




Abstract
In recent years, pembrolizumab has demonstrated significant efficacy in treating tumors characterized by a high tumor mutational burden and high microsatellite instability. Tropomyosin receptor kinase (TRK) inhibitors have shown considerable efficacy against tumors harboring neurotrophic receptor tyrosine kinase (NTRK) fusion genes, highlighting the growing importance of personalized medicine in cancer treatment. Advanced sequencing technologies enable the rapid analysis of numerous genetic abnormalities in tumors, facilitating the identification of patients with positive biomarkers. These advances have increased the likelihood of providing effective, tailored treatments. NTRK fusion genes are present in various cancer types, including sarcomas, and the TRK inhibitors larotrectinib and entrectinib have been effectively used for these malignancies. Consequently, the treatment outcomes for NTRK fusion-positive tumors have improved significantly, reflecting a shift toward more personalized therapeutic approaches. This review focuses on NTRK fusion-positive sarcomas and comprehensively evaluates their epidemiology, clinical features, and radiological and histological characteristics. We also investigated the treatment landscape, including the latest methodologies involving TRK inhibitors, and discussed the long-term efficacy of these inhibitors, and their optimal order of use. Notably, larotrectinib has demonstrated a high response rate in infantile fibrosarcoma, and its efficacy has been confirmed even in advanced cases. However, further research is warranted to optimize treatment duration and subsequent management strategies. The accumulation of clinical cases worldwide will play a pivotal role in refining the treatment approaches for tumors associated with NTRK fusion genes.
Introduction
In recent years, biomarker-based tumor-agnostic treatments such as pembrolizumab for tumor mutational burden (TMB)-high [1] and Microsatellite Instability (MSI)-high [2] tumors and tropomyosin receptor kinase (TRK) inhibitors for neurotrophic receptor tyrosine kinase (NTRK) fusion-positive tumors have been demonstrated. The advent of next-generation sequencing technologies has facilitated comprehensive analysis of a multitude of genetic aberrations in tumors within a relatively short timeframe. Consequently, uncommon biomarkers could be identified and treated effectively. Tumor-agnostic treatment based on biomarkers is anticipated to be highly effective, even in rare cancers, where conventional phase III randomized controlled trials are challenging to conduct. Some drugs have been approved for phase I/II trials [1,3,4].
TRK proteins are predominantly expressed in nervous tissues and regulate several crucial processes, including apoptosis, survival [3], and the development of sympathetic nerve axons [4]. To date, three distinct TRKs have been identified. TRK-A [5], -B [6], and -C [7] are encoded by the NTRK-1, -2, and -3 genes, respectively. When chromosomal rearrangements result in the fusion of different gene regions, leading to the formation of NTRK fusion genes [8], the gene product—the TRK fusion protein—is activated. This phenomenon can be described as the activation of the TRK kinase domain. This process is thought to contribute to tumor formation by influencing signal transduction pathways [9–11]. In 1983, Pulciani et al. [12] identified the TRK oncogenes for the first time in human colon carcinomas. In sarcoma research, NTRK fusion genes were initially identified by Knezevich and colleagues in infantile fibrosarcoma (IFS) [13]. Subsequently, many reports on NTRK fusion genes in sarcomas have been published, including an LMNA-NTRK1 fusion gene [14]. Notably, this case was the first instance of a sarcoma demonstrating responsiveness to TRK inhibitor therapy [14].
In a 2018 phase I/II trial [15], larotrectinib was approved by the Food and Drug Administration (FDA), and subsequent studies confirmed its favorable outcomes [16]. Entrectinib demonstrated a response rate of 57% in metastatic, locally advanced, or unresectable tumors positive for NTRK fusion genes, with an average response duration of 10 months [17], and was approved by the FDA [18].
This article focuses on NTRK fusion-positive sarcomas, excluding those arising from uterine and gastrointestinal stromal tumors (GIST), and provides a clinically oriented and practical review.
Epidemiology
A study of 295,676 patients with cancer revealed that the overall prevalence of NTRK fusion gene–positive tumors was 889 (0.30%) [19]. In adults aged ≥18 years, the prevalence of NTRK fusion gene–positive tumors is 0.28%, whereas in children aged <18 years, it is 1.34% [19]. The frequencies of NTRK fusion gene positivity by histological type of sarcoma are summarized in Table 1 [19] and described as follows: in the adult population, malignant peripheral nerve sheath tumor (MPNST) was the most frequently reported tumor type (2.97%), followed by fibrosarcoma (2.75%), inflammatory myofibroblastic tumor (IMT) (2.27%), soft tissue sarcoma (STS) not otherwise specified (NOS) (1.70%), and myxofibrosarcoma (1.31%). In the pediatric population, fibrosarcoma is the most frequently reported tumor type (52%), followed by solitary fibrous tumors (SFTs), including soft tissue hemangiopericytoma (20%), chondrosarcoma (bone, extraskeletal) (8.3%), undifferentiated soft tissue sarcoma (5.0%), and MPNST (4.65%) [19].
Frequency of NTRK fusion gene positivity by histological type of sarcoma (data are adapted from [19])
| Tumor type | Adults (≥18 years old) | Children (<18 years old) | ||||
|---|---|---|---|---|---|---|
| Total cases | NTRK-positive cases | Frequency (%) | Total cases | NTRK-positive cases | Frequency (%) | |
| Fibrosarcoma | 109 | 3 | 2.75 | 25 | 13 | 52 |
| Soft tissue leiomyosarcoma | 1421 | 4 | 0.28 | 6 | 0 | 0 |
| Malignant peripheral nerve sheath tumor | 337 | 10 | 2.97 | 43 | 2 | 4.65 |
| Inflammatory myofibroblastic tumor | 44 | 1 | 2.27 | 36 | 1 | 2.78 |
| Soft tissue angiosarcoma | 400 | 1 | 0.25 | 9 | 0 | 0 |
| Rhabdomyosarcoma (embryonal, alveolar, pleomorphic, NOS) | 315 (37,51, 33, 194) | 2 (0, 0, 1, 1) | 0.63 | 287 (90, 83, 1, 113) | 1 (0, 0, 0, 1) | 0.35 |
| Osteosarcoma (bone, extraskeletal) | 573 (503, 70) | 3 (2, 1) | 0.52 | 284 (279, 5) | 2 (2, 0) | 0.70 |
| Chondrosarcoma (bone, extraskeletal) | 514 (233, 281) | 1 (1, 0) | 0.19 | 12 (4, 8) | 1 (1, 0) | 8.3 |
| Ewing sarcoma (bone, extraskeletal, soft tissue primitive neuroectoderm tumor) | 410 (71, 292, 47) | 1 (0, 1, 0) | 0.24 | 198 (12, 177, 9) | 1 (0, 0, 1) | 0.51 |
| Synovial sarcoma | 467 | 1 | 0.21 | 31 | 0 | 0 |
| Alveolar soft part sarcoma | 68 | 0 | 0 | 16 | 0 | 0 |
| Soft tissue clear cell sarcoma | 79 | 0 | 0 | 14 | 0 | 0 |
| Bone sarcoma (NOS) | 29 | 0 | 0 | 5 | 0 | 0 |
| Soft tissue sarcoma (NOS) | 2173 | 37 | 1.7 | 146 | 6 | 4.1 |
| Soft tissue sarcoma undifferentiated | 396 | 5 | 1.26 | 20 | 1 | 5.0 |
| Myxofibrosarcoma | 229 | 3 | 1.31 | 1 | 0 | 0 |
| Liposarcoma | 1202 | 13 | 1.08 | 9 | 0 | 0 |
| Soft tissue granular cell tumor | 30 | 0 | 0 | 1 | 0 | 0 |
| Soft tissue solitary fibrous tumor including soft tissue hemangiopericytoma | 277 | 0 | 0 | 5 | 1 | 20 |
| Tumor type | Adults (≥18 years old) | Children (<18 years old) | ||||
|---|---|---|---|---|---|---|
| Total cases | NTRK-positive cases | Frequency (%) | Total cases | NTRK-positive cases | Frequency (%) | |
| Fibrosarcoma | 109 | 3 | 2.75 | 25 | 13 | 52 |
| Soft tissue leiomyosarcoma | 1421 | 4 | 0.28 | 6 | 0 | 0 |
| Malignant peripheral nerve sheath tumor | 337 | 10 | 2.97 | 43 | 2 | 4.65 |
| Inflammatory myofibroblastic tumor | 44 | 1 | 2.27 | 36 | 1 | 2.78 |
| Soft tissue angiosarcoma | 400 | 1 | 0.25 | 9 | 0 | 0 |
| Rhabdomyosarcoma (embryonal, alveolar, pleomorphic, NOS) | 315 (37,51, 33, 194) | 2 (0, 0, 1, 1) | 0.63 | 287 (90, 83, 1, 113) | 1 (0, 0, 0, 1) | 0.35 |
| Osteosarcoma (bone, extraskeletal) | 573 (503, 70) | 3 (2, 1) | 0.52 | 284 (279, 5) | 2 (2, 0) | 0.70 |
| Chondrosarcoma (bone, extraskeletal) | 514 (233, 281) | 1 (1, 0) | 0.19 | 12 (4, 8) | 1 (1, 0) | 8.3 |
| Ewing sarcoma (bone, extraskeletal, soft tissue primitive neuroectoderm tumor) | 410 (71, 292, 47) | 1 (0, 1, 0) | 0.24 | 198 (12, 177, 9) | 1 (0, 0, 1) | 0.51 |
| Synovial sarcoma | 467 | 1 | 0.21 | 31 | 0 | 0 |
| Alveolar soft part sarcoma | 68 | 0 | 0 | 16 | 0 | 0 |
| Soft tissue clear cell sarcoma | 79 | 0 | 0 | 14 | 0 | 0 |
| Bone sarcoma (NOS) | 29 | 0 | 0 | 5 | 0 | 0 |
| Soft tissue sarcoma (NOS) | 2173 | 37 | 1.7 | 146 | 6 | 4.1 |
| Soft tissue sarcoma undifferentiated | 396 | 5 | 1.26 | 20 | 1 | 5.0 |
| Myxofibrosarcoma | 229 | 3 | 1.31 | 1 | 0 | 0 |
| Liposarcoma | 1202 | 13 | 1.08 | 9 | 0 | 0 |
| Soft tissue granular cell tumor | 30 | 0 | 0 | 1 | 0 | 0 |
| Soft tissue solitary fibrous tumor including soft tissue hemangiopericytoma | 277 | 0 | 0 | 5 | 1 | 20 |
Abbreviation: NOS, not otherwise specified.
