





Abstract
Hepatic ischemia–reperfusion (IR) injury is a critical issue during liver transplantation (LT). Recent studies have demonstrated that IL-17a contributes to IR injury and steatohepatitis. However, the underlying mechanism is not understood. This study aimed to examine the role of IL-17a on hepatic IR injury in fatty liver and to investigate the underlying mechanisms. The correlation between serum IL-17a levels and liver function was analyzed in LT patients receiving fatty (n = 42) and normal grafts (n = 44). Rat LT model was applied to validate the clinical findings. IL-17a knockout (KO) and wild-type mice were fed with high-fat diets to induce fatty liver and subjected to hepatic IR injury with major hepatectomy. Frequency of circulating neutrophils and IL-17a expression on PBMCs were analyzed by flow cytometry. Mitochondrial outer membrane permeabilization (MOMP) was examined by a living intravital image system. Serum IL-17a was elevated after human LT, especially with fatty grafts. The aspartate aminotransferase and alanine transaminase levels were increased in recipients with fatty grafts compared with normal grafts. In rat LT model, the intragraft IL-17a expression was significantly higher in fatty grafts than normal ones post-LT. KO of IL-17a in mice notably attenuated liver damage after IR injury in fatty liver, characterized by better-preserved liver architecture, improved liver function, and reduced neutrophil infiltration. MOMP triggered cell death after hepatic IR injury in a caspase-independent way via IL-17a/NF-κB signaling pathway. KO of IL-17a protected the fatty liver against IR injury through the suppression of neutrophil infiltration and mitochondria-driven apoptosis.
1 Introduction
Interruption of liver blood supply during surgical resection, trauma, or transplantation upon reperfusion rapidly leads to liver damage in an antigen-independent way, called hepatic ischemia–reperfusion (IR) injury.1 It causes up to 10% of early organ failure with a higher incidence of both acute and chronic graft rejection after liver transplantation (LT).2,3 During hepatic IR injury, Kupffer cells, CD4+T cells, and neutrophils are critical cellular players in the sequence of events that induce ATP depletion and formation of reactive oxygen species in the ischemic liver.4,5 A variety of cytokines, chemokines, and complement proteins are released and communicate among the cellular players.4 Hepatic IR injury also increases the risks of post-LT tumor recurrence and metastasis by activating hypoxia and angiogenesis signaling pathways for tumor cell growth, migration, and invasion.6,7 On the other hand, due to the shortage of liver donors, using marginal liver grafts, including fatty liver (steatosis less than 30%) in LT is feasible and acceptable in selected patients.8,9 However, donor steatosis increases the risk of postoperative morbidity and mortality.10–12 An emerging consensus suggests that steatosis livers are particularly vulnerable to hepatic IR injury.13–15 But the underlying mechanism is not fully understood.
IL-17a is a member of the Th17 family of cytokines, mainly produced by CD4+Th17 cells, innate lymphoid cells, and typical innate immune cells.16–18 IL-17a plays key regulatory roles in host autoimmunity, various inflammation diseases, and even tumor immunity.19 Recent studies have determined its proinflammatory role in myocardial, intestinal, and hepatic IR injury by regulating parenchymal cell apoptosis and neutrophil infiltration.20–22 Gomes et al.23 showed that metabolic inflammation induces nonalcoholic steatohepatitis in an IL-17a-dependent way, suggesting the role of IL-17a in linking inflammation and fat metabolism. IL-17a signaling activation leads to increased responsiveness to inflammatory stimuli and recruitment of neutrophils to the liver in the condition of overnutrition. It thus induces insulin resistance and fatty acid release in the development and progression of nonalcoholic fatty liver disease to steatohepatitis.24
Given that IL-17a plays the role both in inflammation and fatty liver disease, we hypothesized that activation of the IL-17a signaling might causally contribute to hepatic IR injury in fatty liver. The current study provided, to our knowledge, the first evidence for a critical role for IL-17a signaling in driving hepatic IR injury in fatty liver.
