different roles in the pathogenesis of myocarditis independently or cooperatively during the acute infection, subacute immune response, and cardiac remodeling. The diversity of immune cells imply that a single pathological classification based on the major infiltrating cell types in myocardial tissue is gradually failing to meet the needs of diagnosis and treatment in clinical practice. Cooper283 suggested the identification of the immunophenotype in patients considering a combination of clinical features and high-dimensional immunological data, could be an important milestone on the road to improving the treatment and prognosis of patients with acute myocarditis. To better elaborate, select high-dimensional immunological data, and find the key intersection of the immune network, the application of single-cell sequencing technology provides a viable approach to extensive analysis of cell signaling and immunomodulatory effects in the internal transcriptome of various immune cells and their subsequent immune products.
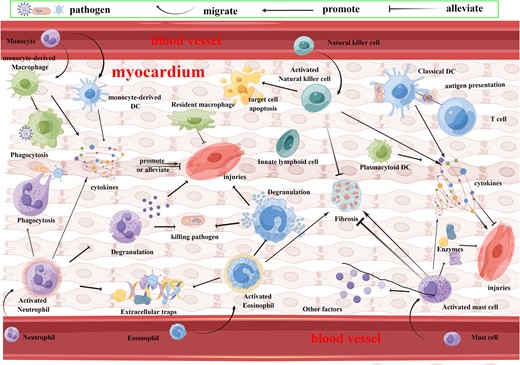
Innate immune cells in myocarditis. Innate immune cells in myocarditis include neutrophils, eosinophils, mast cells, ILCs, monocytes, macrophages, and DCs. When suffering from myocarditis, activated neutrophils kill pathogens through phagocytosis, degranulation, extracellular traps, and cytokines, while activating the myocardial inflammatory response to produce myocardial injuries. Activated eosinophils harm the myocardium and promote fibrosis while killing pathogens through degranulation and extracellular traps. Activated mast cells produce a variety of enzymes and cytokines involved in the pathogenesis of myocarditis, with both pro- and antifibrotic effects. Activated NK cells clear infected target cells while they inhibit myocardial fibrosis. Monocytes migrate into the heart and differentiate into macrophages and DCs. Macrophages can phagocytose pathogens, and they also produce pro- and anti-inflammatory cytokines. DCs can both activate T cells through alloantigen or self-antigen presentation, while they can secrete a variety of cytokines involved in myocarditis. Figure created using FigDraw.
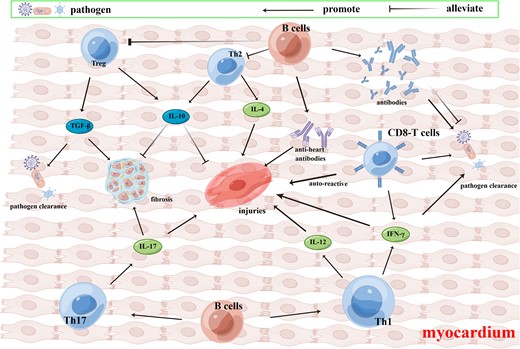
Adaptive immune cells in myocarditis. Adaptive immune cells in myocarditis include CD4 T cells (Th1, Th2, Th17, and Treg cells) and CD8 T cells. When experiencing myocarditis, Th1 cells secrete IL-12 and IFN-γ. IL-12 and IFN-γ promote inflammation and myocardium injuries, while IFN-γ is conducive to pathogen clearance. Th2 cells secrete IL-4 and IL-10, while IL-4 is proinflammatory and IL-10 is anti-inflammatory, antifibrotic, and can alleviate myocardium injuries. Th17 cells secrete IL-17, which promotes myocardium injuries and fibrosis. Treg cells produce IL-10 and TGF-β, and TGF-β can promote pathogen clearance in the acute phase, while it promotes myocardium fibrosis in the chronic phase. CD8 T cells can remove pathogens via granzyme and perforin, as well as produce IFN-γ. Autoreactive CD8 T cells in myocarditis can cause myocardium injuries. B cells secret antibodies to remove pathogens or promote viral entrance via an antibody-dependent enhancement effect. AHAs produced by B cells damage the myocardium. B cells in myocarditis promote Th1 and Th17 differentiation, while they suppress Th2 and Treg differentiation. Figure created using FigDraw.
Author contributions
K.L. reviewed the literature and wrote the manuscript. B.H. revised the manuscript. Both authors read and approved the final manuscript.
Funding
This study was supported by the Jinan Science and Technology Bureau (after subsidy) (no. 202134015) and the Taishan Scholar Foundation of Shandong Province (no. 201511099).
References
Author notes
Conflict of interest The authors declare that they have no conflict of interest.

{kind=link}
{kind=link}