Frequency of NTRK fusion gene positivity by histological type of sarcoma (data are adapted from [19])
| Tumor type | Adults (≥18 years old) | Children (<18 years old) | ||||
|---|---|---|---|---|---|---|
| Total cases | NTRK-positive cases | Frequency (%) | Total cases | NTRK-positive cases | Frequency (%) | |
| Fibrosarcoma | 109 | 3 | 2.75 | 25 | 13 | 52 |
| Soft tissue leiomyosarcoma | 1421 | 4 | 0.28 | 6 | 0 | 0 |
| Malignant peripheral nerve sheath tumor | 337 | 10 | 2.97 | 43 | 2 | 4.65 |
| Inflammatory myofibroblastic tumor | 44 | 1 | 2.27 | 36 | 1 | 2.78 |
| Soft tissue angiosarcoma | 400 | 1 | 0.25 | 9 | 0 | 0 |
| Rhabdomyosarcoma (embryonal, alveolar, pleomorphic, NOS) | 315 (37,51, 33, 194) | 2 (0, 0, 1, 1) | 0.63 | 287 (90, 83, 1, 113) | 1 (0, 0, 0, 1) | 0.35 |
| Osteosarcoma (bone, extraskeletal) | 573 (503, 70) | 3 (2, 1) | 0.52 | 284 (279, 5) | 2 (2, 0) | 0.70 |
| Chondrosarcoma (bone, extraskeletal) | 514 (233, 281) | 1 (1, 0) | 0.19 | 12 (4, 8) | 1 (1, 0) | 8.3 |
| Ewing sarcoma (bone, extraskeletal, soft tissue primitive neuroectoderm tumor) | 410 (71, 292, 47) | 1 (0, 1, 0) | 0.24 | 198 (12, 177, 9) | 1 (0, 0, 1) | 0.51 |
| Synovial sarcoma | 467 | 1 | 0.21 | 31 | 0 | 0 |
| Alveolar soft part sarcoma | 68 | 0 | 0 | 16 | 0 | 0 |
| Soft tissue clear cell sarcoma | 79 | 0 | 0 | 14 | 0 | 0 |
| Bone sarcoma (NOS) | 29 | 0 | 0 | 5 | 0 | 0 |
| Soft tissue sarcoma (NOS) | 2173 | 37 | 1.7 | 146 | 6 | 4.1 |
| Soft tissue sarcoma undifferentiated | 396 | 5 | 1.26 | 20 | 1 | 5.0 |
| Myxofibrosarcoma | 229 | 3 | 1.31 | 1 | 0 | 0 |
| Liposarcoma | 1202 | 13 | 1.08 | 9 | 0 | 0 |
| Soft tissue granular cell tumor | 30 | 0 | 0 | 1 | 0 | 0 |
| Soft tissue solitary fibrous tumor including soft tissue hemangiopericytoma | 277 | 0 | 0 | 5 | 1 | 20 |
| Tumor type | Adults (≥18 years old) | Children (<18 years old) | ||||
|---|---|---|---|---|---|---|
| Total cases | NTRK-positive cases | Frequency (%) | Total cases | NTRK-positive cases | Frequency (%) | |
| Fibrosarcoma | 109 | 3 | 2.75 | 25 | 13 | 52 |
| Soft tissue leiomyosarcoma | 1421 | 4 | 0.28 | 6 | 0 | 0 |
| Malignant peripheral nerve sheath tumor | 337 | 10 | 2.97 | 43 | 2 | 4.65 |
| Inflammatory myofibroblastic tumor | 44 | 1 | 2.27 | 36 | 1 | 2.78 |
| Soft tissue angiosarcoma | 400 | 1 | 0.25 | 9 | 0 | 0 |
| Rhabdomyosarcoma (embryonal, alveolar, pleomorphic, NOS) | 315 (37,51, 33, 194) | 2 (0, 0, 1, 1) | 0.63 | 287 (90, 83, 1, 113) | 1 (0, 0, 0, 1) | 0.35 |
| Osteosarcoma (bone, extraskeletal) | 573 (503, 70) | 3 (2, 1) | 0.52 | 284 (279, 5) | 2 (2, 0) | 0.70 |
| Chondrosarcoma (bone, extraskeletal) | 514 (233, 281) | 1 (1, 0) | 0.19 | 12 (4, 8) | 1 (1, 0) | 8.3 |
| Ewing sarcoma (bone, extraskeletal, soft tissue primitive neuroectoderm tumor) | 410 (71, 292, 47) | 1 (0, 1, 0) | 0.24 | 198 (12, 177, 9) | 1 (0, 0, 1) | 0.51 |
| Synovial sarcoma | 467 | 1 | 0.21 | 31 | 0 | 0 |
| Alveolar soft part sarcoma | 68 | 0 | 0 | 16 | 0 | 0 |
| Soft tissue clear cell sarcoma | 79 | 0 | 0 | 14 | 0 | 0 |
| Bone sarcoma (NOS) | 29 | 0 | 0 | 5 | 0 | 0 |
| Soft tissue sarcoma (NOS) | 2173 | 37 | 1.7 | 146 | 6 | 4.1 |
| Soft tissue sarcoma undifferentiated | 396 | 5 | 1.26 | 20 | 1 | 5.0 |
| Myxofibrosarcoma | 229 | 3 | 1.31 | 1 | 0 | 0 |
| Liposarcoma | 1202 | 13 | 1.08 | 9 | 0 | 0 |
| Soft tissue granular cell tumor | 30 | 0 | 0 | 1 | 0 | 0 |
| Soft tissue solitary fibrous tumor including soft tissue hemangiopericytoma | 277 | 0 | 0 | 5 | 1 | 20 |
Abbreviation: NOS, not otherwise specified.
In a Japanese analysis, 91 (74 adults and 17 children) (0.20%) of 46,621 patients with cancer were found to be NTRK fusion gene positive [20]. This positivity was observed in 0.57% of STSs in adults and 5.06% in children [20]. Furthermore, a meta-analysis demonstrated that NTRK fusion gene positivity was present in 0.69% of STSs, and 0.16% of bone sarcomas [21].
Clinical presentation
These tumors are observed in a broad range of age groups, with a higher prevalence in younger individuals, even when IFS is excluded. For instance, among spindle cell sarcomas with NTRK1 gene fusions exhibiting a myo-/hemangiopericytic growth pattern, two out of four cases involved children aged 11 months and 2 years [22]. Similarly, three of seven spindle cell tumors with NTRK3 gene rearrangements (excluding IFS) have been reported in children aged 1, 6, and 11 years [23].
These tumors can arise in nearly every anatomical location. For example, 10 of 14 lipofibromatosis-like neural tumors (LPF-NTs) harboring NTRK1 gene fusions originated from the extremities, while the remaining four were identified in axial sites such as the flank, scalp, buttock, and neck/face [24]. Additionally, two out of seven spindle cell tumors with NTRK3 gene rearrangements (excluding IFS) arose in intracavitary locations, including the mesentery and retroperitoneum [23]. Regarding gastrointestinal involvement, 1 of 15 NTRK1/2-rearranged spindle cell tumors originated from the stomach [25]. Among the cases of IFS, 7 out of 50 occurred in the abdomen, two in the retroperitoneum, and two in the kidney [26]. Some variants occur superficially [24].
Certain variants may exhibit a slow clinical course, although they show rapid growth during the initial stages [26]. The sites of occurrence and degree of malignancy varied considerably.
Radiological features
IFSs are characterized by heterogeneous signals on CT and MRI [27]. Necrosis (63%) and hemorrhage (47%) were identified within the tumor, whereas perilesional invasion was noted in 68% of cases [27]. However, calcification was observed in 13% of cases, and fibrosis was absent (0%) [27]. Furthermore, aberrant vascularization patterns have been consistently observed [27]. In a case series of six NTRK-rearranged spindle cell neoplasms (NTRK-RSCNs), the presence of intra- and peritumoral flow voids and infiltrative features was notable, while fatty and fibrous components, central necrosis, and lobular structures were observed inconsistently [28] (Figs. 1 and 2). NTRK-RSCNs are less vascularized than alveolar soft part sarcoma, with infiltrative features distinguishing them from SFTs [28]. Additionally, they may exhibit the “tail sign,” which is characteristic of myxofibrosarcomas [29].
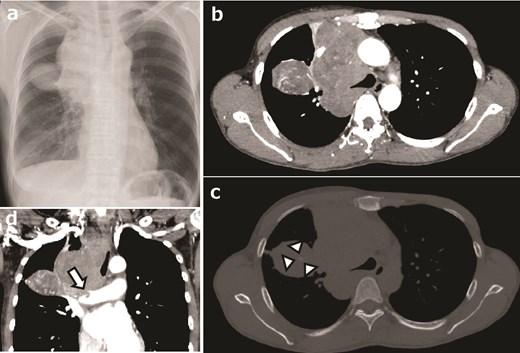
NTRK-positive sarcoma of the right anteromediasternum in a 48-year-old woman. (a) Chest radiograph showing a mass lesion in the upper right chest area, expanding from the mediasternum. (b) Contrast-enhanced computed tomography (CT) image showing a lobular lesion with heterogeneously enhanced signals, highly vascularized intra- and peritumoral areas, without apparent necrosis or hemorrhage. (c) Bone window plain CT image showing a slightly calcified area (marks). (d) Coronal plane CT image showing tumor invasion into the right pulmonary artery (arrow).
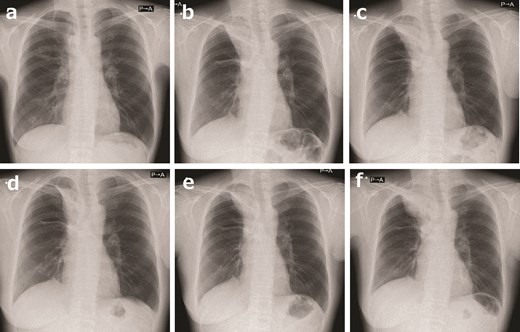
A 48-year-old woman with an NTRK-positive sarcoma in the right anteromediasternum was treated with a TRK inhibitor. (a) After conventional chemotherapy consisting of vincristine, doxorubicin, and cyclophosphamide alternating with ifosfamide and etoposide (VDC/IE) combined with radiotherapy, the tumor had shrunk immensely. (b) Although conventional chemotherapy was continued, the tumor began to grow. In ~2 years, the tumor was much larger. This occurrence allowed for the crucial opportunity to perform a second biopsy, during which the NTRK fusion was finally identified; the patient commenced entrectinib. (c) After 5 months, the tumor began to regrow. The patient commenced larotrectinib. (d) After 1 month, larotrectinib shrunk the tumor a little. (e) After 1 year of larotrectinib, the tumor grew slightly. (f) After 2 years and 8 months of larotrectinib, the tumor progressed, and the patient stopped taking larotrectinib.