2 Methods
2.1 Clinical samples
From January 2010 to January 2017, 86 patients (fatty graft: n = 42; normal graft: n = 44) who underwent LT in Department of Surgery, Queen Mary Hospital, Hong Kong were included in the current study. The ethical approval [UW11-100 (HKU/HA HKW IRB)] was obtained from the University of Hong Kong and Hospital Authority-Hong Kong Western Cluster and the consents were signed by the LT patients. The levels of fatty change ranged from 5% to 20% with the average of 9.5%. The blood samples were collected before and after LT for detecting the IL-17a levels in serum. The graft samples were collected at 2 h after portal vein reperfusion after LT for total RNA extraction. Informed consent was signed from each patient before operation. alanine transaminase (ALT) and aspartate aminotransferase (AST) levels of the recipients after LT were collected.
2.2 Rat orthotopic LT
Sprague Dawley rats (250–350 g, male) were purchased from the Laboratory Animal Unit, The University of Hong Kong. Rat LTs were conducted in 2 groups: normal graft group (n = 12 pairs) and fatty graft group (n = 12 pairs). All the recipient rats were fed with normal diet. The donors in fatty graft group were fed with high-fat diet (a purified Test Diet 58G8 with 45% energy from fat) for 2 weeks to develop fatty livers, whereas the donors in normal graft group were fed with normal diet. Liver tissues of 6 recipients per group were sampled at 2, 6, and 24 h after LT. The details of surgical procedure of rat LT have been described in our previous article.25,26
2.3 Mouse hepatic IR injury and major hepatectomy
Inbred male C57BL/6N (6 weeks old, male) mice were purchased from the Laboratory Animal Unit, the University of Hong Kong. The IL-17a knockout (KO) mice of C57BL/6N background were kindly provided by Dr. Yoichiro Iwakura at The Institute of Medical Science, The University of Tokyo, Japan. The experimental protocol was approved by the committee on the Use of Live Animals in Teaching and Research, the University of Hong Kong (4055-16). Fatty liver in mice was induced by feeding with high-fat diet for 2 weeks. The degree of fatty changes in the liver were determined by Oil-Red-O staining (Supplemental Fig. 1). IL-17a KO and wild-type (WT) mice with or without fatty liver were subjected to partial hepatic IR injury and major hepatectomy to mimic the pathologic damage in the graft injury during LT. The procedure of mouse hepatic IR injury and major hepatectomy was described previously.27,28 Briefly, the branch of hepatic artery and portal vein to the right and triangle lobes were clamped for 45 min by a micro vessel clamp, followed by release of the clamp. Major hepatectomy of the left and caudate lobes was performed during the ischemia duration. The mice were sacrificed at 6 h after portal vein reperfusion. The circulating blood and liver tissues underwent hepatic IR injury were sampled.
2.4 Mitochondrial outer membrane permeabilization evaluation in mouse after hepatic IR injury
Mitochondrial outer membrane permeabilization (MOMP) evaluation was performed using a living intravital image system at 6 h after hepatic IR injury in the IL-17a KO and WT mice with fatty liver. After anesthesia, the mice were injected with rhodamine123 (to assay mitochondrial membrane potential in populations of apoptotic cells), propidium iodide (to stain nucleic acids released from dead cells), and Hoechst (to stain nuclear in live cells) in PBS solution (i.p.). The lobe of the liver after IR injury was exposed and observed under the Confocol microscopy.
2.5 ELISA for human and mouse IL-17a levels in serum
The human (No. ab216167; Abcam, Cambridge, UK) and mouse (No. ab199081; Abcam, Cambridge, UK) IL-17a ELISA kits were used to detect the serum IL-17a levels according to the manufacturer’s introduction. The 96-well plate was precoated with anti-human/anti-mouse IL-17a antibody. The human/mouse serum samples (50 μl/well) with dilution buffer were added into the wells and the plate was incubated for 1 h at room temperature. The plate was washed for 3 times with washing buffer. Then secondary antibody conjugated with HRP was added for 30 min incubation at room temperature. Hundred microliters of 3,3′,5,5′-tetramethylbenzidine development solution was added to each well, followed by 100 μL stop solution. Finally, the plate was read at both 450 and 570 nm in an absorbance microplate reader. Standard curve was made with the standard IL-17a samples and all experimental values fell on the standard curve.