Histological features
Recently, specific morphological patterns have been reported to be associated with soft tissue tumors containing NTRK fusion genes [30]. They can be classified into six main categories: LPF-NTs, spindle cell tumors with S100 and CD34 co-reactivity resembling MPNST, IFS with canonical ETV6-NTRK3 fusions, IFS-like lesions with related fusion kinases, adult-type fibrosarcoma, and spindle cell sarcomas with hemangiopericytic or myopericytoma-like pattern [30]. In addition, Suurmeijer et al. [31] and the World Health Organization classification [32] were also considered, and NTRK-positive sarcomas are summarized in Table 2 [13,16,22–25,27,28,30,33–56].
Other cases have also been reported, such as pediatric angiosarcoma with the KHDRBS1-NTRK3 fusion gene [57] and superficial spindle cell sarcoma in adults, resembling fibrosarcomatous dermatofibrosarcoma protuberans with the EML4-NTRK3 fusion gene [58].
Morphological changes linked to fusion partners have also been reported. For example, the NTRK fusion partners TPR and KANK1 have been associated with higher-grade morphology and more aggressive behavior [59].
Pan-TRK immunohistochemistry
Pan-TRK immunostaining was considered positive if there was any staining above the background level or if >1% of the cells exhibited some degree of staining [60]. However, weak membrane or nuclear staining may be overlooked, potentially reducing the diagnostic sensitivity [60].
Solomon et al. [61] performed pan-TRK immunohistochemistry (IHC) in 66 patients with positive NTRK fusions. Sensitivity was 96.2% (26/27) for NTRK1 fusion cases, 100% (5/5) for NTRK2 fusion cases, and 79.4% (27/34) for NTRK3 fusion cases. The sensitivity of detecting NTRK3 fusion was lower than that of detecting NTRK1 and NTRK2 [61]. Specificity varied widely by tissue and was particularly low in the salivary gland (52%), sarcoma (74.4%), and breast carcinoma (82.1%) [61]. Both sensitivity (80%) and specificity were suboptimal for sarcomas, suggesting that either DNA or RNA sequencing (DNA/RNA-seq) should be considered when a fusion gene is suspected [61]. Furthermore, a report indicated that pan-TRK IHC was negative in one case of colorectal cancer [62] and one of renal cancer [63] despite the presence of ETV6-NTRK3 fusion. Generally, pan-TRK immunostaining may be weak in tumors with NTRK3 rearrangements [63].
According to Van Bockstal et al. [64], 2,457 tumors (92%) were pan-TRK negative and 212 (8%) were pan-TRK positive. Twenty-two pan-TRK-positive tumors harbored NTRK fusions (0.8%), representing 10% of all pan-TRK-positive tumors [64]. Cytoplasmic immunoreactivity is the most common among the cellular compartments, followed by membrane immunoreactivity [64]. Nuclear pan-TRK positivity was the least common but most frequently associated with NTRK fusion (33%) [64]. Membrane and cytoplasmic immunoreactivity are associated with NTRK fusion in 3 of 94 cases (3%) and 20 of 161 cases (12%), respectively [64]. Additionally, diffuse and strong cytoplasmic staining may indicate an NTRK1/NTRK2 fusion, whereas nuclear staining may indicate an NTRK3 fusion [65]. However, if the cytoplasmic staining is weak, molecular testing is recommended [65].
Caution should also be exercised when considering angiomatoid fibrous histiocytomas, which are easily stained with pan-TRK IHC [66].
Histological classification of NTRK fusion gene–positive sarcoma
| Tumor type | Clinical features | |
|---|---|---|
| Infantile fibrosarcoma (IFS) | IFS with canonical ETV6-NTRK3 fusions | Histological features: Interlacing fascicles of spindle cells with focal necrosis, mitoses, and a focal hemangiopericytomatous vascular pattern [33]. Clinically and pathologically, they may mimic vascular/lymphatic malformations [34] Immunohistochemical features: Most cases are positive for vimentin [33] and pan-TRK immunostaining [35] Genetic features: ETV6-NTRK3 [13] Radiological features: CT/MRI shows aberrant vascularization pattern, necrosis, and hemorrhage [27] Prognosis: Generally favorable, better prognosis compared to adult-type fibrosarcoma [36] Differential diagnosis: Vascular/lymphatic malformations [34], infantile myofibromatosis [37] TRK inhibitor: Larotrectinib has shown a 96% response rate (27/28) for IFS [16] |
| IFS-like lesions with related fusion kinases (IFS-like tumors) | Histological features: Monomorphic spindle cells arranged in long, intersecting fascicles [30] Immunohistochemical features: These tumors demonstrated a heterogeneous immunoprofile. While no single marker was consistently expressed across all cases, variable positivity was observed for CD34, S100, smooth muscle actin (SMA), and CD30 [38] Genetic features: A number of fusion genes have been identified, including EML4-NTRK3 [39] and LMNA-NTRK1 [40], BRAF [41], and MET [42] Radiological features: It is possible for tumors to infiltrate adjacent structures [42] Prognosis: These tumors exhibited locally aggressive behavior [38]. Metastases were rare, with lung metastases reported in 1 of 6 cases [38] Differential diagnosis: N.A. TRK inhibitor: One patient showed partial response to lartrectinib with a 93% reduction [43], all three patients with IFS with NTRK1 fusions responded to larotrectinib [16]. One IFS (case 15) with a SPECC1L-NTRK3 fusion was treated with larotrectinib and achieved complete tumor regression [44] | |
| Neurotrophic receptor tyrosine kinase (NTRK)-rearranged spindle cell neoplasm | Lipofibromatosis-like neural tumor (LPF-NT) | Histological features: Spindle-shaped cell tumors with a notably infiltrative growth pattern have been observed in the superficial soft tissues of children and young adults [24,45] Immunohistochemical features: Co-express S100 and CD34 [24,45] Genetic features: The NTRK1 fusion gene is associated [24,45] Radiological features: LPF-LT lesions are frequently characterized as relatively well-defined masses primarily involving subcutaneous tissues, with some cases exhibiting diffuse thickening and minimal extension into deeper soft tissues [46–48] In one reported case, magnetic resonance imaging (MRI) findings included areas of T2 hyperintensity, intermediate T1 signal intensity, and occasional small fat components [46] Prognosis: LPF-LT is predominantly a benign or locally aggressive neoplasm with rarely metastases or disease-related mortality reported in typical cases [24] Differential diagnosis: Lipofibromatosis and low-grade MPNSTs [30] TRK inhibitor: One patient with LPF-NT who had difficulty in primary surgical resection took preoperative entrectinib, and 45% reduction of tumor lead to tumor resection [48] |
| Spindle cell tumors with S100 and CD34 co-reactivity resembling malignant peripheral nerve sheath tumor (MPNST) | Histological features: They resemble MPNSTs but with keloid-like collagen around connective tissue and vessels [25,30]. These tumors show low cellularity, a low mitotic count, and lacked necrosis, although some cases demonstrate a malignant phenotype [25] Immunohistochemical features: Often co-expresses S100 and CD34 [25] Genetic features: Often with NTRK1/2, RAF1 or BRAF fusion genes [25] Radiological features: N.A. Prognosis: Hypocellular tumors are classified as low grade, exhibit a favorable prognosis, and show slow cell growth [25]. However, if the tumor shows increased cell density and mitotic activity, there is an increased likelihood of distant metastasis [25] Differential diagnosis: Low-grade or intermediate-grade MPNSTs [25] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including 6 MPNST, responded to larotrectinib [49] | |
| Adult-type fibrosarcoma | Histological features: The histology shows sweeping fascicles of spindle cells that may exhibit storiform architecture, herringbone patterns, hemangiopericytomatous vessels, and mild nuclear atypia in some cases [23,50] Immunohistochemical features: CD34 immunostaining was positive in the majority of cases, whereas S100 was negative, and pan-TRK immunostaining was positive in many cases [23,50] Genetic features: It may be associated with NTRK3 [23,50] fusion genes Radiological features: N.A. Prognosis: The prognosis varies widely, from low to high malignancy [23,50] Differential diagnosis: Differential diagnoses include MPNST, fibrosarcomatous dermatofibrosarcoma protuberans, solitary fibrous tumor, and synovial sarcoma [23] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including 1 adult-type fibrosarcoma, responded to larotrectinib [49] | |
| Spindle cell sarcomas with hemangiopericytic or myopericytoma-like pattern | Histological features: Histologically, the tumor exhibits a growth pattern similar to hemangiopericytoma or myopericytoma, often with high mitotic activity [22] Immunohistochemical features: Immunohistochemically, some cases of SMA and CD34 positivity were observed [22] Genetic features: It is associated with the NTRK1 fusion gene [22] Radiological features: Typical MRI characteristics include isointensity on T1-weighted images, high signal intensity on T2-weighted images, intense contrast enhancement, and the absence of fatty components or lobular architecture [28] Prognosis: Low-grade sarcomas with myopericytoma or hemangiopericytoma-like morphology often exhibit a more favorable prognosis, though some cases may show aggressive behavior [22,28] Differential diagnosis: Myopericytoma and hemangiopericytoma, solitary fibrous tumor, IFS, myofibromatosis [22] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including two with myopericytoma, responded to larotrectinib [49] | |
| Inflammatory myofibroblastic tumors (IMTs) harboring fusions involving ALK and other kinases | Histological features: Histological features are highly diverse, ranging from inflammatory pseudotumors with few spindle cells and prominent inflammation to highly cellular myofibroblastic proliferations or sarcomatous neoplasms lacking significant inflammation or fibromyxoid stroma [51] Immunohistochemical features: Immunohistochemically, several cases showed positive staining for ALK or ROS1 [51] Genetic features: Cases of IMT with ALK gene rearrangement, ROS1 [51], RET [51] rearrangement, and ETV6-NTRK3 fusion [52,53] have been reported Radiological features: IMTs typically appear as homogeneous or heterogeneous lesions with variable enhancement on contrast-enhanced computed tomography or gadolinium-enhanced MRI, reflecting fibrotic tissue. T1- and T2-weighted MRI sequences often show low signal intensity due to the tumor’s fibrotic composition [54] Prognosis: Three out of four cases reported in two studies have maintained long-term disease-free survival [52,53] Differential Diagnosis: Rhabdomyosarcoma, fibrosarcoma, synovial sarcoma [53,54] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including two with IMT, responded to entrectinib [49] | |
| Spindle cell/sclerosing rhabdomyosarcoma (ssRMS) | Histological features: ssRMS exhibits three histological patterns—fibrosarcoma-like, VGLL2-type, and Triton-like—characterized by fibromatous-like appearance with sparse tumor cells in abundant sclerotic stroma, moderate atypia, and rare mitotic figures [55] Immunohistochemical features: Heterogeneous staining with desmin, myogenin [55] Genetic features: TPM3-NTRK1, SYPL1-BRAF, and TOP2B-RAF1 have been reported [55] Radiological features: The ssRMS show slightly high intensity on T2-weighted and iso- or high intensity on T1-weighted images compared to muscle. On post-contrast images, ssRMS showed hetero- or homogeneous enhancement, as described in a case report [56] Prognosis: Fusion-positive ssRMS has a very good outcome [55] Differential diagnosis: N.A. TRK inhibitor: N.A. | |
| Tumor type | Clinical features | |
|---|---|---|
| Infantile fibrosarcoma (IFS) | IFS with canonical ETV6-NTRK3 fusions | Histological features: Interlacing fascicles of spindle cells with focal necrosis, mitoses, and a focal hemangiopericytomatous vascular pattern [33]. Clinically and pathologically, they may mimic vascular/lymphatic malformations [34] Immunohistochemical features: Most cases are positive for vimentin [33] and pan-TRK immunostaining [35] Genetic features: ETV6-NTRK3 [13] Radiological features: CT/MRI shows aberrant vascularization pattern, necrosis, and hemorrhage [27] Prognosis: Generally favorable, better prognosis compared to adult-type fibrosarcoma [36] Differential diagnosis: Vascular/lymphatic malformations [34], infantile myofibromatosis [37] TRK inhibitor: Larotrectinib has shown a 96% response rate (27/28) for IFS [16] |
| IFS-like lesions with related fusion kinases (IFS-like tumors) | Histological features: Monomorphic spindle cells arranged in long, intersecting fascicles [30] Immunohistochemical features: These tumors demonstrated a heterogeneous immunoprofile. While no single marker was consistently expressed across all cases, variable positivity was observed for CD34, S100, smooth muscle actin (SMA), and CD30 [38] Genetic features: A number of fusion genes have been identified, including EML4-NTRK3 [39] and LMNA-NTRK1 [40], BRAF [41], and MET [42] Radiological features: It is possible for tumors to infiltrate adjacent structures [42] Prognosis: These tumors exhibited locally aggressive behavior [38]. Metastases were rare, with lung metastases reported in 1 of 6 cases [38] Differential diagnosis: N.A. TRK inhibitor: One patient showed partial response to lartrectinib with a 93% reduction [43], all three patients with IFS with NTRK1 fusions responded to larotrectinib [16]. One IFS (case 15) with a SPECC1L-NTRK3 fusion was treated with larotrectinib and achieved complete tumor regression [44] | |
| Neurotrophic receptor tyrosine kinase (NTRK)-rearranged spindle cell neoplasm | Lipofibromatosis-like neural tumor (LPF-NT) | Histological features: Spindle-shaped cell tumors with a notably infiltrative growth pattern have been observed in the superficial soft tissues of children and young adults [24,45] Immunohistochemical features: Co-express S100 and CD34 [24,45] Genetic features: The NTRK1 fusion gene is associated [24,45] Radiological features: LPF-LT lesions are frequently characterized as relatively well-defined masses primarily involving subcutaneous tissues, with some cases exhibiting diffuse thickening and minimal extension into deeper soft tissues [46–48] In one reported case, magnetic resonance imaging (MRI) findings included areas of T2 hyperintensity, intermediate T1 signal intensity, and occasional small fat components [46] Prognosis: LPF-LT is predominantly a benign or locally aggressive neoplasm with rarely metastases or disease-related mortality reported in typical cases [24] Differential diagnosis: Lipofibromatosis and low-grade MPNSTs [30] TRK inhibitor: One patient with LPF-NT who had difficulty in primary surgical resection took preoperative entrectinib, and 45% reduction of tumor lead to tumor resection [48] |
| Spindle cell tumors with S100 and CD34 co-reactivity resembling malignant peripheral nerve sheath tumor (MPNST) | Histological features: They resemble MPNSTs but with keloid-like collagen around connective tissue and vessels [25,30]. These tumors show low cellularity, a low mitotic count, and lacked necrosis, although some cases demonstrate a malignant phenotype [25] Immunohistochemical features: Often co-expresses S100 and CD34 [25] Genetic features: Often with NTRK1/2, RAF1 or BRAF fusion genes [25] Radiological features: N.A. Prognosis: Hypocellular tumors are classified as low grade, exhibit a favorable prognosis, and show slow cell growth [25]. However, if the tumor shows increased cell density and mitotic activity, there is an increased likelihood of distant metastasis [25] Differential diagnosis: Low-grade or intermediate-grade MPNSTs [25] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including 6 MPNST, responded to larotrectinib [49] | |
| Adult-type fibrosarcoma | Histological features: The histology shows sweeping fascicles of spindle cells that may exhibit storiform architecture, herringbone patterns, hemangiopericytomatous vessels, and mild nuclear atypia in some cases [23,50] Immunohistochemical features: CD34 immunostaining was positive in the majority of cases, whereas S100 was negative, and pan-TRK immunostaining was positive in many cases [23,50] Genetic features: It may be associated with NTRK3 [23,50] fusion genes Radiological features: N.A. Prognosis: The prognosis varies widely, from low to high malignancy [23,50] Differential diagnosis: Differential diagnoses include MPNST, fibrosarcomatous dermatofibrosarcoma protuberans, solitary fibrous tumor, and synovial sarcoma [23] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including 1 adult-type fibrosarcoma, responded to larotrectinib [49] | |
| Spindle cell sarcomas with hemangiopericytic or myopericytoma-like pattern | Histological features: Histologically, the tumor exhibits a growth pattern similar to hemangiopericytoma or myopericytoma, often with high mitotic activity [22] Immunohistochemical features: Immunohistochemically, some cases of SMA and CD34 positivity were observed [22] Genetic features: It is associated with the NTRK1 fusion gene [22] Radiological features: Typical MRI characteristics include isointensity on T1-weighted images, high signal intensity on T2-weighted images, intense contrast enhancement, and the absence of fatty components or lobular architecture [28] Prognosis: Low-grade sarcomas with myopericytoma or hemangiopericytoma-like morphology often exhibit a more favorable prognosis, though some cases may show aggressive behavior [22,28] Differential diagnosis: Myopericytoma and hemangiopericytoma, solitary fibrous tumor, IFS, myofibromatosis [22] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including two with myopericytoma, responded to larotrectinib [49] | |
| Inflammatory myofibroblastic tumors (IMTs) harboring fusions involving ALK and other kinases | Histological features: Histological features are highly diverse, ranging from inflammatory pseudotumors with few spindle cells and prominent inflammation to highly cellular myofibroblastic proliferations or sarcomatous neoplasms lacking significant inflammation or fibromyxoid stroma [51] Immunohistochemical features: Immunohistochemically, several cases showed positive staining for ALK or ROS1 [51] Genetic features: Cases of IMT with ALK gene rearrangement, ROS1 [51], RET [51] rearrangement, and ETV6-NTRK3 fusion [52,53] have been reported Radiological features: IMTs typically appear as homogeneous or heterogeneous lesions with variable enhancement on contrast-enhanced computed tomography or gadolinium-enhanced MRI, reflecting fibrotic tissue. T1- and T2-weighted MRI sequences often show low signal intensity due to the tumor’s fibrotic composition [54] Prognosis: Three out of four cases reported in two studies have maintained long-term disease-free survival [52,53] Differential Diagnosis: Rhabdomyosarcoma, fibrosarcoma, synovial sarcoma [53,54] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including two with IMT, responded to entrectinib [49] | |
| Spindle cell/sclerosing rhabdomyosarcoma (ssRMS) | Histological features: ssRMS exhibits three histological patterns—fibrosarcoma-like, VGLL2-type, and Triton-like—characterized by fibromatous-like appearance with sparse tumor cells in abundant sclerotic stroma, moderate atypia, and rare mitotic figures [55] Immunohistochemical features: Heterogeneous staining with desmin, myogenin [55] Genetic features: TPM3-NTRK1, SYPL1-BRAF, and TOP2B-RAF1 have been reported [55] Radiological features: The ssRMS show slightly high intensity on T2-weighted and iso- or high intensity on T1-weighted images compared to muscle. On post-contrast images, ssRMS showed hetero- or homogeneous enhancement, as described in a case report [56] Prognosis: Fusion-positive ssRMS has a very good outcome [55] Differential diagnosis: N.A. TRK inhibitor: N.A. | |
Abbreviation: N.A., not available.