2.6 H&E staining and immunohistochemical staining
The human graft tissues from LT surgery and mouse liver tissues from hepatic IR injury model were collected. The tissue samples were fixed with formalin and embedded with paraffin. Paraffin sections (4 mm thick) were dewaxed in xylene, rinsed in grade alcohol, and rehydrated in tap water. The sections were stained with H&E for histologic examination, called H&E staining. For immunohistochemical (IHC) staining, the sections were placed in citric buffer (pH 6.0) and heated to 100°C for 20 min in a microwave. Afterwards, the sections were treated with 10% FBS for 30 min followed by primary antibodies (at 1:100 dilution) for 1 h at 4°C. Then, the sections underwent blocking with 3% peroxidase for 30 min and proper secondary antibody for 1 h. Positive staining signals were developed with 3,3′-diaminobenzidine substrate solution (DakoCytomation, Glostrup, DK). The sections were finally stained with hematoxylin solution. Anti-mouse Ly-6G antibody (purchased from Abcam) was used for neutrophil staining.
2.7 TUNEL assay
TUNEL assay was performed to detect DNA fragmentation during hepatocellular apoptosis in mouse liver. The tissue slides were incubated with 1 μg/ml proteinase K for 15 min at room temperature. After 2 times of PBS washes, the slides were incubated with TUNEL solution for 60 min at 37°C in the dark. The slides were then washed with PBS for 3 times followed by incubation with a chromogenic HRP substrate solution for 30 min at room temperature. The brown signals were generated in apoptosis cells.
2.8 Oil-Red-O staining
The fatty liver tissues were fixed with O.C.T. compound. The samples were cut into 5 nm-thick sections. The sections were stained with a saturated oil-red-O solution diluted in isopropanol and then incubated at room temperature for 1 h. The oil-red-O solution was washed with 70% ethanol then followed by 3 times of PBS washes. Light microscope was applied to observe the red staining for cytoplasm lipid.
2.9 Real-time RT-PCR
RT-PCR was performed to detect targeted gene expressions at mRNA levels. Total RNA was extracted from clinical tissue samples and animal models using Trizol reagents (ThermoFisher Scientific, Waltham, MA, US) according to the manufacturer’s instruction. Reverse transcription was performed using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA, US) for cDNA synthesis. The qPCR reaction mixture per well was prepared as follows: 2 μl cDNA, 10 μl SYBR Green, 0.1 μl primer mix (10 pmol/μl) and 7.9 μl distilled H2O. The PCR running program was set up as follows: 50°C, 2 min for 1 cycle; 95°C, 10 min for 1 cycle; 95°C, 15 s with 60°C, 1 min for 40 cycles; 95°C, 15 s for 1 cycle; 60°C, 1 min for 1 cycle; and 95°C, 15 s for 1 cycle. The RT-PCR results were analyzed by ViiATM 7 software and then calculated using the 2^−△△T method. Gene expression levels were showed as the fold changes relative to the normal liver. Primer sequences were provided in the supplementary documents (Supplemental Table 1).
2.10 Flow cytometry
Flow cytometry was performed to analyze the profile of IL-17a expression on T cells, NK cells and neutrophils in mice. The fresh blood samples were collected from the circulation of the mice. The red blood cells were eliminated by RBC lysis buffer. The immune cells were analyzed by CytoFLEX Model S (Beckman, Brea, CA, US) using the following antibodies: PE-anti-mouse CD45 (Biolegend, San Diego, CA, US; dilution 1:200), APC-anti-mouse IL-17a (Biolegend; dilution 1:200), FITC-anti-mouse Ly-6G (Biolegend; dilution 1:200), APC-A750-anti-mouse CD11b (Biolegend; dilution 1:200), FITC-anti-mouse F4/80 (Biolegend; dilution 1:200), FITC-anti-mouse NK1.1 (Biolegend; dilution 1:200), PB450-anti-mouse CD3 (Biolegend; dilution 1:200), and FITC-anti-mouse CD4 (Biolegend; dilution 1:200). FACS gating strategy was outlined in Supplemental Fig. 2).