Histological classification of NTRK fusion gene–positive sarcoma
| Tumor type | Clinical features | |
|---|---|---|
| Infantile fibrosarcoma (IFS) | IFS with canonical ETV6-NTRK3 fusions | Histological features: Interlacing fascicles of spindle cells with focal necrosis, mitoses, and a focal hemangiopericytomatous vascular pattern [33]. Clinically and pathologically, they may mimic vascular/lymphatic malformations [34] Immunohistochemical features: Most cases are positive for vimentin [33] and pan-TRK immunostaining [35] Genetic features: ETV6-NTRK3 [13] Radiological features: CT/MRI shows aberrant vascularization pattern, necrosis, and hemorrhage [27] Prognosis: Generally favorable, better prognosis compared to adult-type fibrosarcoma [36] Differential diagnosis: Vascular/lymphatic malformations [34], infantile myofibromatosis [37] TRK inhibitor: Larotrectinib has shown a 96% response rate (27/28) for IFS [16] |
| IFS-like lesions with related fusion kinases (IFS-like tumors) | Histological features: Monomorphic spindle cells arranged in long, intersecting fascicles [30] Immunohistochemical features: These tumors demonstrated a heterogeneous immunoprofile. While no single marker was consistently expressed across all cases, variable positivity was observed for CD34, S100, smooth muscle actin (SMA), and CD30 [38] Genetic features: A number of fusion genes have been identified, including EML4-NTRK3 [39] and LMNA-NTRK1 [40], BRAF [41], and MET [42] Radiological features: It is possible for tumors to infiltrate adjacent structures [42] Prognosis: These tumors exhibited locally aggressive behavior [38]. Metastases were rare, with lung metastases reported in 1 of 6 cases [38] Differential diagnosis: N.A. TRK inhibitor: One patient showed partial response to lartrectinib with a 93% reduction [43], all three patients with IFS with NTRK1 fusions responded to larotrectinib [16]. One IFS (case 15) with a SPECC1L-NTRK3 fusion was treated with larotrectinib and achieved complete tumor regression [44] | |
| Neurotrophic receptor tyrosine kinase (NTRK)-rearranged spindle cell neoplasm | Lipofibromatosis-like neural tumor (LPF-NT) | Histological features: Spindle-shaped cell tumors with a notably infiltrative growth pattern have been observed in the superficial soft tissues of children and young adults [24,45] Immunohistochemical features: Co-express S100 and CD34 [24,45] Genetic features: The NTRK1 fusion gene is associated [24,45] Radiological features: LPF-LT lesions are frequently characterized as relatively well-defined masses primarily involving subcutaneous tissues, with some cases exhibiting diffuse thickening and minimal extension into deeper soft tissues [46–48] In one reported case, magnetic resonance imaging (MRI) findings included areas of T2 hyperintensity, intermediate T1 signal intensity, and occasional small fat components [46] Prognosis: LPF-LT is predominantly a benign or locally aggressive neoplasm with rarely metastases or disease-related mortality reported in typical cases [24] Differential diagnosis: Lipofibromatosis and low-grade MPNSTs [30] TRK inhibitor: One patient with LPF-NT who had difficulty in primary surgical resection took preoperative entrectinib, and 45% reduction of tumor lead to tumor resection [48] |
| Spindle cell tumors with S100 and CD34 co-reactivity resembling malignant peripheral nerve sheath tumor (MPNST) | Histological features: They resemble MPNSTs but with keloid-like collagen around connective tissue and vessels [25,30]. These tumors show low cellularity, a low mitotic count, and lacked necrosis, although some cases demonstrate a malignant phenotype [25] Immunohistochemical features: Often co-expresses S100 and CD34 [25] Genetic features: Often with NTRK1/2, RAF1 or BRAF fusion genes [25] Radiological features: N.A. Prognosis: Hypocellular tumors are classified as low grade, exhibit a favorable prognosis, and show slow cell growth [25]. However, if the tumor shows increased cell density and mitotic activity, there is an increased likelihood of distant metastasis [25] Differential diagnosis: Low-grade or intermediate-grade MPNSTs [25] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including 6 MPNST, responded to larotrectinib [49] | |
| Adult-type fibrosarcoma | Histological features: The histology shows sweeping fascicles of spindle cells that may exhibit storiform architecture, herringbone patterns, hemangiopericytomatous vessels, and mild nuclear atypia in some cases [23,50] Immunohistochemical features: CD34 immunostaining was positive in the majority of cases, whereas S100 was negative, and pan-TRK immunostaining was positive in many cases [23,50] Genetic features: It may be associated with NTRK3 [23,50] fusion genes Radiological features: N.A. Prognosis: The prognosis varies widely, from low to high malignancy [23,50] Differential diagnosis: Differential diagnoses include MPNST, fibrosarcomatous dermatofibrosarcoma protuberans, solitary fibrous tumor, and synovial sarcoma [23] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including 1 adult-type fibrosarcoma, responded to larotrectinib [49] | |
| Spindle cell sarcomas with hemangiopericytic or myopericytoma-like pattern | Histological features: Histologically, the tumor exhibits a growth pattern similar to hemangiopericytoma or myopericytoma, often with high mitotic activity [22] Immunohistochemical features: Immunohistochemically, some cases of SMA and CD34 positivity were observed [22] Genetic features: It is associated with the NTRK1 fusion gene [22] Radiological features: Typical MRI characteristics include isointensity on T1-weighted images, high signal intensity on T2-weighted images, intense contrast enhancement, and the absence of fatty components or lobular architecture [28] Prognosis: Low-grade sarcomas with myopericytoma or hemangiopericytoma-like morphology often exhibit a more favorable prognosis, though some cases may show aggressive behavior [22,28] Differential diagnosis: Myopericytoma and hemangiopericytoma, solitary fibrous tumor, IFS, myofibromatosis [22] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including two with myopericytoma, responded to larotrectinib [49] | |
| Inflammatory myofibroblastic tumors (IMTs) harboring fusions involving ALK and other kinases | Histological features: Histological features are highly diverse, ranging from inflammatory pseudotumors with few spindle cells and prominent inflammation to highly cellular myofibroblastic proliferations or sarcomatous neoplasms lacking significant inflammation or fibromyxoid stroma [51] Immunohistochemical features: Immunohistochemically, several cases showed positive staining for ALK or ROS1 [51] Genetic features: Cases of IMT with ALK gene rearrangement, ROS1 [51], RET [51] rearrangement, and ETV6-NTRK3 fusion [52,53] have been reported Radiological features: IMTs typically appear as homogeneous or heterogeneous lesions with variable enhancement on contrast-enhanced computed tomography or gadolinium-enhanced MRI, reflecting fibrotic tissue. T1- and T2-weighted MRI sequences often show low signal intensity due to the tumor’s fibrotic composition [54] Prognosis: Three out of four cases reported in two studies have maintained long-term disease-free survival [52,53] Differential Diagnosis: Rhabdomyosarcoma, fibrosarcoma, synovial sarcoma [53,54] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including two with IMT, responded to entrectinib [49] | |
| Spindle cell/sclerosing rhabdomyosarcoma (ssRMS) | Histological features: ssRMS exhibits three histological patterns—fibrosarcoma-like, VGLL2-type, and Triton-like—characterized by fibromatous-like appearance with sparse tumor cells in abundant sclerotic stroma, moderate atypia, and rare mitotic figures [55] Immunohistochemical features: Heterogeneous staining with desmin, myogenin [55] Genetic features: TPM3-NTRK1, SYPL1-BRAF, and TOP2B-RAF1 have been reported [55] Radiological features: The ssRMS show slightly high intensity on T2-weighted and iso- or high intensity on T1-weighted images compared to muscle. On post-contrast images, ssRMS showed hetero- or homogeneous enhancement, as described in a case report [56] Prognosis: Fusion-positive ssRMS has a very good outcome [55] Differential diagnosis: N.A. TRK inhibitor: N.A. | |
| Tumor type | Clinical features | |
|---|---|---|
| Infantile fibrosarcoma (IFS) | IFS with canonical ETV6-NTRK3 fusions | Histological features: Interlacing fascicles of spindle cells with focal necrosis, mitoses, and a focal hemangiopericytomatous vascular pattern [33]. Clinically and pathologically, they may mimic vascular/lymphatic malformations [34] Immunohistochemical features: Most cases are positive for vimentin [33] and pan-TRK immunostaining [35] Genetic features: ETV6-NTRK3 [13] Radiological features: CT/MRI shows aberrant vascularization pattern, necrosis, and hemorrhage [27] Prognosis: Generally favorable, better prognosis compared to adult-type fibrosarcoma [36] Differential diagnosis: Vascular/lymphatic malformations [34], infantile myofibromatosis [37] TRK inhibitor: Larotrectinib has shown a 96% response rate (27/28) for IFS [16] |
| IFS-like lesions with related fusion kinases (IFS-like tumors) | Histological features: Monomorphic spindle cells arranged in long, intersecting fascicles [30] Immunohistochemical features: These tumors demonstrated a heterogeneous immunoprofile. While no single marker was consistently expressed across all cases, variable positivity was observed for CD34, S100, smooth muscle actin (SMA), and CD30 [38] Genetic features: A number of fusion genes have been identified, including EML4-NTRK3 [39] and LMNA-NTRK1 [40], BRAF [41], and MET [42] Radiological features: It is possible for tumors to infiltrate adjacent structures [42] Prognosis: These tumors exhibited locally aggressive behavior [38]. Metastases were rare, with lung metastases reported in 1 of 6 cases [38] Differential diagnosis: N.A. TRK inhibitor: One patient showed partial response to lartrectinib with a 93% reduction [43], all three patients with IFS with NTRK1 fusions responded to larotrectinib [16]. One IFS (case 15) with a SPECC1L-NTRK3 fusion was treated with larotrectinib and achieved complete tumor regression [44] | |
| Neurotrophic receptor tyrosine kinase (NTRK)-rearranged spindle cell neoplasm | Lipofibromatosis-like neural tumor (LPF-NT) | Histological features: Spindle-shaped cell tumors with a notably infiltrative growth pattern have been observed in the superficial soft tissues of children and young adults [24,45] Immunohistochemical features: Co-express S100 and CD34 [24,45] Genetic features: The NTRK1 fusion gene is associated [24,45] Radiological features: LPF-LT lesions are frequently characterized as relatively well-defined masses primarily involving subcutaneous tissues, with some cases exhibiting diffuse thickening and minimal extension into deeper soft tissues [46–48] In one reported case, magnetic resonance imaging (MRI) findings included areas of T2 hyperintensity, intermediate T1 signal intensity, and occasional small fat components [46] Prognosis: LPF-LT is predominantly a benign or locally aggressive neoplasm with rarely metastases or disease-related mortality reported in typical cases [24] Differential diagnosis: Lipofibromatosis and low-grade MPNSTs [30] TRK inhibitor: One patient with LPF-NT who had difficulty in primary surgical resection took preoperative entrectinib, and 45% reduction of tumor lead to tumor resection [48] |
| Spindle cell tumors with S100 and CD34 co-reactivity resembling malignant peripheral nerve sheath tumor (MPNST) | Histological features: They resemble MPNSTs but with keloid-like collagen around connective tissue and vessels [25,30]. These tumors show low cellularity, a low mitotic count, and lacked necrosis, although some cases demonstrate a malignant phenotype [25] Immunohistochemical features: Often co-expresses S100 and CD34 [25] Genetic features: Often with NTRK1/2, RAF1 or BRAF fusion genes [25] Radiological features: N.A. Prognosis: Hypocellular tumors are classified as low grade, exhibit a favorable prognosis, and show slow cell growth [25]. However, if the tumor shows increased cell density and mitotic activity, there is an increased likelihood of distant metastasis [25] Differential diagnosis: Low-grade or intermediate-grade MPNSTs [25] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including 6 MPNST, responded to larotrectinib [49] | |
| Adult-type fibrosarcoma | Histological features: The histology shows sweeping fascicles of spindle cells that may exhibit storiform architecture, herringbone patterns, hemangiopericytomatous vessels, and mild nuclear atypia in some cases [23,50] Immunohistochemical features: CD34 immunostaining was positive in the majority of cases, whereas S100 was negative, and pan-TRK immunostaining was positive in many cases [23,50] Genetic features: It may be associated with NTRK3 [23,50] fusion genes Radiological features: N.A. Prognosis: The prognosis varies widely, from low to high malignancy [23,50] Differential diagnosis: Differential diagnoses include MPNST, fibrosarcomatous dermatofibrosarcoma protuberans, solitary fibrous tumor, and synovial sarcoma [23] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including 1 adult-type fibrosarcoma, responded to larotrectinib [49] | |
| Spindle cell sarcomas with hemangiopericytic or myopericytoma-like pattern | Histological features: Histologically, the tumor exhibits a growth pattern similar to hemangiopericytoma or myopericytoma, often with high mitotic activity [22] Immunohistochemical features: Immunohistochemically, some cases of SMA and CD34 positivity were observed [22] Genetic features: It is associated with the NTRK1 fusion gene [22] Radiological features: Typical MRI characteristics include isointensity on T1-weighted images, high signal intensity on T2-weighted images, intense contrast enhancement, and the absence of fatty components or lobular architecture [28] Prognosis: Low-grade sarcomas with myopericytoma or hemangiopericytoma-like morphology often exhibit a more favorable prognosis, though some cases may show aggressive behavior [22,28] Differential diagnosis: Myopericytoma and hemangiopericytoma, solitary fibrous tumor, IFS, myofibromatosis [22] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including two with myopericytoma, responded to larotrectinib [49] | |
| Inflammatory myofibroblastic tumors (IMTs) harboring fusions involving ALK and other kinases | Histological features: Histological features are highly diverse, ranging from inflammatory pseudotumors with few spindle cells and prominent inflammation to highly cellular myofibroblastic proliferations or sarcomatous neoplasms lacking significant inflammation or fibromyxoid stroma [51] Immunohistochemical features: Immunohistochemically, several cases showed positive staining for ALK or ROS1 [51] Genetic features: Cases of IMT with ALK gene rearrangement, ROS1 [51], RET [51] rearrangement, and ETV6-NTRK3 fusion [52,53] have been reported Radiological features: IMTs typically appear as homogeneous or heterogeneous lesions with variable enhancement on contrast-enhanced computed tomography or gadolinium-enhanced MRI, reflecting fibrotic tissue. T1- and T2-weighted MRI sequences often show low signal intensity due to the tumor’s fibrotic composition [54] Prognosis: Three out of four cases reported in two studies have maintained long-term disease-free survival [52,53] Differential Diagnosis: Rhabdomyosarcoma, fibrosarcoma, synovial sarcoma [53,54] TRK inhibitor: Sixteen out of 30 patients (53%) with soft tissue sarcoma (STS), including two with IMT, responded to entrectinib [49] | |
| Spindle cell/sclerosing rhabdomyosarcoma (ssRMS) | Histological features: ssRMS exhibits three histological patterns—fibrosarcoma-like, VGLL2-type, and Triton-like—characterized by fibromatous-like appearance with sparse tumor cells in abundant sclerotic stroma, moderate atypia, and rare mitotic figures [55] Immunohistochemical features: Heterogeneous staining with desmin, myogenin [55] Genetic features: TPM3-NTRK1, SYPL1-BRAF, and TOP2B-RAF1 have been reported [55] Radiological features: The ssRMS show slightly high intensity on T2-weighted and iso- or high intensity on T1-weighted images compared to muscle. On post-contrast images, ssRMS showed hetero- or homogeneous enhancement, as described in a case report [56] Prognosis: Fusion-positive ssRMS has a very good outcome [55] Differential diagnosis: N.A. TRK inhibitor: N.A. | |
Abbreviation: N.A., not available.
Molecular genetic features
Detection method
Many reviews have examined the methods for detecting NTRK, including reverse transcription–polymerase chain reaction (PCR), fluorescence in situ hybridization, IHC, and next-generation sequencing. Most of these studies concluded that DNA/RNA-seq is ultimately beneficial, with many placing particular emphasis on RNA-seq [67–69].
DNA sequencing identifies NTRK fusions and other genetic abnormalities, including TMB-high, and MSI [69]. However, limitations, such as false negatives caused by large intronic regions or unknown partners, must be considered [69]. RNA sequencing (RNA-seq), in contrast, offers higher sensitivity [69] by analyzing spliced mRNA, enabling partner-agnostic detection. However, challenges related to sample quality and potential limitations in detecting regulatory sequence replacements [70,71].
Different RNA-seq methods, including anchored multiplex PCR, amplicon-based multiplex PCR, and hybrid capture, demonstrate high performance in detecting NTRK fusions in clinical samples, with unique strengths and weaknesses [72,73]. For example, amplicon-based multiplex PCR is highly sensitive but less effective for rare or novel partners compared to hybrid capture and anchored multiplex PCR [73].
Circulating tumor DNA (ctDNA) analysis in a retrospective study of 37 patients with NTRK1 fusions in advanced solid tumors showed that NTRK1 fusions detected by ctDNA were confirmed in tissues in 88% of cases [74]. Liquid biopsy using ctDNA serves as a rapid and noninvasive screening tool that may improve the identification of patients who can benefit from TRK-targeted therapies and potentially identify both on- and off-target resistance mechanisms [74].
About the fusion partner
Various fusion partners for NTRK fusions in IFS and other sarcomas have been identified, with an increasing number expected (Table 3) [15,20,25,38,40,44,50,57–59,75–86]. Breakpoints in NTRK1, NTRK2, and NTRK3 are partially understood, as illustrated in Fig. 3 [20,25,38,40,44,50,57,58, 73,77,78,80,85,86].
NTRK fusion partners in infantile fibrosarcoma and sarcoma
| Infantile fibrosarcoma (IFS) | Sarcoma (excluding IFS, vagina, uterine, GIST) | |
|---|---|---|
| NTRK1 | LMNA [20,40], TPM3 [15,38], TPR [15,75], SQSTM1 [43], MIR584F1 [75] | TPM3 [15,20,79–82], CPSF6 [80], IGR [80], GAS2L1 [80], SQSTM1 [80], PDE4DIP [15], LMNA [15,20,77,80], PHF20 [20,83], PEAR1 [20], MEF2D [20], KIRREL1 [20], DCTN1 [84], IRF2BP2 [76,77], VAMP4 [81], GON4L [81], CCDC171 [81], TSNAX [81], PDIA3 [82], ZNF382 [82], TPR [82] |
| NTRK2 | Not available | STRN [15,85], SPECC1L [25], KANK1 [59], MAMDC2 [82] |
| NTRK3 | ETV6 [13,44,76,77], EML4 [78], SPECC1L [44] | TFG [44,86], ETV6 [15],TPM4 [15], EML4 [58], STRN3 [44,50], STRN [50], KHDRBS1 [57], MEF2A [76], ITFG1 [76], RBPMS [77,82], MCTP2 [81], FANCI [81], RAB14 [81], AKAP13 [82], IQGAP [82], FAM19A2 [82], SPECC1L [82], KIF7 [82] |
| Infantile fibrosarcoma (IFS) | Sarcoma (excluding IFS, vagina, uterine, GIST) | |
|---|---|---|
| NTRK1 | LMNA [20,40], TPM3 [15,38], TPR [15,75], SQSTM1 [43], MIR584F1 [75] | TPM3 [15,20,79–82], CPSF6 [80], IGR [80], GAS2L1 [80], SQSTM1 [80], PDE4DIP [15], LMNA [15,20,77,80], PHF20 [20,83], PEAR1 [20], MEF2D [20], KIRREL1 [20], DCTN1 [84], IRF2BP2 [76,77], VAMP4 [81], GON4L [81], CCDC171 [81], TSNAX [81], PDIA3 [82], ZNF382 [82], TPR [82] |
| NTRK2 | Not available | STRN [15,85], SPECC1L [25], KANK1 [59], MAMDC2 [82] |
| NTRK3 | ETV6 [13,44,76,77], EML4 [78], SPECC1L [44] | TFG [44,86], ETV6 [15],TPM4 [15], EML4 [58], STRN3 [44,50], STRN [50], KHDRBS1 [57], MEF2A [76], ITFG1 [76], RBPMS [77,82], MCTP2 [81], FANCI [81], RAB14 [81], AKAP13 [82], IQGAP [82], FAM19A2 [82], SPECC1L [82], KIF7 [82] |
Abbreviation: GIST, gastrointestinal stromal tumor.