2.11 Western blot
Total protein was extracted from clinical tissue samples and animal models using protein lysis buffer with 1% protease inhibitor. Primary monoclonal antibodies against β-Actin, cleaved caspase-3, caspase-3, Bad, Bcl-2, phosphorylated NF-κB, and NF-κB were purchased from Cell Signaling Technology (Danvers, MA, USA). The protein samples were denatured by boiling at 100°C for 10 min with loading buffer. The Western blot was conducted with equal amounts of protein samples along with 1 molecular weight marker in the SDS-PAGE gel. The gel was run in the running buffer (25 mM Tris base, 190 mM glycine, and 0.1% SDS) for 3 h at 100 V. Subsequently, the gel was run in the transfer buffer (25 mM Tris base, 190 mM glycine, and 20% methanol) for 1 h at 90 V. The proteins were transferred to the polyvinylidene fluoride or polyvinylidene difluoride (PVDF) membrane. The PVDF membrane was blocked with 5% nonfat dry milk for 2 h at room temperature followed by incubation in primary antibodies (1:1000 dilution) overnight at 4°C and secondary antibodies (1:2000 dilution) for 2 h at room temperature. Finally, protein bands were detected using ECL reagent (GE Life Sciences, Chicago, IL, US) and developed using darkroom development techniques.
2.12 Statistical analysis
All data were shown as the mean ± standard deviation. Differences between experimental and control groups were assessed by Mann–Whitney analysis using GraphPad Prism 5.0 software. The correlation between 2 experimental data sets was analyzed by Person Correlation Coefficient. A P value less than 0.05 was considered statistically significant.
3 Results
3.1 IL-17a was up-regulated after human LT with fatty graft
To evaluate the role of IL-17a on fatty graft injury in LT, the serum IL-17a levels in LT recipients were detected at 2 h after human LT by ELISA. The level of serum IL-17a in recipients with fatty grafts (n = 42) was significantly higher than that in recipients with normal grafts (n = 44) (Fig. 1A). The liver function after LT was compared between LT recipients receiving fatty grafts and normal grafts. Higher levels of serum AST and ALT were observed in recipients with fatty grafts than those in recipients with normal grafts (Fig. 1B), possibly implying that IL-17a participated in mediating fatty graft injury. The mRNA levels of intragraft IL-17a and proinflammatory molecular mediators at 2 h after LT was examined by RT-PCR. The expression of IL-17a in fatty grafts were significantly higher than those in normal grafts (P = 0.044), as well as TLR4 (P = 0.029), IL-1β (P = 0.047), IL-6 (P = 0.019), CXCL1 (P = 0.041), and CXCL2 (P = 0.032) mRNA levels (Fig. 1C). There was a positive correlation between intragraft mRNA expression of IL-17a and CXCL1 in fatty grafts (Fig. 1D), suggesting IL-17a might play a pivotal role in fatty graft injury during LT by promoting neutrophil infiltration.
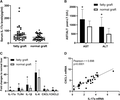
IL-17a was up-regulated after human orthotopic liver transplantation with fatty graft. (A) The serum IL-17a levels in LT recipients were detected at 2 h after LT by ELISA. (B) The serum AST/ALT levels at 2 h after LT were compared between LT recipients with fatty grafts and normal grafts. (C) The mRNA levels of intragraft IL-17a, TLR4, IL-1β, IL-6, CXCL1, and CXCL2 at 2 h after LT were examined by RT-PCR. The data were present as the fold changes to normal liver from healthy donors. (D) The correlation between intragraft IL-17a mRNA and CXCL1 mRNA expression in fatty grafts was analyzed using Person Correlation Coefficient. *P < 0.05 between groups of fatty graft and normal graft
3.2 IL-17a promoted graft injury in rat orthotopic LT with fatty graft
To validate the association between intragraft IL-17a expression and fatty liver graft injury, we established rat orthotopic LT models with fatty and normal grafts. The intragraft IL-17a mRNA expressions were detected by RT-PCR at 2, 6, and 24 h after LT. The IL-17a expression was up-regulated after LT with the peak at 6 h in both fatty and normal graft groups (Fig. 2A). The IL-17a expression was significantly higher in the fatty graft group compared to normal graft group both at 6 and 24 h after LT (Fig. 2A). The intragraft expressions of TLR4, IL-1β, IL-6, CXCL1, and CXCL2 at 6 h after rat LT were detected by RT-PCR. As expected, the intragraft mRNA levels of TLR4, IL-1β, IL-6, CXCL1, and CXCL2 were up-regulated in the group of fatty grafts compared with normal grafts, respectively (Fig. 2B). H&E staining of the graft tissues showed that poorer liver architecture was found in fatty grafts compared with normal ones at 6 h after LT, characterized by more hepatic necrosis and inflammatory cell infiltration (Fig. 2C). Thus, IL-17a was critical for IR-triggered proinflammatory response at early phase after LT, especially with fatty grafts.