NTRK fusion partners in infantile fibrosarcoma and sarcoma
| Infantile fibrosarcoma (IFS) | Sarcoma (excluding IFS, vagina, uterine, GIST) | |
|---|---|---|
| NTRK1 | LMNA [20,40], TPM3 [15,38], TPR [15,75], SQSTM1 [43], MIR584F1 [75] | TPM3 [15,20,79–82], CPSF6 [80], IGR [80], GAS2L1 [80], SQSTM1 [80], PDE4DIP [15], LMNA [15,20,77,80], PHF20 [20,83], PEAR1 [20], MEF2D [20], KIRREL1 [20], DCTN1 [84], IRF2BP2 [76,77], VAMP4 [81], GON4L [81], CCDC171 [81], TSNAX [81], PDIA3 [82], ZNF382 [82], TPR [82] |
| NTRK2 | Not available | STRN [15,85], SPECC1L [25], KANK1 [59], MAMDC2 [82] |
| NTRK3 | ETV6 [13,44,76,77], EML4 [78], SPECC1L [44] | TFG [44,86], ETV6 [15],TPM4 [15], EML4 [58], STRN3 [44,50], STRN [50], KHDRBS1 [57], MEF2A [76], ITFG1 [76], RBPMS [77,82], MCTP2 [81], FANCI [81], RAB14 [81], AKAP13 [82], IQGAP [82], FAM19A2 [82], SPECC1L [82], KIF7 [82] |
| Infantile fibrosarcoma (IFS) | Sarcoma (excluding IFS, vagina, uterine, GIST) | |
|---|---|---|
| NTRK1 | LMNA [20,40], TPM3 [15,38], TPR [15,75], SQSTM1 [43], MIR584F1 [75] | TPM3 [15,20,79–82], CPSF6 [80], IGR [80], GAS2L1 [80], SQSTM1 [80], PDE4DIP [15], LMNA [15,20,77,80], PHF20 [20,83], PEAR1 [20], MEF2D [20], KIRREL1 [20], DCTN1 [84], IRF2BP2 [76,77], VAMP4 [81], GON4L [81], CCDC171 [81], TSNAX [81], PDIA3 [82], ZNF382 [82], TPR [82] |
| NTRK2 | Not available | STRN [15,85], SPECC1L [25], KANK1 [59], MAMDC2 [82] |
| NTRK3 | ETV6 [13,44,76,77], EML4 [78], SPECC1L [44] | TFG [44,86], ETV6 [15],TPM4 [15], EML4 [58], STRN3 [44,50], STRN [50], KHDRBS1 [57], MEF2A [76], ITFG1 [76], RBPMS [77,82], MCTP2 [81], FANCI [81], RAB14 [81], AKAP13 [82], IQGAP [82], FAM19A2 [82], SPECC1L [82], KIF7 [82] |
Abbreviation: GIST, gastrointestinal stromal tumor.
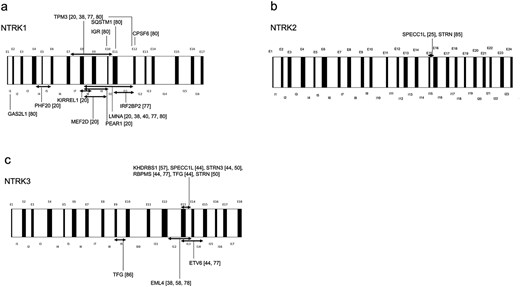
The breakpoint locations in the DNA of NTRK fusion gene–positive sarcomas are as follows: (a) NTRK1, (b) NTRK2, and (c) NTRK3, as inferred from the findings in the cited literature. Abbreviations: I, intron; E, exon.
Related to other genetic abnormalities
NTRK fusions are usually mutually exclusive with common proto-oncogenic driver alterations that activate MAPK signaling, such as KRAS, BRAF, EGFR, ALK, RET, and ROS1 [87]. Although TRK inhibitors demonstrate significant efficacy in TRK fusion-positive cancers, the effect of immunotherapy is generally limited, particularly in cases without other predictive biomarkers such as MSI-high [88]. In colorectal cancer, NTRK fusions are occasionally associated with MSI or TMB-high [19,89]. However, this association is inconsistent across NTRK fusion gene–positive tumors in general [20].
In sarcomas, NTRK fusions are mutually exclusive with Ewing sarcoma–specific fusions. On the other hand, they can coexist with MDM2/CDK4 amplification, for which testing is recommended despite the clinical significance that remains unclear [90].
Bone sarcoma
In 113 osteosarcoma samples, NTRK fusions were identified in three cases but lacked functional protein expression, suggesting passenger rather than driver mutations [91]. Specific fusions such as NTRK2-CLEC16A [20], STRN3-NTRK3 [92], and STRN-NTRK3 [50] have been identified in bone sarcomas, but responses to TRK inhibitors remain unknown. The World Sarcoma Network recommends pan-TRK IHC screening for sarcomas with complex genomes [93], which is generally considered to include osteosarcomas.
Natural history and prognostic factor
A comparison of 15,223 patients with cancer with and without NTRK fusion genes revealed a slightly higher risk of death in patients with NTRK fusions [hazard ratio (HR) 1.47, 95% confidence interval (CI) 0.39–5.57]; however, this difference was not statistically significant [89]. Although there was no statistically significant difference in the progression risk between patients with NTRK-positive and -negative tumors, the analysis was confounded by the inclusion of patients receiving TRK inhibitors, which may have affected the assessment of progression risk [94]. Another study reported a 50% increase in the risk of death in NTRK fusion gene–positive tumors with no prior TRK inhibitor use (HR 1.51, 95% CI: 1.01–2.29) [95], suggesting a potential decrease in mortality and progression risk following the introduction of TRK inhibitors.
The prognostic characteristics of each histological type of NTRK fusion-positive sarcoma are shown in Table 2. There is a relationship between histological grade and prognosis; however, even with the same fusion gene, the prognosis can vary, with deep-seated lesions generally having a worse prognosis than superficial ones [96]. Overall, the prognosis and risk of metastasis for NTRK fusion gene–positive tumors vary according to the histological grade and tissue morphology, and further research is needed.
Management of NTRK fusion-positive sarcoma
Local therapy
In a study of six patients with NTRK-RSCN, five underwent wide excision and one underwent marginal excision [28]. During postoperative follow-up, none of the patients who underwent wide excision experienced recurrence or metastasis, whereas the patient who underwent marginal excision developed local recurrence and distant metastasis, ultimately resulting in death [28]. These findings indicated that wide excision may reduce the risk of recurrence [28].
Although TRK inhibitors are effective, there are few reports on radiotherapy as a local treatment. A 6-year-old girl with an inoperable NTRK3 fusion gene–positive sarcoma achieved partial response after larotrectinib treatment, followed by a complete response after proton beam radiotherapy [97]. This case report demonstrates that NTRK fusion-positive sarcomas can be safely treated with larotrectinib and radiotherapy [97].
TRK inhibitor as neoadjuvant chemotherapy
To date, the only clinical trial that included patients receiving a TRK inhibitor as perioperative treatment is the phase I/II trial of larotrectinib (NCT02637687) [43,98]. Of the enrolled pediatric patients, five had NTRK fusion-positive sarcomas (three IFS and two STS), experienced tumor shrinkage after preoperative larotrectinib treatment (median six courses), and all underwent surgery [43]. Three cases were complete (R0), one had microscopic residual disease (R1), and one had macroscopic residual disease (R2) [43]. Three patients with R0 disease remained recurrence-free for 7–15 months postoperatively [43]. Two patients with R1 and R2 disease continued treatment with larotrectinib after surgery and remained relapse-free without metastases [43]. The patient who underwent R2 resection had a tumor in the right upper thigh [43]. After an initial attempt at surgical resection, the tumor recurred, and subsequent systemic chemotherapy was ineffective [43]. Larotrectinib was initiated, leading to a partial response, and subsequent R2 surgical resection revealed a 40%–60% viable tumor with significant treatment-related morphological changes [43]. Larotrectinib was resumed, resulting in a second partial response and a second R2 resection with a viable tumor at the margin [43]. The patient subsequently received adjuvant radiotherapy and ongoing adjuvant larotrectinib, maintaining a complete response at the time of the latest follow-up [43].
Furthermore, although these are case reports, the usefulness of TRK inhibitors as adjuvant therapy has been reported in grade 3 NTRK-related spindle cell sarcoma in the distal left lower leg of a 30-year-old man, where the use of larotrectinib in the neoadjuvant setting avoided limb amputation [99]. It has also been reported in a high-grade mesenchymal neoplasm in the right gluteal region of a 35-year-old woman, which was positive for STRN-NTRK2 fusions [100]. Due to the location and extent of the tumor, it was initially deemed unresectable or would have required a morbid upfront surgery, such as hemipelvectomy [100]. However, treatment with larotrectinib led to significant tumor shrinkage, enabling radical resection of the right gluteal soft tissue sarcoma and right ilium, which was uneventful [100]. The resection margins were tumor-free, achieving a complete surgical outcome [100]. A low-grade sarcoma in the right upper arm of an 8-year-old girl presented with local recurrence, where reoperation would likely have required amputation, but treatment with larotrectinib achieved complete radiographic remission, enabling local resection and confirming complete pathological remission [101].
In a phase I/II study in pediatric patients, an objective response rate of 14 out of 15 patients with TRK fusions (93%) was achieved, and the median time to response was 1.7 months (interquartile range 1.0–2.9) [98]. Moreover, some patients exhibit clear responses within a few days [98].
Thus, it is not yet clear whether TRK inhibitors influence prognosis or metastasis. However, when the primary lesion is unresectable due to its size or location, and rapid tumor shrinkage is clinically necessary, preoperative TRK inhibitors may be considered.
TRK inhibitor for advanced cases
A report by Drilon et al. [15] on treatment with larotrectinib for advanced cases of NTRK fusion gene–positive tumors included 55 patients, ranging from infants to adults. The overall response rate (ORR) was 75% [complete response (CR): 7 patients (13%); partial response (PR): 34 patients (62%)] [15]. Responses were observed irrespective of tumor type or age [15]. After 1 year, 71% of the patients (no. at risk = 10) responded to treatment, and 55% (no. at risk = 12) were progression-free [15]. The side effects were mild and mainly included anemia and elevated liver enzyme levels [15].
In an integrated analysis of three phase I/II trials of larotrectinib, 121 (79%; 95% CI: 72–85) of evaluable 153 patients showed an objective response, with 24 (16%) achieving CR, 97 (63%) achieving PR, 19 (12%) with stable disease (SD), and 9 (6%) with progressive disease (PD) [16]. The median duration of response (DoR) was 35.2 months [95% CI: 22.8–not estimable (NE)], median progression-free survival was 28.3 months (95% CI: 22.1–NE), and median overall survival was 44.4 months (95% CI: 36.5–NE) [16].
In a subsequent analysis focusing on patients with sarcoma, 36 cases of NTRK fusion-positive sarcomas, all treated with larotrectinib therapy, comprised 30 soft tissue sarcomas (83%), 2 malignant bone tumors (chondrosarcoma, NOS) (6%), and 4 GISTs (11%) [49]. Among 32 cases, excluding GIST, CR was observed in 6 patients (19%), PR in 11 (34%), SD in 12 (38%), and PD in 2 (6%), with 1 (3%) not determined, resulting in an ORR of 53% (17 patients) [49]. The progression-free survival (PFS) rate at 36 months for patients with soft tissue sarcomas was 56% (95% CI: 37–75%; no. at risk = 4) and the overall survival rate at 36 months was 71% (95% CI: 53–90%; no. at risk = 9) [49].
In an integrated analysis of entrectinib in 54 adults with advanced or metastatic solid tumors harboring NTRK fusion genes across three clinical trials (ALKA-372-001, STARTRK-1, and STARTRK-2), ORR was 57% (31 patients; 95% CI: 43.2–70.8), with 4 patients (7%) achieving CR and 27 (50%) achieving PR [17]. The median DoR was 10.4 months (95% CI: 7.1–NE), and the median PFS was 11.2 months (95% CI: 8.0–14.9), with weight gain (10%) and anemia (12%) being the most common grade 3 or 4 adverse effects [17].
In a report that expanded the number of patients to 121, comprising the NTRK efficacy-evaluable population with advanced or metastatic solid tumors, who had received ≥1 entrectinib dose and followed them up for a median of 25.8 months, the ORR was 61.2% (74 patients), CR was achieved in 19 patients (15.7%), and PR was achieved in 55 (45.5%) [82]. High response rates were observed in sarcoma [82]. The median PFS was 13.8 months (95% CI: 10.1–19.9), and the median OS was 33.8 months (95% CI: 23.4–46.4) [82]. Fifteen of the 26 patients with sarcoma (57.7%) responded, including 4 patients (15.4%) with CR and 11 (42.3%) with PR, with a median DoR of 15.0 months (95% CI: 4.6–NE); the median PFS was 10.1 months (95% CI: 6.3–13.7) and the median OS was 18.7 months (95% CI: 14.5–NE) [82].
We encountered a case of long-term stable disease treated with a TRK inhibitor [83] (Fig. 1, 2). Possibly, TRK inhibitors can achieve long-term stable disease without a significant response.
NTRK fusion-positive sarcoma can be genetically classified into simple genomic sarcoma (SGS) and complex genomic sarcoma (CGS) [81]. SGS is characterized by relatively simple genomic alterations, such as NTRK-RSCN, while CGS, associated with complex chromosomal abnormalities (e.g. leiomyosarcoma and MPNST) and genomic instability, may show limited response to TRK inhibitors, warranting further investigation [81].
Timing of TRK inhibitor use in advanced cases
The efficacy of TRK inhibitors as a first-line treatment for NTRK fusion-positive solid tumors has been demonstrated, with high response rates and mild adverse events, although direct comparative studies with standard treatments have not been conducted [67]. The use of TRK inhibitors is recommended as a first-line treatment to avoid the lost opportunity for therapeutic intervention for a patient who would benefit from TRK inhibitors owing to disease progression [67].
An analysis using larotrectinib in the STS model showed that it prolongs estimate lifetime life-years by 5.56 years and increases quality-adjusted life-years by 1.99 years compared with standard treatment (doxorubicin/ifosfamide) [102].
Furthermore, in an analysis using the Growth Modulation Index (GMI), which compares time-to-tumor progression (TTP) under a new therapy (period B) to prior treatment (period A) using GMI = TTP (B)/TTP (A), a GMI >1.33 indicates significant tumor growth modulation [103]. In the analysis, 65% of the patients treated with larotrectinib achieved a GMI ≥1.33 [104], and 65.8% of those treated with entrectinib achieved almost the same criteria [105]. These results suggest that both drugs have a clinical effect that is superior to that of previous treatments.
In an integrated analysis of the NAVIGATE and SCOUT trials, rapid improvements in health-related quality of life were observed in 68% of adults and 71% of pediatric patients within 2 months of initiating larotrectinib [106], suggesting the benefits of using TRK inhibitors as a first-line treatment for NTRK fusion-positive tumors.
Overview of tissue types
Infantile fibrosarcoma
The standard treatment for IFS is surgery alone or surgery combined with chemotherapy. In very young patients, observation may be an option because spontaneous regression may occur [26]. If complete resection is possible, long-term survival is expected, and reoperation is effective in the event of recurrence [36]. Although the recurrence rate is high, the risk of metastasis is low, and the overall prognosis is good [36].
In recent years, TRK inhibitors have shown a very high response rate (96%) to IFS [16] and are used particularly for metastatic and inoperable tumors. Orbach et al. [107] stated that both conventional chemotherapy and TRK inhibitors are considered options for localized disease in patients with infantile fibrosarcoma, depending on the discretion of the physician and parents. However, further research is needed to compare conventional chemotherapy with the first-line treatment. Although conventional chemotherapy is challenging to manage in young patients [107], TRK inhibitors are expected to have an early effect on tumor reduction [98].
NTRK-rearranged spindle cell neoplasm
NTRK-rearranged spindle cell neoplasms have not yet been definitively defined. However, this study refers to LPF-NT, spindle cell tumors with S100 and CD34 co-reactivity resembling MPNST, adult-type fibrosarcoma, spindle cell sarcomas with hemangiopericytic or myopericytoma-like patterns, IMTs harboring fusions involving ALK and other kinases, and spindle cell/sclerosing rhabdomyosarcoma (ssRMS).
Lipofibromatosis-like neural tumor
As shown in Table 2, LPF-NT is a spindle cell tumor that occurs mainly in the soft tissues of children and young adults [24]. Histologically, spindle cells express S100 and CD34 [24,45] and show a characteristic growth pattern of infiltration between adipose tissues [24]. These tumors are associated with NTRK1 gene fusions [24,45], and their clinical course is often relatively benign [24]. LPF-NT was locally invasive in 5 of 12 cases, but there were no cases of metastasis or death; hence, it is thought to be locally invasive but with low metastatic potential [24]. In a case of LPF-NT in which surgical resection of the primary lesion was difficult, enrectinib was administered preoperatively, and the tumor shrank by 45%, making tumor resection possible [48]. Based on these findings, TRK inhibitors may be considered in unresectable cases.
Spindle cell tumors with S100 and CD34 co-reactivity resembling MPNSTs
Spindle cell tumors with S100 and CD34 co-reactivity resemble MPNSTs but exhibit unique histological features, including keloid-like collagen around connective tissues and blood vessels, as previously described [25,30]. The behavior of these tumors varies with cell density and mitotic activity, ranging from low grade with favorable prognosis to higher grade with an increased risk of distant metastasis [25]. Immunohistochemically, they often co-express S100 and CD34, which aids their identification [25]. Genetically, they frequently harbor fusion genes such as NTRK1/2, RAF1, and BRAF, which are important for oncogenesis and they may be amenable to targeted therapy [25].
Adult-type fibrosarcoma
Adult-type fibrosarcoma is histologically characterized by sweeping fascicles of spindle cells that may exhibit storiform architecture, herringbone patterns, hemangiopericytomatous vessels, and mild nuclear atypia in some cases [23,50]. Immunohistochemical analysis revealed positivity for CD34 and pan-TRK in most cases [23,50]. Genetically, this tumor is associated with the NTRK3 [23,50] fusion genes. Although radiological features are not well described, the prognosis varies widely, from low to high malignancy [23,50].
Spindle cell sarcomas with hemangiopericytic or myopericytoma-like patterns
Spindle cell sarcomas with hemangiopericytic or myopericytoma-like patterns are histologically characterized by a growth pattern resembling that of hemangiopericytomas or myopericytomas, often accompanied by high mitotic activity [22]. Immunohistochemically, some cases are positive for smooth muscle actin (SMA) and CD34 [22]. Genetically, these tumors are associated with NTRK1 gene fusion [22]. Radiological imaging reveals hyperintense mass on T2-weighted MRI with intense contrast enhancement and no fatty components [28]. Low-grade sarcomas with myopericytoma or hemangiopericytoma-like morphology generally demonstrate favorable outcomes; however, occasional metastasis may occur [22,28].
Inflammatory myofibroblastic tumors
IMTs are known to harbor fusions involving ALK and other kinases [51–53]. Their histological features are highly diverse, ranging from inflammatory pseudotumors with a paucity of lesional spindle cells to highly cellular tumors, often with prominent myofibroblasts [51]. Musculoskeletal IMTs typically show a nonhomogeneous and solid appearance [54]. While long-term disease-free survival is often observed, it should be noted that some cases may still experience progression of metastases [52,53].
Spindle cell/sclerosing rhabdomyosarcoma
Histologically, ssRMS is characterized by three patterns: fibrosarcoma-like, VGLL2-type, and Triton-like [55]. These patterns exhibit a “fibromatous-like” appearance, characterized by few tumor cells on an abundant sclerotic stroma, with moderate atypia and containing very few mitotic figures [55]. Genetically, ssRMS is associated with fusion genes such as TPM3-NTRK1, SYPL1-BRAF, and TOP2B-RAF1 [55]. Prognostically, fusion-positive ssRMS has a very good outcome [55].
Future perspectives
The long-term safety and efficacy of TRK inhibitors must be clarified through ongoing research. Specifically, long-term follow-up studies are necessary to evaluate its impact on neurocognitive function and development in pediatric patients [108]. Further studies are needed to determine whether TRK inhibitors are as effective as perioperative chemotherapy. Additionally, determining the optimal treatment duration for responders, the best treatment line, drug selection, and strategies to address resistance mutations in advanced stages are key priorities [108]. Furthermore, given that the sequence of TRK inhibitors may influence their efficacy [83,109], research is needed to establish the optimal order of administration.
In addition, data on the differential activities of TRK inhibitors against various NTRK fusion partners are limited [110]. Resistance mechanisms, including on-target mutations, such as TRKA G595R and TRKC G623R [108,111], as well as off-target mechanisms, such as BRAF- and KRAS-activating mutations [112], present significant challenges. Although second-generation TRK inhibitors are under development [113,114], they have not yet been approved for routine clinical use.
Advances in the detection of NTRK fusion genes and TRK inhibitors have revolutionized the treatment and prognosis of these tumors. However, owing to the rarity of NTRK fusion gene–positive tumors, large-scale comparative studies are challenging [115]. International collaboration is essential for addressing these limitations [115]. While providing personalized medicine, accumulating global evidence is crucial for improving patient outcomes worldwide.
Acknowledgements
The authors acknowledge Editage (www.editage.com) for the English language editing.
Conflict of interest: None declared.
Funding
This study was funded in part by the National Cancer Center Research and Development Fund [grant no.: 2023-J-03] and the Agency for Medical Research and Development [grant no.: JP22ck0106764].

{kind=link}
{kind=link}
{kind=link}