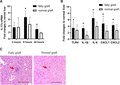
IL-17a was up-regulated after rat orthotopic liver transplantation with fatty graft and promotes graft inflammation. (A) The intragraft IL-17a mRNA expressions were detected by RT-PCR at 2, 6, and 24 h after rat LT. (B) The intragraft expressions of TLR4, IL-1β, IL-6, CXCL1, and CXCL2 at 6 h after rat LT were detected by RT-PCR. *P < 0.05 between groups of fatty graft and normal graft. (C) Representative H&E stained liver sections from graft tissues of fatty graft and normal graft at 6 h after rat LT. Red arrows pointed to the hepatocellular necrosis areas where were replaced by infiltrated inflammatory cells. Bar = 200 μm
3.3 IL-17a contributed to mouse hepatic IR injury in fatty liver by mediating neutrophil infiltration
To document the role of IL-17a on hepatic inflammation in fatty liver against IR injury, IL-17a KO and WT mice with or without fatty liver were subjected to 45 min of warm ischemia followed by reperfusion and major hepatectomy. The serum IL-17a levels in IL-17a WT mice were detected by ELISA at 6 h after hepatic IR injury. We observed serum IL-17a concentrations in mice with fatty liver were significantly higher than those with normal liver after IR injury (Fig. 3A). Then, the hepatocellular damage, liver function, and proinflammatory gene expressions in the liver were measured after hepatic IR injury. More infiltrated inflammatory cells were observed in the ischemia lobe with larger area of hepatic necrosis in IL-17a WT mice with fatty liver compared to normal liver after IR injury (Fig. 3B, upper panel), suggesting fatty liver was more vulnerable to hepatic IR injury. In contrast, liver architecture was better preserved in IL-17 KO mice compared with WT ones with fatty liver (Fig. 3B, left panel). Furthermore, serum ALT level was significantly decreased in IL-17 KO mice compared with WT mice in response to hepatic IR injury in both fatty and normal liver (Fig. 3C). We further detected the hepatic TLR4, IL-1β, IL-6, CXCL1, and CXCL2 mRNA expressions in the present mouse model by RT-PCR. The induction of TLR4, IL-1β, IL-6, CXCL1, and CXCL2 were down-regulated in IL-17a KO mice with fatty liver compared to WT mice at 6 h after hepatic IR injury (Fig. 3D). CXCL1 and CXCL2 are important chemotactic factors for neutrophil infiltration, thus we stained the neutrophils which initiated inflammation response during hepatic IR injury by IHC staining with anti-mouse Ly-6G antibody. The numbers of infiltrated Ly-6G+ cells were significantly higher in mice with fatty liver than those with normal 1 after hepatic IR injury (Fig. 4A). KO of IL-17a significantly decreased the number of infiltrated neutrophils both in fatty and normal livers (Fig. 4A). In normal IL-17a WT mice, the neutrophils in total CD45+ PBMCs were 4–8%, whereas the frequency of Ly-6G+CD11b+ cells increased up to around 48.0% at 6 h after hepatic IR injury in fatty liver (Fig. 4B). Notably, the frequency of Ly-6G+CD11b+ cells was about 4.1% in normal IL-17a KO mice and 4.3% at 6 h after hepatic IR injury in fatty liver (Fig. 4B). These findings confirmed the effects of IL-17a on hepatic IR injury with fatty liver by inducing neutrophil infiltration and that KO of IL-17a could preserve the liver function against IR-induced inflammatory response.
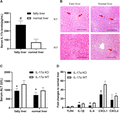
Knockout (KO) of IL-17a attenuated hepatic ischemia–reperfusion (IR) injury in mouse model with fatty liver. (A) The serum IL-17a levels in IL-17 WT mice with fatty liver and normal livers were examined by ELISA at 6 h after hepatic IR injury. (B) Representative H&E stained liver sections from liver tissues of IL-17 WT and KO mice with fatty liver and normal liver at 6 h after hepatic IR injury. Red arrows pointed to the hepatocellular necrosis areas. Bar = 200 μm. (C) To compare the liver function between IL-17a KO and WT mice in response to IR injury, serum ALT level at 6 h after hepatic IR injury was detected using the mouse ALT kit. (D) Hepatic TLR4, IL-1β, IL-6, CXCL1, and CXCL2 mRNA expressions at 6 h after IR injury in IL-17 KO and WT mice with fatty liver were detected by RT-PCR. (E) The neutrophils were stained by anti-mouse Ly-6G antibody in the groups of IL-17a KO and WT mice with or without fatty liver after hepatic IR injury. Bar = 100 μm. The bar chart represented the numbers of positive Ly-6G cells per field in each group. #P < 0.05 between groups of fatty liver and normal liver. *P < 0.05 between groups of IL-17a KO and WT mice

Knockout of IL-17a decreased neutrophil infiltration after hepatic ischemia–reperfusion (IR) injury in mouse model with fatty liver. (A) The neutrophils were stained by anti-mouse Ly-6G antibody in the groups of IL-17a KO and WT mice with or without fatty liver after hepatic IR injury. Bar = 100 μm. The bar chart (right) represented the numbers of positive Ly-6G cells per field in each group. #P < 0.05 between groups of fatty liver and normal liver. *P < 0.05 between groups of IL-17a KO and WT mice. (B) Representative dot-plots of Ly-6G+CD11b+ neutrophils from CD45+PBMCs in IL-17 WT and KO mice with or without hepatic IR injury. The bar chart (right) shows the mean percentage of Ly-6G+CD11b+ neutrophils in CD45+PBMCs (n = 6 in each group). ***P < 0.001
3.4 IL-17a was mainly derived from neutrophils during hepatic IR injury in fatty liver
Having confirmed the critical roles of IL-17a in hepatic IR injury and the correlation between IL-17a and neutrophil infiltration, we next explored the source of IL-17a. As mentioned above, IL-17a is produced by unconventional T cells, members of innate lymphoid cells, as well as typical innate immune cells, such as neutrophils and macrophages located in the epithelial barriers in respond to various inflammatory diseases.16–18 We performed flow cytometry to analyze the IL-17a expression on circulating CD4+ T cells, Ly-6G+ cells, F4/80+ cells, and NK cells in IL-17 WT mice with fatty liver before and at 6 h after hepatic IR injury. It was found that IL-17a was expressed on Ly-6G+ neutrophils and some of CD4+ T cells, F4/80+ cells and few NK cells (Fig. 5). Moreover, there was a significant increase of IL-17a expression on Ly-6G+ neutrophils, CD4+ T cells, F4/80+ cells after hepatic IR injury. In the current model of hepatic IR injury in fatty liver, Ly-6G+ neutrophils were the main source of IL-17a.
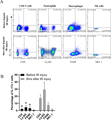
IL-17a was mainly produced by neutrophils during hepatic IR injury in the fatty liver. (A) Representative dot-plots of IL-17a expression on gated CD4+ cells, Ly-6G+ cells, F4/80+ cells, and NK1.1+ cells in IL-17a WT mice with fatty liver before and at 6 h after hepatic IR injury. (B) Mean percentage of intracellular IL-17+ staining on CD4+ cells, Ly-6G+ cells, F4/80+ cells, and NK1.1+ cells from the PBMCs of IL-17a WT mice with fatty liver (n = 6 in each group). *P < 0.05 between the groups before and after hepatic IR injury
3.5 IL-17a KO suppressed mitochondria-mediated apoptotic pathways in fatty liver
Activation of apoptotic pathways was involved in IR injury, we detected hepatic apoptosis by Tunel staining. It was observed that IL-17a deficiency suppressed the activation of hepatic apoptosis in both fatty and normal liver groups at 6 h after IR injury (Fig. 6A). The apoptosis is started from the impaired mitochondrial function. During apoptosis, MOMP basically leads to cell death in a way of no return, even in the absence of caspase activity.29,30 We further evaluated the MOMP using a living intravital image system. In the IL-17a WT mice, mitochondria were severely dysfunctional after hepatic IR injury, especially in the group of fatty liver, characterized by more MOMP signals (loss of green) in the injured hepatocellular areas with few nonviable hepatocytes, which were stained as red, as compared with IL-17a KO mice at 6 h after IR injury (Fig. 6B). These findings indicated that IL-17a promoted hepatocellular apoptosis by hepatic MOMP-induced apoptosis during hepatic IR injury in fatty liver.
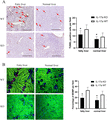
IL-17a knockout suppressed activation of mitochondria-mediated apoptosis after hepatic IR injury in fatty liver. (A) Representative figures of Tunel staining in the groups of IL-17a KO and WT mice with or without fatty liver after hepatic IR injury. Bar = 400 μm. The bar chart (right) represented the numbers of positive Tunel cells per field in each group. (B) Hepatic mitochondrial outer membrane permeabilization (MOMP) was evaluated using a living intravital image system. Representative figures of MOMP (loss of green) captured in the groups of IL-17a KO and WT mice with or without fatty liver after hepatic IR injury. Bar = 200 μm. The bar chart represented the percentages of MOMP per field in each group. *P < 0.05 between groups of IL-17a KO and WT mice
3.6 Activation of IL-17a/NF-κB signaling pathway promoted MOMP-induced inflammation
MOMP typically leads to cell death in a caspase-dependent or -independent way. Thus, we analyzed the cleaved caspase-3 expression by Western bolt. In the IL-17a WT mice, the cleaved caspase-3 expressions were notably blunted both in fatty liver and normal liver after IR injury as compared with KO mice (Fig. 7). We further found that the expression of proapoptosis Bad was up-regulated and the expression of antiapoptosis Bcl-2 was blunted in IL-17a WT mice after IR injury as compared with IL-17a KO mice (Fig. 7), suggesting the MOMP triggered cells death via a caspase-independent manner in the present mouse model. Unlike MOMP-induced caspase-dependent apoptosis, cells undergoing caspase-independent cell death displays a proinflammatory phenotype dependent on NF-κB activity in the dying cells.30 Given IL-17a can induce the activation of NF-κB signaling,31 the expression of phosphorylated NF-κB and NF-κB were detected by Western blot in the present mouse model. Strikingly, IL-17a deficiency significantly decreased the activation of the phosphorylated NF-κB at 6 h after hepatic IR injury in fatty liver and normal liver (Fig. 7). Our data demonstrated that KO of IL-17a decreased NF-κB activation, thus attenuated MOMP-triggered inflammation.

IL-17a activated NF-κB signaling pathway after hepatic IR injury in fatty liver. Western blots were performed on whole liver protein extracts with antibodies specific to cleaved caspase-3, total caspase-3, Bad, Bcl-2, phosphorylated NF-κB, and total NF-κB. β-actin expression was used as the loading control. Representative autoradiographs of (A) cleaved caspase-3, total caspase-3, (B) Bad, Bcl-2, phosphorylated NF-κB, and total NF-κB were shown in each group
4 Discussion
We have defined a role for IL-17a in the regulation of hepatic IR injury in fatty liver. The IL-17a expression was up-regulated after human LT, especially in fatty graft compared to normal graft. There was a positive correlation between intragraft mRNA expression of IL-17a and CXCL1, indicating IL-17a could regulate the intragraft infiltration of neutrophils. In contrast, KO of IL-17a in mice fed with high-fat diet showed an attenuated IR-triggered hepatic injury (i.e., preserved liver architecture, improved liver function, and reduced neutrophil infiltration). Our findings demonstrated that IL-17a accelerated the hepatic IR injury in fatty liver in both clinical samples and experimental animal models.
Accumulating studies have shown evidence that using liver grafts with steatosis increases the risks of complications after LT.14 However, the underlying mechanisms of the increased susceptibility to IR injury in steatosis livers are not fully understood. Recent findings have focused on the pathogenetic role of IL-17a in the regulation of adipogenesis and obesity-associated sequelae. IL-17a induces insulin resistance and fatty acid release, subsequently stored in the liver as triglycerides, accelerating the progression of nonalcoholic fatty liver disease (NAFLD) to nonalcoholic steatohepatitis (NASH).23,32,33 Consistent to the present study, the degree of fatty changes in the liver was much lower in IL-17a-deficient mice receiving high-fat diet for 2 weeks as compared with WT mice (Supplemental Fig. 1), suggesting that IL-17a was required for the adipogenesis in hepatocytes. Of note, IL-17a drives neutrophil chemokine expression by IL-17 receptor A positive cells in the liver,34 and the increase of hepatic neutrophil/lymphocyte ratio is a new marker for predicting the progression of NAFLD to NASH.35 Our data demonstrated the number of infiltrated neutrophils into the liver and the expressions of neutrophil chemokines CXCL1 and CXCL2 were dramatically reduced in IL-17a KO mice after IR injury compared with WT mice (Figs. 3D and 4A). CXCL1 and CXCL2 acts in a sequential way to attract neutrophils through venular circulation. CXCL1 mainly comes from TNF-stimulated endothelial cells, whereas CXCL2 is dominantly produced by neutrophils.36 TNF-α was produced by Kupffer cells within the duration of liver ischemia before reperfusion,37 then up-regulated CXCL1 expression and neutrophil infiltration. In our mouse hepatic IR injury model, the IL-17a was mainly produced by Ly-6G+ neutrophils (Fig. 5). IL-17a is involved in the expression of TNF-α in the subacute phase after hepatic IR injury,38 that amplifies TNF-α/CXCL1 induced neutrophil responses. These findings suggested that there was a positive loop among IL-17a, TNF-α, CXCL1, and neutrophil infiltration. The IL-17a signaling was activated before IR injury in fatty liver, that might explain why steatosis livers were more vulnerable to IR injury. However, as shown in Figs. 4A and 6B, in absence of IL-17a, the hepatic neutrophil infiltration and apoptosis were still induced by high-fat-diet, implicating IL-17a independent mechanisms for increased neutrophil infiltration and apoptosis in fatty liver upon hepatic IR injury, which needs further investigation.
Except for the amplification effects of neutrophil-induced inflammatory responses, IL-17a promoted the mitochondria-driven cell death, which was an underlying cause of IR injury. Mitochondria regulate caspase activation and cell death through an event termed MOMP, which is often considered a point of no return because it typically leads to cell death, even in the absence of caspase activity.29 MOMP independent of caspase is highly regulated through interactions between proapoptotic and antiapoptotic members of the Bcl-2 family.29 In current study, we first observed MOMP was increased after hepatic IR injury in IL-17a WT group (Fig. 6B). We further demonstrated the up-regulation of proapoptosis Bad but not cleaved caspase-3 in IL-17a WT mice rather than IL-17a KO mice after IR injury (Fig. 7), implying the MOMP triggered cells death via a caspase-independent manner in the present mouse model. Caspase-3 is frequently activated during cell apoptosis, dependent or independent of cytochrome c release from the mitochondria.39,40 Cruz reported that the activation of caspase-3 is down-regulated by IL-17a activity.39 Consistent with Cruz’s study, we found the cleaved caspase-3 expression was elevated in IL-17 KO group instead of WT group, indicating caspase-3 signaling was not activated after hepatic IR injury. Unlike mitochondrial apoptosis, cells undergoing caspase-independent cell death display a proinflammatory phenotype dependent on NF-κB activity in the dying cells.30 The NF-κB activation was increased in IL-17a WT mice instead of IL-17a KO mice after IR injury (Fig. 7), indicating IL-17a was required for the activation of NK-κB signaling and the NK-κB-dependent MOMP-triggered inflammation. Hereby, KO of IL-17a attenuated mitochondria-driven cell apoptosis and inflammation by inhibition of NK-κB activation.
In conclusion, in the present study, IL-17a was demonstrated for the first time to be involved in regulation of hepatic IR injury in fatty liver, through the recruitment of the neutrophils and increases in mitochondria-driven apoptosis. Understanding the cellular and molecular events during hepatic IR injury in fatty liver is fundamental to developing therapeutic strategies in order to benefit more LT patients.
Authorship
X.X.Y. and K.M. designed the present study. X.X.Y., C.X.L., K.T.P.N., J.L., H.L., and W.Y.Z. and F.X. conducted the experiments and analyzed the data. X.C.L., C.M.L., L.W.L., and K.M. were responsible for project administration. X.X.Y. drafted the manuscript. K.M. edited and revised the manuscript. All the authors reviewed the manuscript.
Abbreviations
- ALT
alanine transaminase
- AST
aspartate aminotransferase
- IHC
Immunohistochemical
- KO
knockout
- MOMP
mitochondrial outer membrane permeabilization
- PVDF
polyvinylidene fluoride or polyvinylidene difluoride
Acknowledgments
This study was financially supported by General Research Fund (17122517) and Theme-based Research Funding (T12-703/19R) from Research Grant Council, Hong Kong.
Disclosures
The authors declare no conflicts of interest.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}